Ab initio electronic structure calculation of oxygen vacancies in rutile titanium dioxide Faruque M. Hossain a,b , G. E. Murch a, ⁎ , L. Sheppard b , J. Nowotny b a Diffusion in Solids Group, School of Engineering, The University of Newcastle, Callaghan, NSW 2308, Australia b Centre for Materials Research in Energy Conversion, School of Materials Science and Engineering, The University of New South Wales, NSW 2052, Australia Received 2 February 2006; received in revised form 28 November 2006; accepted 21 December 2006 Abstract The electronic structure of rutile TiO 2 − x is studied using first-principles density functional theory (DFT) calculations. Nonstoichiometry in rutile TiO 2 due to defects in the form of oxygen vacancies leads to a considerable change in the electronic structure. In this paper, we calculate the band structure, density of states, and orbital energy distribution in a reduced (oxygen deficient) TiO 2 − x for different concentrations of oxygen vacancies (x). Energy levels are found to appear inside the forbidden energy region either as an isolated form of bands at different energy levels or merged with the conduction band depending on the value of x and the size of the super cells. © 2007 Elsevier B.V. All rights reserved. Keywords: Electronic structure; Titanium dioxide; Nonstoichiometry; Point defects 1. Introduction Since the pioneering work of Fujishima and Honda [1], TiO 2 has received special attention as a prime candidate material for photo-electrochemical water-splitting and other photo-catalytic applications [2–4]. TiO 2 holds considerable promise due to its chemical stability in aqueous environments and under high energy illumination [3]. However, due to its large band gap, ∼ 3.0–3.2 eV (for rutile and anatase phases respectively), titanium dioxide lacks sensitivity to visible light which is necessary for high performance under solar illumination. This is especially the case for the generation of hydrogen from water to reach commercial efficiencies [3]. Accordingly, efforts have been made to increase the visible light sensitivity through band gap reduction [3,4]. However, an understanding of how the band gap can be manipulated in practice is limited, and made more difficult by discrepancies in the reported band gap literature. In terms of band gap, the rutile phase is preferable due to the lower band gap energy (3.05 eV) than the anatase (3.2 eV) phase. The rutile phase is also the more extensively studied phase of TiO 2 . There are many possibilities of band gap reduction using extrinsic doping with lattice matched foreign atoms, but these doped materials suffer from thermal instability and are associated with higher carrier recombination centers. Such extrinsic doping techniques cannot adequately increase the photo-electrochemical water-splitting efficiency and the effi- ciency of other photo-catalytic devices. It is instructive in this context to note that intrinsic defects such as oxygen vacancies, titanium interstitials, and titanium vacancies in the bulk or on the surface of TiO 2 may introduce defect energy levels inside the band gap. Larger aggregates of such point defects may form energy bands inside the band gap, which in turn may enhance the quantum efficiency of photo-catalytic devices. It is also a matter of concern that the quantum efficiency alone cannot increase the overall efficiency of these devices, unless the electrical conductivity of the electrode material fabricated of TiO 2 is also increased. Hence, to achieve better perfor- mance of such devices both the quantum efficiency and the electrical conductivity should increase concurrently. Pure TiO 2 at room temperature is an electrical insulator. It is well known Solid State Ionics 178 (2007) 319 – 325 www.elsevier.com/locate/ssi ⁎ Corresponding author. E-mail addresses: [email protected] (F.M. Hossain), [email protected] (G.E. Murch). 0167-2738/$ - see front matter © 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.ssi.2006.12.015

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

(2007) 319–325www.elsevier.com/locate/ssi
Solid State Ionics 178
Ab initio electronic structure calculation of oxygenvacancies in rutile titanium dioxide
Faruque M. Hossain a,b, G. E. Murch a,⁎, L. Sheppard b, J. Nowotny b
a Diffusion in Solids Group, School of Engineering, The University of Newcastle, Callaghan, NSW 2308, Australiab Centre for Materials Research in Energy Conversion, School of Materials Science and Engineering, The University of New South Wales, NSW 2052, Australia
Received 2 February 2006; received in revised form 28 November 2006; accepted 21 December 2006
Abstract
The electronic structure of rutile TiO2− x is studied using first-principles density functional theory (DFT) calculations. Nonstoichiometry inrutile TiO2 due to defects in the form of oxygen vacancies leads to a considerable change in the electronic structure. In this paper, we calculate theband structure, density of states, and orbital energy distribution in a reduced (oxygen deficient) TiO2− x for different concentrations of oxygenvacancies (x). Energy levels are found to appear inside the forbidden energy region either as an isolated form of bands at different energy levels ormerged with the conduction band depending on the value of x and the size of the super cells.© 2007 Elsevier B.V. All rights reserved.
