Analytica Chimica Acta 584 (2007) 295–301 A solid paraffin-based carbon paste electrode modified with 2-aminothiazole organofunctionalized silica for differential pulse adsorptive stripping analysis of nickel in ethanol fuel Regina M. Takeuchi a,∗ , Andr´ e L. Santos a , Pedro M. Padilha b , Nelson R. Stradiotto a a Departamento de Qu´ ımica Anal´ ıtica, Instituto de Qu´ ımica, UNESP, CP 355, 14801-970 Araraquara, SP, Brazil b Departamento de Qu´ ımica e Bioqu´ ımica—IB/UNESP, CP 510, 18618-000 Botucatu, SP, Brazil Received 13 September 2006; received in revised form 21 November 2006; accepted 27 November 2006 Available online 3 December 2006 Abstract A solid paraffin-based carbon paste electrode modified with 2-aminothiazole organofunctionalized silica (SiAt-SPCPE) was applied to Ni 2+ determination in commercial ethanol fuel samples. The proposed method comprised four steps: (1) Ni 2+ preconcentration at open circuit potential directly in the ethanol fuel sample, (2) transference of the electrode to an electrochemical cell containing DMG, (3) differential pulse voltammogram registering and (4) surface regeneration by polishing the electrode. The proposed method combines the high Ni 2+ adsorption capacity presented by 2-aminothiazole organofunctionalized silica with the electrochemical properties of the Ni(DMG) 2 complex, whose electrochemical reduction provides the analytical signal. All experimental parameters involved in the proposed method were optimized. Using a preconcentration time of 20 min, it was obtained a linear range from 7.5 × 10 −9 to 1.0 × 10 −6 mol L −1 with detection limit of 2.0 × 10 −9 mol L −1 . Recovery values between 96.5 and 102.4% were obtained for commercial samples spiked with 1.0 mol L −1 Ni 2+ and the developed electrode was totally stable in ethanolic solutions. The contents of Ni 2+ found in the commercial samples using the proposed method were compared to those obtained by graphite furnace atomic absorption spectroscopy by using the F- and t-test. Neither the F- nor t-values exceeded the critical values at 95% confidence level, confirming that there are not statistical differences between the results obtained by both methods. These results indicate that the developed electrode can be successfully employed to reliable Ni 2+ determination in commercial ethanol fuel samples without any sample pretreatment or dilution step. © 2006 Elsevier B.V. All rights reserved. Keywords: Nickel; Ethanol fuel; Solid paraffin-based carbon paste electrodes; Organofunctionalized silica; 2-Aminothiazole; Differential pulse stripping voltammetry 1. Introduction Ethanol obtained from sugar cane has been used as an auto- motive fuel in Brazil since the 1970s [1]. This fuel was initially used as an additive in gasoline, which enhances its octane number, replacing the organolead compounds [2,3]. Nowadays, ethanol is used in Brazil predominantly as a transport fuel with both economic and environment benefits [4]. Not only it presents an attractive alternative and renewable biological fuel source, but also allows a significant decrease of CO 2 emission levels in Brazil. For all this, ethanol will surely be a key fuel source worldwide in the very near future. ∗ Corresponding author at: Av. Prof. Francisco Degni s/n, CEP14800-900, Araraquara, SP, Brazil. Tel.: +55 16 3301 6621; fax: +55 16 3322 7932. E-mail address: [email protected] (R.M. Takeuchi). The determination of metals in ethanol fuel is especially difficult due to their low concentrations and lack of certified ref- erence samples. However, even in low concentrations, metallic species can cause damage to automobile engines [5] and to the environment [6]. The main sources of metal ions are associated to the ethanol production process, storage and transportation [7]. The determination of metals by electrothermal atomic absorp- tion spectroscopy in ethanol fuel is well established [6,8,9] and flame atomic absorption spectroscopy (FAAS) can be used after a preconcentration step [10–12]. Stripping voltammetric techniques have become very pop- ular for metal ions analysis [13,14]. These techniques provide high analytical performance with relatively low instrumental costs and some works have shown that stripping voltammetric techniques can be successfully applied to metal ions determi- nation in ethanol fuel [15–18]. The introduction of a chemical 0003-2670/$ – see front matter © 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.aca.2006.11.069

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

A
ddrbp
rffbdr©
K
1
munebabiw
A
0d
Analytica Chimica Acta 584 (2007) 295–301
A solid paraffin-based carbon paste electrode modified with2-aminothiazole organofunctionalized silica for differentialpulse adsorptive stripping analysis of nickel in ethanol fuel
Regina M. Takeuchi a,∗, Andre L. Santos a, Pedro M. Padilha b, Nelson R. Stradiotto a
a Departamento de Quımica Analıtica, Instituto de Quımica, UNESP, CP 355, 14801-970 Araraquara, SP, Brazilb Departamento de Quımica e Bioquımica—IB/UNESP, CP 510, 18618-000 Botucatu, SP, Brazil
Received 13 September 2006; received in revised form 21 November 2006; accepted 27 November 2006Available online 3 December 2006
bstract
A solid paraffin-based carbon paste electrode modified with 2-aminothiazole organofunctionalized silica (SiAt-SPCPE) was applied to Ni2+
etermination in commercial ethanol fuel samples. The proposed method comprised four steps: (1) Ni2+ preconcentration at open circuit potentialirectly in the ethanol fuel sample, (2) transference of the electrode to an electrochemical cell containing DMG, (3) differential pulse voltammogramegistering and (4) surface regeneration by polishing the electrode. The proposed method combines the high Ni2+ adsorption capacity presentedy 2-aminothiazole organofunctionalized silica with the electrochemical properties of the Ni(DMG)2 complex, whose electrochemical reductionrovides the analytical signal.