Keywords: Electronic structure; Titanium dioxide; Nonstoichiometry; Point defects
1. Introduction
Since the pioneering work of Fujishima and Honda [1], TiO2
has received special attention as a prime candidate material forphoto-electrochemical water-splitting and other photo-catalyticapplications [2–4]. TiO2 holds considerable promise due to itschemical stability in aqueous environments and under highenergy illumination [3]. However, due to its large band gap,∼3.0–3.2 eV (for rutile and anatase phases respectively),titanium dioxide lacks sensitivity to visible light which isnecessary for high performance under solar illumination. This isespecially the case for the generation of hydrogen from water toreach commercial efficiencies [3]. Accordingly, efforts havebeen made to increase the visible light sensitivity through bandgap reduction [3,4]. However, an understanding of how theband gap can be manipulated in practice is limited, and mademore difficult by discrepancies in the reported band gapliterature. In terms of band gap, the rutile phase is preferable due
⁎ Corresponding author.E-mail addresses: [email protected] (F.M. Hossain),
[email protected] (G.E. Murch).
0167-2738/$ - see front matter © 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.ssi.2006.12.015
to the lower band gap energy (3.05 eV) than the anatase (3.2 eV)phase. The rutile phase is also the more extensively studiedphase of TiO2.
There are many possibilities of band gap reduction usingextrinsic doping with lattice matched foreign atoms, but thesedoped materials suffer from thermal instability and areassociated with higher carrier recombination centers. Suchextrinsic doping techniques cannot adequately increase thephoto-electrochemical water-splitting efficiency and the effi-ciency of other photo-catalytic devices. It is instructive in thiscontext to note that intrinsic defects such as oxygen vacancies,titanium interstitials, and titanium vacancies in the bulk or onthe surface of TiO2 may introduce defect energy levels insidethe band gap. Larger aggregates of such point defects mayform energy bands inside the band gap, which in turn mayenhance the quantum efficiency of photo-catalytic devices. Itis also a matter of concern that the quantum efficiency alonecannot increase the overall efficiency of these devices, unlessthe electrical conductivity of the electrode material fabricatedof TiO2 is also increased. Hence, to achieve better perfor-mance of such devices both the quantum efficiency and theelectrical conductivity should increase concurrently. Pure TiO2
at room temperature is an electrical insulator. It is well known

Fig. 1. A two dimensional view of the 2×2×2 super-cell structure of rutile TiO2
(Ti16O30; 6.25% oxygen vacancy). Two oxygen atoms are removed fromneighboring octahedra of a stoichiometric super cell (Ti16O32).
320 F.M. Hossain et al. / Solid State Ionics 178 (2007) 319–325
that the electrical conductivity of TiO2 varies with the dopanttypes, O/Ti atomic ratio, and its preparation conditions (heattreatment and variation in oxygen partial pressure). In the caseof reduced rutile TiO2, it has been suggested [5] that oxygenvacancies and Ti3+ interstitials may be the dominant defects inthis system. However, it is not clearly evident that intrinsicdoping produces isolated energy levels inside the band gap orproduce energy states that merge with the valence orconduction band to produce a concrete band gap reduction.There have been few attempts of intrinsic doping to increasethe conductivity; however, no specific steps have been takento investigate the energy band structure along the forbiddenenergy region of TiO2.
Experimental results of Cronemeyer [6] and Ghosh et al. [7]show the effect of intrinsic point defects in forming localenergy levels inside the band gap of rutile TiO2. Infraredabsorption spectroscopy studies of Cronemeyer [6] showedthat the oxygen vacancies produce two energy levels at0.75 eV and 1.18 eV below the conduction band edge for thefirst and second O-vacancy ionizations respectively. Hisweight-loss experiment shows that there must be a transitionfrom singly ionized O vacancies to doubly ionized O vacanciesat room temperature as the number of O vacancies increases toa high concentration. The second ionization occurs due to theinteraction of the individual donor centers for a high con-centration of O vacancies. However, Ghosh et al. [7] identifiedeight (0.27, 0.28, 0.39, 0.48, 0.56, 0.62, 0.76, and 0.87 eVbelow the conduction band edge) discrete energy levels on aslightly reduced rutile TiO2 sample and proposed that at leastone level would be for an O vacancy.
There have been few theoretical attempts that can becompared with the experimental results of Cronemeyer andGhosh and colleagues. Munnix et al. [8] have investigated therole of ideal, undistorted oxygen vacancies in a small unit cell ofrutile TiO2 by a scattering theoretical method using a classicalGreen's function. However, neither lattice constant nor ionssurrounding O vacancies were allowed to relax throughout theircalculations. They found that bulk oxygen vacancies have nocontribution in inducing any energy levels inside the band gapof rutile TiO2, rather lead to a pinning of the Fermi level near theconduction band edge. In contrast, using ab initio self-consistent pseudopotential total-energy calculations, Rama-morthy et al. [9] found that bulk oxygen vacancies introducedefect energy levels inside the band gap at 0.3 eV below theedge of the conduction band. They modeled a bulk-likedefective super cell by removing an oxygen atom from thecentral layer in the slab geometry without consideration ofvacuum layers and allowed the entire slab to relax. It may beconcluded based on results of two calculations [Munnix et al.and Ramamorthy et al.] that the structural relaxation in thepresence of oxygen vacancies is responsible for introducingdefect energy levels inside the band gap of bulk rutile TiO2. Onthe other hand, a semi-empirical self-consistent method usingthe tight-binding model of Yu and Halley [10] found that asingle oxygen vacancy in a unit cell imposes isolated energystates at 0.7 eV below the conduction band and two vacanciesimpose two individual energy levels (shallow and deep)
depending on the position of oxygen vacancy in the octahedron.Recent electronic structure calculations of Cho et al. [11] onpoint defects in rutile TiO2− x showed that the oxygen vacancyis very sensitive to the super-cell size and does not introduceany energy levels within the band gap. Although they examinedatomic relaxations around the vacancy, nonetheless their resultsdisagree with the experimental results of Cronemeyer [6]and Ghosh et al. [7] and the theoretical investigations ofRamamorthy et al. [9] and Yu and Halley [10] as discussedabove.
There is therefore a considerable degree of controversy in theexperimental observations and different methods of theoreticalcalculation. In this paper, we theoretically investigate the natureand activity of defect states developed by oxygen vacancies athigh concentrations (up to 12.5%) in rutile TiO2 using the first-principles total energy density functional (DFT) method. Wealso carry out calculations to study the effect of super-cell sizeand the higher concentration of oxygen vacancies in bulk rutileTiO2 in order to detect the position of vacancy-induced energylevels and investigate the tendency of band formation inside theband gap.
2. Method of calculation
Two different sizes (2×2×2 and 2×2×3) of super cell ofrutile TiO2 were used in this calculation. For the 2×2×2 super-cell oxygen atoms (1, 2, 3, and 4) from neighboring octahedra ofthe structure were removed in sequential order to model adifferent concentration of oxygen vacancies (3.125, 6.25, 9.375,and 12.5%) respectively. Similarly, for the 2×2×3 super-cell 1,3, 4 and 6 oxygen atoms were removed to make 2.08%, 6.25%,8.33% and 12.5% oxygen vacancies respectively. Fig. 1 showsan example of a 6.25% oxygen deficient 2×2×2 super-cellstructure, where two oxygen atoms are removed fromneighboring octahedra. Calculations are based on the density
321F.M. Hossain et al. / Solid State Ionics 178 (2007) 319–325
functional theory (DFT) in the generalized gradient approx-imation (GGA-PBE) proposed by Perdew et al. [12] andemployed Vanderbilt ultrasoft pseudopotentials (USP) [13]for electron–ion interactions. Electron wave functions at eachk-point were expanded in terms of a discrete plane-wave basisset, and the sum of electronic eigenvalues was minimizedusing a conjugate-gradient based approach as implemented inthe computer code CASTEP [14–16]. All atoms in 2×2×2and 2×2×3 stoichiometric super cell were allowed to relax
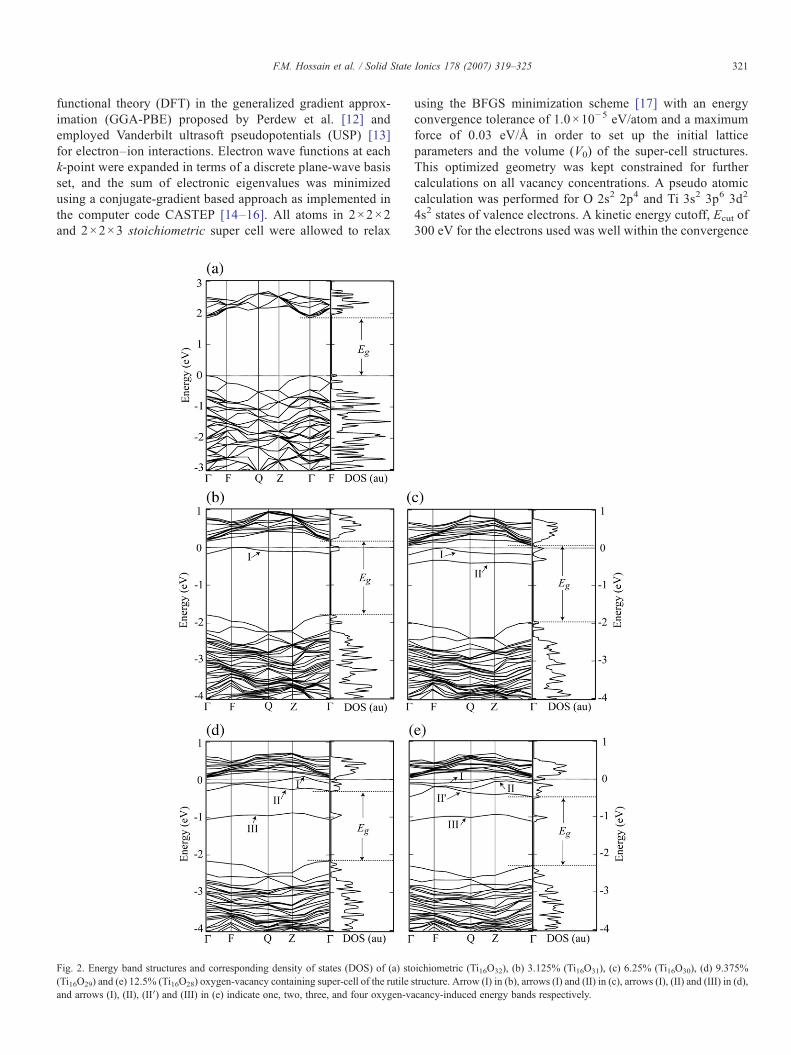
Fig. 2. Energy band structures and corresponding density of states (DOS) of (a) sto(Ti16O29) and (e) 12.5% (Ti16O28) oxygen-vacancy containing super-cell of the rutile sand arrows (I), (II), (II′) and (III) in (e) indicate one, two, three, and four oxygen-va
using the BFGS minimization scheme [17] with an energyconvergence tolerance of 1.0×10−5 eV/atom and a maximumforce of 0.03 eV/Å in order to set up the initial latticeparameters and the volume (V0) of the super-cell structures.This optimized geometry was kept constrained for furthercalculations on all vacancy concentrations. A pseudo atomiccalculation was performed for O 2s2 2p4 and Ti 3s2 3p6 3d2
4s2 states of valence electrons. A kinetic energy cutoff, Ecut of300 eV for the electrons used was well within the convergence
ichiometric (Ti16O32), (b) 3.125% (Ti16O31), (c) 6.25% (Ti16O30), (d) 9.375%tructure. Arrow (I) in (b), arrows (I) and (II) in (c), arrows (I), (II) and (III) in (d),cancy-induced energy bands respectively.