All experimental parameters involved in the proposed method were optimized. Using a preconcentration time of 20 min, it was obtained a linearange from 7.5 × 10−9 to 1.0 × 10−6 mol L−1 with detection limit of 2.0 × 10−9 mol L−1. Recovery values between 96.5 and 102.4% were obtainedor commercial samples spiked with 1.0 �mol L−1 Ni2+ and the developed electrode was totally stable in ethanolic solutions. The contents of Ni2+
ound in the commercial samples using the proposed method were compared to those obtained by graphite furnace atomic absorption spectroscopy
y using the F- and t-test. Neither the F- nor t-values exceeded the critical values at 95% confidence level, confirming that there are not statisticalifferences between the results obtained by both methods. These results indicate that the developed electrode can be successfully employed toeliable Ni2+ determination in commercial ethanol fuel samples without any sample pretreatment or dilution step.2006 Elsevier B.V. All rights reserved.
ganof
desetT
eywords: Nickel; Ethanol fuel; Solid paraffin-based carbon paste electrodes; Or
. Introduction
Ethanol obtained from sugar cane has been used as an auto-otive fuel in Brazil since the 1970s [1]. This fuel was initially
sed as an additive in gasoline, which enhances its octaneumber, replacing the organolead compounds [2,3]. Nowadays,thanol is used in Brazil predominantly as a transport fuel with
oth economic and environment benefits [4]. Not only it presentsn attractive alternative and renewable biological fuel source,ut also allows a significant decrease of CO2 emission levelsn Brazil. For all this, ethanol will surely be a key fuel sourceorldwide in the very near future.∗ Corresponding author at: Av. Prof. Francisco Degni s/n, CEP14800-900,raraquara, SP, Brazil. Tel.: +55 16 3301 6621; fax: +55 16 3322 7932.
E-mail address: [email protected] (R.M. Takeuchi).
tfla
uhctn
003-2670/$ – see front matter © 2006 Elsevier B.V. All rights reserved.oi:10.1016/j.aca.2006.11.069
unctionalized silica; 2-Aminothiazole; Differential pulse stripping voltammetry
The determination of metals in ethanol fuel is especiallyifficult due to their low concentrations and lack of certified ref-rence samples. However, even in low concentrations, metallicpecies can cause damage to automobile engines [5] and to thenvironment [6]. The main sources of metal ions are associatedo the ethanol production process, storage and transportation [7].he determination of metals by electrothermal atomic absorp-
ion spectroscopy in ethanol fuel is well established [6,8,9] andame atomic absorption spectroscopy (FAAS) can be used afterpreconcentration step [10–12].