322 F.M. Hossain et al. / Solid State Ionics 178 (2007) 319–325
of a total-energy calculation. For k-point sampling, symme-trized (2×2×3) and (2×2×2) Monkhorst–Pack uniformmeshes [18] in the Brillouin zone were employed for2×2×2 and 2×2×3 super cells respectively.
3. Results and discussion
The electronic structures (band structure and DOS) werecalculated on relaxed geometry of 2×2×2 and 2×2×3stoichiometric super cells including 3.125% and 6.25% vacancycontaining 2×2×2 super cell. The results for 2×2×2 super cellare specifically illustrated as shown in Fig. 2 and elaboratelydiscussed. A synopsis of this result is tabulated in Table 1 withthe results obtained for 2×2×3 super cell for a quick view andcomparison. Fig. 2(a) shows the position of the Fermi level, Ef,at the top of the valence band comprised of the 2p band of O−2,which is completely filled and the conduction band comprisedof 3d band of Ti+4 which is completely empty, representing aninsulating crystal with an intrinsic energy gap of 2.0 eV. Thisband gap energy is much lower than the experimentallyobserved band gap of 3.05 eV, due to the well-knowndrawbacks of DFT using GGA [19]. The density of stateswere also calculated using a much lower smearing width(Gaussian broadening applied to the eigenvalues) of5.0×10−3 eV than the kT value for the detection of isolatedenergy states. No energy states were formed inside the bandgap.
As mentioned above, the lattice parameters and cell volumefor all structures of different oxygen-vacancy concentrationswere kept constrained to that of the relaxed stoichiometric supercell, hence it is worth testing the structural optimization and thecorresponding change of electronic structure within anacceptable tolerance at least at lower vacancy concentrations.Only the 2×2×2 super cell was taken into consideration forrelaxation purposes. First, our test structure containing a singleoxygen vacancy (Ti16O31) was optimized with a relaxed cellvolume change [(V0−V) /V0] of 7.6%. All octahedra (TiO6)
Table 1Effective band gap and vacancy-induced energy levels inside the band gap fordifferent concentration of oxygen vacancies for a 2×2×2 and a 2×2×3 supercell of rutile TiO2
% oxygenvacancies
Effective band gap, Eg=Ec
−Ev (eV)Vacancy-induced energy bandbelow Ec (eV)
2×2×2 super cell0 1.85 –3.125 1.95 0.226.25 2.04 0.13, 0.419.375 1.87 0.26, 0.93 (⁎Et)12.5 1.84 0.44, 0.98 (⁎Et)
2×2×3 super cell0 1.85 –2.08 1.92 0.136.25 2.07 (−)0.46 to (−)0.128.33 1.68 (−)0.26 to (+)0.2112.5 1.61 (−)0.41 to (+)0.18
⁎Et = deep level defect states; (−) and (+) indicate energy levels below and abovethe Fermi level.
deform systematically with an elongation along the [ 1̄ 10]crystal direction. Second, our test structure containing twooxygen vacancies (Ti16O30) was also successfully optimizedwith a relaxed cell volume change [(V0−V) /V0] of 24.2% thatshowed a random deformation of octahedra. We did not proceedto test the structural optimization for the rutile structure ofhigher oxygen-vacancy concentrations (three and four oxygenvacancies in the 2×2×2 super cells) due to such irregulardeformations. However, in order to verify the reliability of ourcalculations on unrelaxed structures we compare these two testelectronic structures on the relaxed geometry with thepreviously reported theoretical calculations.