Stripping voltammetric techniques have become very pop-lar for metal ions analysis [13,14]. These techniques provide
igh analytical performance with relatively low instrumentalosts and some works have shown that stripping voltammetricechniques can be successfully applied to metal ions determi-ation in ethanol fuel [15–18]. The introduction of a chemical
2 Chim
msldwmms[iimce
sniDuatica[pahwboed
cvaeffrtri(utaps
tbDto
topeawe
2
2
cMpgpuestatsSttvdSs
2
evpGweSmmrTs
2
p
96 R.M. Takeuchi et al. / Analytica
odifier able to preconcentrate metallic ions at electrodeurface either by complexation or electrostatic attraction canead to more sensitive electroanalytical procedures with loweretection limit values. Silica and organofunctionalized silicaith groups containing S, N or O atoms are very efficientaterials to promote metal ions preconcentration [19]. Theseaterials have been widely used to preconcentrate metals for
ubsequent determination by AAS in several kinds of matrices20–22]. The extremely advantageous silica properties, such as,ts high adsorption capacity, chemical stability and the possibil-ty of functionalization with a large variety of functional groups
ake these materials excellent modifier agents to constructhemically modified electrodes [23,24], which have been widelymployed for metal determination in different matrices [25–29].
Carbon paste electrodes (CPEs) are easily modified, since nopecific interactions between modifier and electrode surface areecessary, thus this type of electrode has been widely employedn electroanalysis of organic and inorganic compounds [30].espite great success in using CPEs in electroanalysis, theirse in totally non-aqueous media is limited due to the bindergent dissolution. Several strategies have been used to improvehe chemical/mechanical properties of CPEs, all of them replac-ng mineral oil, the most used binder agent, by another agentapable of producing more rigid composites. Several promisinglternative binder agents can be found in literature, such as epoxy31,32], silicone [33], Teflon® [34], polyurethane [35]. Solidaraffin is another promising binder agent introduced by Petitnd Kauffman [36,37], which presents low cost and can be easilyandled allowing the electrode preparation in a simple and fastay. Recently, our group has demonstrated that a solid paraffin-ased carbon paste electrode modified with 2-aminothiazolerganofunctionalized silica (SiAt-SPCPE) is totally stable inthanolic solutions and it can be successfully employed to Cu2+
etermination directly in commercial ethanol fuel samples [16].Most of stripping procedures suggested for electrochemi-
al determination of Ni2+ are based on adsorptive strippingoltammetry using complexing agents; this procedure replacesdvantageously the usual metal ion reduction/re-oxidationmployed in anodic stripping analysis. The main difficultiesound in the electrodeposition of Ni2+ are its strong tendency toorm intermetallic compounds [38] and the fact that irreversibleeduction of Ni(II) to Ni(0) requires potentials more negativehan −1.1 V versus SCE, potentials in which the H+ ions are alsoeduced on different types of carbon [39]. Among the complex-ng agents used on nickel stripping analysis, dimethylglyoximeDMG) has been the most widely employed. DMG has beensed to prepare chemically modified CPEs [39–41] or in solu-ion, where the Ni(DMG)2 complex is formed and subsequentlyccumulated onto electrode surface by applying a controlledotential. This last method often employs metallic electrodes,uch as mercury [42,43], bismuth [44,45] or lead films [46].
In this context, this work describes the use of SiAt-SPCPEso Ni2+ determination in commercial ethanol fuel samples
y differential pulse adsorptive stripping voltammetry usingMG as complexing agent. The proposed method combineshe high Ni2+ adsorption capacity presented by 2-aminothiazolerganofunctionalized silica, which efficiently extracts Ni2+ from
pcao
ica Acta 584 (2007) 295–301
he ethanolic solutions, with the electrochemical propertiesf the Ni(DMG)2 complex, whose electrochemical reductionrovides the analytical signal. This is a more efficient strat-gy for Ni2+ determination in ethanol fuel in comparison toDMG-based conventional CPE whose stability is limited toater:ethanol mixtures containing in the maximum 10% of
thanol [41].
. Experimental
.1. Reagents
Stock solutions of nickel were prepared by dissolving nickelhloride (Sigma, 99.99%) in analytical purity grade ethanol fromerck. Stock solutions of DMG (J.T. Baker, 99.7%) were pre-
ared by dissolving the complexing agent in analytical purityrade ethanol. Water used to prepare all aqueous solutions wasurified via Milli-Q system. Ammonium chloride (Merck) wassed to prepare the supporting electrolyte. The pH of supportinglectrolyte was adjusted at desired value by adding 2.0 mol L−1
odium hydroxide (Merck) or 2.0 mol L−1 HCl (Merck) solu-ion. Silica gel (Merck) with specific surface area between 486nd 520 m2 g−1 and average pore diameter of 0.6 nm was utilizedo construct both the SiAt-SPCPE and the non-functionalizedilica modified solid paraffin-based carbon paste electrode (Si-PCPE). 2-Aminothiazole (Aldrich) was employed to achieve
he silica functionalization. The 2-aminothiazole organofunc-ionalized silica was synthesized according to the procedure pre-iously described in literature [12]. Spectroscopic carbon pow-er from Merck was used to obtain the SPCPEs and paraffin fromynth was used as the binder agent. The commercial ethanol fuelamples were acquired from different local gas stations.