For the relaxed Ti16O31 super-cell structure, an energy bandwith a peak at 0.2 eV below Fermi level is formed associatedwith single oxygen vacancy that merges with the conductionband. This vacancy-induced energy band mixing with theconduction band is confirmed using a scissors operator, whichcan rigidly shift unoccupied conduction band states away fromthe occupied energy states. In this case, an applied scissorsoperator fails to make a rigid shift of the conduction band,because vacancy-induced energy states pin the Fermi level upabove the conduction band edge. The orbitals associated withthis vacancy-induced energy level are localized to the Ti atomssurrounding the oxygen vacancy. The estimated band gap isincreased to ≈2.0 eV from stoichiometric case of 1.85 eV, dueto energy states disappearance from the top of the valence bandassociated with the removed oxygen atom. This result is insemi-quantitative agreement with the previous ab initio self-consistent pseudopotential total-energy calculations of Rama-morthy et al. [9] and the recent theoretical calculations of Choet al. [11]. There seems to be a little controversy with the resultsof Cho et al., which arises due to their estimated band gapenergy of 1.7 eV and their consideration of a single k-point (Γpoint). In our calculation on the other hand, we have used a(2×2×3) mesh for k-point integration and our estimated bandgap is 1.85 eV, which is very close to the previously reportedresults of Glassford et al. [20] and Lee et al. [21]. If we compareour band structure with Fig. 2(d) of Cho et al. by aligning thevalence band maximum, it may then be realized that the positionof oxygen-vacancy-induced energy state will align at the sameenergy level. However, a large variation of dispersion in thedefect state may occur due to the difference in the considerationof k-points.
For the relaxed Ti16O30 super cell structure, two isolatedenergy bands with a peak at 0.07 eVand 0.3 eV below the Fermilevel are formed that are related to double oxygen vacancies.These two energy bands are found isolated from the conductionband, which is also tested by applying a scissors operator. Theestimated band gap is increased ≈0.5 eV due to furtherreduction of energy states from the top of the valence band forhigher oxygen-vacancy concentration. This result for doubleoxygen vacancies is also comparable with the results of theapical divacancy of Cho et al. [11]. They identified an in-gapstate located about 0.2 eV below the conduction bandminimum, which is equivalent to our results of 0.3 eV belowthe Fermi level. It is worthwhile mentioning once again thatsuch a little discrepancy arises from the difference in band gap
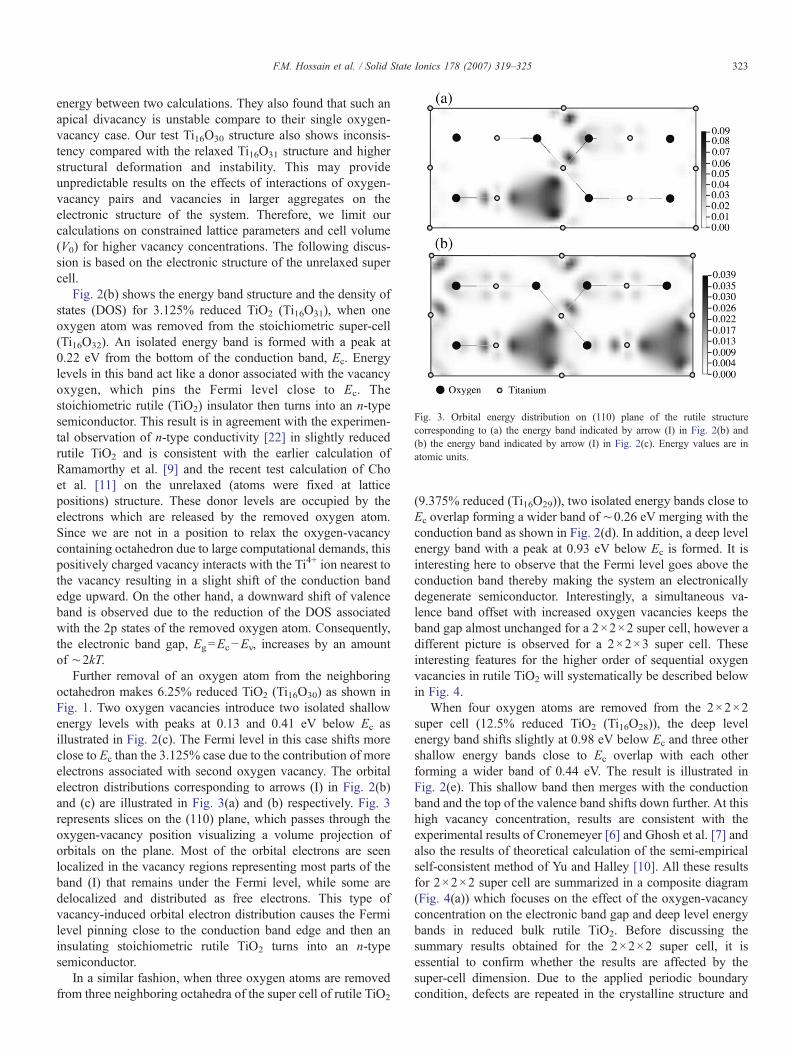
Fig. 3. Orbital energy distribution on (110) plane of the rutile structurecorresponding to (a) the energy band indicated by arrow (I) in Fig. 2(b) and(b) the energy band indicated by arrow (I) in Fig. 2(c). Energy values are inatomic units.
323F.M. Hossain et al. / Solid State Ionics 178 (2007) 319–325
energy between two calculations. They also found that such anapical divacancy is unstable compare to their single oxygen-vacancy case. Our test Ti16O30 structure also shows inconsis-tency compared with the relaxed Ti16O31 structure and higherstructural deformation and instability. This may provideunpredictable results on the effects of interactions of oxygen-vacancy pairs and vacancies in larger aggregates on theelectronic structure of the system. Therefore, we limit ourcalculations on constrained lattice parameters and cell volume(V0) for higher vacancy concentrations. The following discus-sion is based on the electronic structure of the unrelaxed supercell.
Fig. 2(b) shows the energy band structure and the density ofstates (DOS) for 3.125% reduced TiO2 (Ti16O31), when oneoxygen atom was removed from the stoichiometric super-cell(Ti16O32). An isolated energy band is formed with a peak at0.22 eV from the bottom of the conduction band, Ec. Energylevels in this band act like a donor associated with the vacancyoxygen, which pins the Fermi level close to Ec. Thestoichiometric rutile (TiO2) insulator then turns into an n-typesemiconductor. This result is in agreement with the experimen-tal observation of n-type conductivity [22] in slightly reducedrutile TiO2 and is consistent with the earlier calculation ofRamamorthy et al. [9] and the recent test calculation of Choet al. [11] on the unrelaxed (atoms were fixed at latticepositions) structure. These donor levels are occupied by theelectrons which are released by the removed oxygen atom.Since we are not in a position to relax the oxygen-vacancycontaining octahedron due to large computational demands, thispositively charged vacancy interacts with the Ti4+ ion nearest tothe vacancy resulting in a slight shift of the conduction bandedge upward. On the other hand, a downward shift of valenceband is observed due to the reduction of the DOS associatedwith the 2p states of the removed oxygen atom. Consequently,the electronic band gap, Eg=Ec−Ev, increases by an amountof ∼2kT.
Further removal of an oxygen atom from the neighboringoctahedron makes 6.25% reduced TiO2 (Ti16O30) as shown inFig. 1. Two oxygen vacancies introduce two isolated shallowenergy levels with peaks at 0.13 and 0.41 eV below Ec asillustrated in Fig. 2(c). The Fermi level in this case shifts moreclose to Ec than the 3.125% case due to the contribution of moreelectrons associated with second oxygen vacancy. The orbitalelectron distributions corresponding to arrows (I) in Fig. 2(b)and (c) are illustrated in Fig. 3(a) and (b) respectively. Fig. 3represents slices on the (110) plane, which passes through theoxygen-vacancy position visualizing a volume projection oforbitals on the plane. Most of the orbital electrons are seenlocalized in the vacancy regions representing most parts of theband (I) that remains under the Fermi level, while some aredelocalized and distributed as free electrons. This type ofvacancy-induced orbital electron distribution causes the Fermilevel pinning close to the conduction band edge and then aninsulating stoichiometric rutile TiO2 turns into an n-typesemiconductor.
In a similar fashion, when three oxygen atoms are removedfrom three neighboring octahedra of the super cell of rutile TiO2
(9.375% reduced (Ti16O29)), two isolated energy bands close toEc overlap forming a wider band of ∼0.26 eV merging with theconduction band as shown in Fig. 2(d). In addition, a deep levelenergy band with a peak at 0.93 eV below Ec is formed. It isinteresting here to observe that the Fermi level goes above theconduction band thereby making the system an electronicallydegenerate semiconductor. Interestingly, a simultaneous va-lence band offset with increased oxygen vacancies keeps theband gap almost unchanged for a 2×2×2 super cell, however adifferent picture is observed for a 2×2×3 super cell. Theseinteresting features for the higher order of sequential oxygenvacancies in rutile TiO2 will systematically be described belowin Fig. 4.
When four oxygen atoms are removed from the 2×2×2super cell (12.5% reduced TiO2 (Ti16O28)), the deep levelenergy band shifts slightly at 0.98 eV below Ec and three othershallow energy bands close to Ec overlap with each otherforming a wider band of 0.44 eV. The result is illustrated inFig. 2(e). This shallow band then merges with the conductionband and the top of the valence band shifts down further. At thishigh vacancy concentration, results are consistent with theexperimental results of Cronemeyer [6] and Ghosh et al. [7] andalso the results of theoretical calculation of the semi-empiricalself-consistent method of Yu and Halley [10]. All these resultsfor 2×2×2 super cell are summarized in a composite diagram(Fig. 4(a)) which focuses on the effect of the oxygen-vacancyconcentration on the electronic band gap and deep level energybands in reduced bulk rutile TiO2. Before discussing thesummary results obtained for the 2×2×2 super cell, it isessential to confirm whether the results are affected by thesuper-cell dimension. Due to the applied periodic boundarycondition, defects are repeated in the crystalline structure and
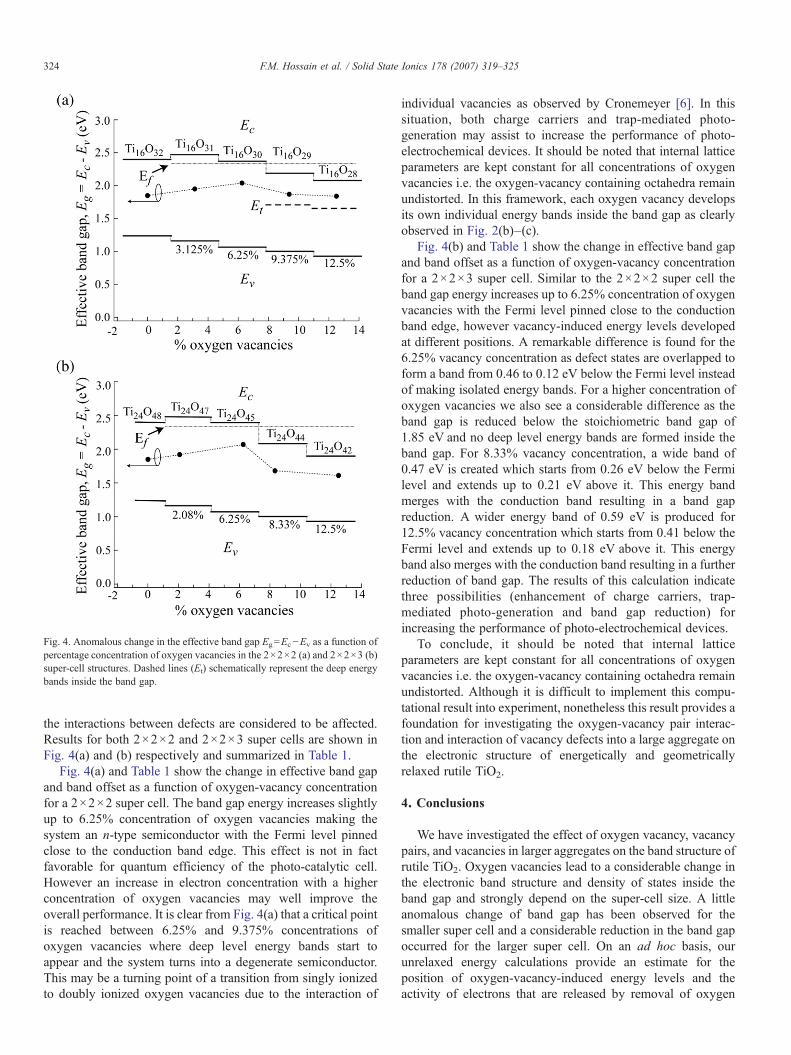
Fig. 4. Anomalous change in the effective band gap Eg=Ec−Ev as a function ofpercentage concentration of oxygen vacancies in the 2×2×2 (a) and 2×2×3 (b)super-cell structures. Dashed lines (Et) schematically represent the deep energybands inside the band gap.
324 F.M. Hossain et al. / Solid State Ionics 178 (2007) 319–325
the interactions between defects are considered to be affected.Results for both 2×2×2 and 2×2×3 super cells are shown inFig. 4(a) and (b) respectively and summarized in Table 1.
Fig. 4(a) and Table 1 show the change in effective band gapand band offset as a function of oxygen-vacancy concentrationfor a 2×2×2 super cell. The band gap energy increases slightlyup to 6.25% concentration of oxygen vacancies making thesystem an n-type semiconductor with the Fermi level pinnedclose to the conduction band edge. This effect is not in factfavorable for quantum efficiency of the photo-catalytic cell.However an increase in electron concentration with a higherconcentration of oxygen vacancies may well improve theoverall performance. It is clear from Fig. 4(a) that a critical pointis reached between 6.25% and 9.375% concentrations ofoxygen vacancies where deep level energy bands start toappear and the system turns into a degenerate semiconductor.This may be a turning point of a transition from singly ionizedto doubly ionized oxygen vacancies due to the interaction of
individual vacancies as observed by Cronemeyer [6]. In thissituation, both charge carriers and trap-mediated photo-generation may assist to increase the performance of photo-electrochemical devices. It should be noted that internal latticeparameters are kept constant for all concentrations of oxygenvacancies i.e. the oxygen-vacancy containing octahedra remainundistorted. In this framework, each oxygen vacancy developsits own individual energy bands inside the band gap as clearlyobserved in Fig. 2(b)–(c).
Fig. 4(b) and Table 1 show the change in effective band gapand band offset as a function of oxygen-vacancy concentrationfor a 2×2×3 super cell. Similar to the 2×2×2 super cell theband gap energy increases up to 6.25% concentration of oxygenvacancies with the Fermi level pinned close to the conductionband edge, however vacancy-induced energy levels developedat different positions. A remarkable difference is found for the6.25% vacancy concentration as defect states are overlapped toform a band from 0.46 to 0.12 eV below the Fermi level insteadof making isolated energy bands. For a higher concentration ofoxygen vacancies we also see a considerable difference as theband gap is reduced below the stoichiometric band gap of1.85 eV and no deep level energy bands are formed inside theband gap. For 8.33% vacancy concentration, a wide band of0.47 eV is created which starts from 0.26 eV below the Fermilevel and extends up to 0.21 eV above it. This energy bandmerges with the conduction band resulting in a band gapreduction. A wider energy band of 0.59 eV is produced for12.5% vacancy concentration which starts from 0.41 below theFermi level and extends up to 0.18 eV above it. This energyband also merges with the conduction band resulting in a furtherreduction of band gap. The results of this calculation indicatethree possibilities (enhancement of charge carriers, trap-mediated photo-generation and band gap reduction) forincreasing the performance of photo-electrochemical devices.
To conclude, it should be noted that internal latticeparameters are kept constant for all concentrations of oxygenvacancies i.e. the oxygen-vacancy containing octahedra remainundistorted. Although it is difficult to implement this compu-tational result into experiment, nonetheless this result provides afoundation for investigating the oxygen-vacancy pair interac-tion and interaction of vacancy defects into a large aggregate onthe electronic structure of energetically and geometricallyrelaxed rutile TiO2.
4. Conclusions
We have investigated the effect of oxygen vacancy, vacancypairs, and vacancies in larger aggregates on the band structure ofrutile TiO2. Oxygen vacancies lead to a considerable change inthe electronic band structure and density of states inside theband gap and strongly depend on the super-cell size. A littleanomalous change of band gap has been observed for thesmaller super cell and a considerable reduction in the band gapoccurred for the larger super cell. On an ad hoc basis, ourunrelaxed energy calculations provide an estimate for theposition of oxygen-vacancy-induced energy levels and theactivity of electrons that are released by removal of oxygen

325F.M. Hossain et al. / Solid State Ionics 178 (2007) 319–325
atoms. This result concludes that the intrinsic defects in a formof oxygen vacancies with a larger vacancy aggregate can beexpected to decrease the band gap energy, increase chargecarriers, and introduce intermediate bands inside the band gap.Therefore, these vacancies may enhance the overall efficiencyof a TiO2-based photo-electrochemical device. Further inves-tigations are necessary on the optimized structure for suchoxygen vacancies in the bulk rutile TiO2 to observe how theoctahedra in rutile structure distort and how released electronsfrom removed oxygen atoms redistribute in the reduced TiO2− x
structure.
Acknowledgment
We wish to thank the Australian Research Council(Discovery and Linkage Project Grants Schemes) for its supportof this research.
References
[1] A. Fujishima, K. Honda, Nature (London) 37 (1972) 238.[2] A.L. Linsebigler, G.Q. Lu, J.T. Yates, Chem. Rev. 95 (1995) 735.[3] T. Bak, J. Nowotny, M. Rekas, C.C. Sorrell, Int. J. Hydrogen Energy 27
(2002) 991.
[4] S.U.M. Khan, M. Al-Shahry, W.B. Ingler Jr., Science 297 (2002) 2243.[5] P.F. Chester, J. Appl. Phys. 32 (1961) 2233.[6] D.C. Cronemeyer, Phys. Rev. 113 (1959) 1222.[7] A.K. Ghosh, F.G. Wakim, R.R. Addiss Jr., Phys. Rev. 184 (1969) 979.[8] S. Munnix, M. Schmeits, Solid State Commun. 50 (1984) 1087.[9] M. Ramamorthy, R.D. King-Smith, D. Vanderbilt, Phys. Rev., B 49 (1994)
7709.[10] N. Yu, J.W. Halley, Phys. Rev., B 51 (1995) 4768.[11] E. Cho, S. Han, H.-S. Ahn, K.-R. Lee, S.K. Kim, C.S. Hwang, Phys. Rev.,
B 73 (2006) 193202.[12] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.[13] D. Vanderbilt, Phys. Rev., B 41 (1990) 7892.[14] M.C. Payne, M.P. Teter, D.C. Allan, T.A. Arias, J.D. Joannopoulos, Rev.
Mod. Phys. 64 (1992) 1045.[15] L.J. Clarke, I. Stich, M.C. Payne, Comput. Phys. Commun. 72 (1992) 14.[16] M.D. Segall, P.L.D. Lindan, M.J. Probert, C.J. Pickard, P.J. Hasnip, S.J.
Clark, M.C. Payne, J. Phys., Condens. Matter 14 (2002) 2717.[17] T.H. Fischer, J. Almlof, J. Phys. Chem. 96 (1992) 9768.[18] H.J. Monkhorst, J.D. Pack, Phys. Rev., B 13 (1976) 5188 Phys. Rev., B 16
(1977) 1748.[19] C. Stampfl, C.G. Van de Wall, Phys. Rev., B 59 (1999) 5521.[20] K.M. Glassford, J.R. Chelikowsky, Phys. Rev., B 46 (1992) 1284.[21] C. Lee, P. Ghosez, X. Gonze, Phys. Rev., B 50 (1994) 13379.[22] R.R. Hashiguti, E. Yagi, Phys. Rev., B 49 (1994) 7251.
Related Documents