.2. Apparatus
A HANNA Instruments pH-meter model HI 8417 wasmployed for pH measurements. Differential pulse strippingoltammetry was performed with an AUTOLAB PGSTAT 30otentiostat coupled to a microcomputer and controlled byPES 4.9 software. A one-compartment electrochemical cellith a platinum wire auxiliary electrode and an Ag/AgClsat ref-
rence electrode were used in all voltammetric measurements.iAt-SPCPE was used as working electrode. GFAAS experi-ents were carried out by using a spectrometer from Varianodel Spectra A 640Z containing a Zeeman background cor-
ector and a nickel hollow cathode lamp from Perkin-Elmer.he analytical procedure adopted in GFAAS analysis was theame used by Tartarotti et al. [41].
.3. Electrode preparation
Unmodified SPCPEs, used for comparison purpose, wererepared by the sluggish addition of carbon powder in melted
araffin in the proportion of 60%:40% (w/w). Modifiedarbon pastes were prepared by substituting correspondingmounts of the carbon powder by silica or 2-aminothiazolerganofunctionalized silica to obtain the desired composition.
Chimica Acta 584 (2007) 295–301 297
TamTbicAiitu
2
csci
icitdtesmng
twcm
3
awptfNcN
wNsSSp(
Fig. 1. Differential pulse voltammograms registered in 0.1 mol L−1 NH4Cl solu-tion + 2.5 mmol L−1 DMG (pH 5.0) after preconcentration in a 25 �mol L−1
N5(
aSotSiTitmSv
3
ceteiISosae
w
Dfc
R.M. Takeuchi et al. / Analytica
he carbon powder/modifier mixture was homogenized inmortar and pestle, subsequently the mixture was added toelted paraffin whose percentage was always kept at 40%.he new mixture was again homogenized in a thermostatizedath (65–75 ◦C). The resulting carbon pastes were then packedn a cylindrical plastic tube (internal diameter of 4.8 mm) andonnected to a copper wire to provide the electric contact.fter the solidification of paraffin, the composite, obtained
n a cylindrical form, was removed from the plastic tube andmmersed in melted paraffin to isolate the lateral parts. Beforeheir use, the electrodes were polished on A4 paper 80 gm−2
ntil the complete removal of paraffin from the surface.
.4. Analytical procedure
The analytical procedure comprised four steps: (1) Ni2+ pre-oncentration at open circuit potential directly in the ethanol fuelample, (2) transference of the electrode to an electrochemicalell containing DMG, (3) differential pulse voltammogram reg-stering and (4) surface regeneration by polishing the electrode.
For the preconcentration step, the SiAt-SPCPE was immersedn a stirred 10 mL Ni2+ solution in analytical-grade ethanol,ontaining a known amount of this metal for a controlled timenterval in open circuit potential conditions. The electrode washen removed from the preconcentration cell, briefly rinsed witheionized water, and placed in an electrochemical cell con-aining 10 mL of 0.1 mol L−1 NH4Cl solution as supportinglectrolyte and DMG. After each measurement, the electrodeurface was renewed by polishing the electrode. All measure-ents were performed without deoxygenating the solution since
o difference in voltammetric response was observed in deoxy-enated or non-deoxygenated solution.
For Ni2+ determination in commercial ethanol fuel samples,he preconcentration step was performed directly in the sample,ithout any pretreatment or dilution step. The Ni2+ quantifi-
ation in these samples was performed by standard additionethod.
. Results and discussion
We have initially tried to use conventional CPEs with Nujol®
s binder agent. However, we have observed a swelling effecthen these electrodes were in contact with ethanolic solutions,robably due to binder dissolution, demonstrating that conven-ional CPEs are inappropriate to nickel determination in ethanoluel samples. To overcome this problem, we have replacedujol® by solid paraffin in order to obtain a more rigid and
ompact electrode, which can be successfully employed fori2+ determination directly in commercial ethanol fuel samples.Fig. 1 presents differential pulse voltammograms registered
ith different SPCPEs after preconcentration in a 25 �mol L−1
i2+ ethanolic solution for 5 min. The voltammograms pre-ented in this figure were registered using a non-modified
PCPE (curve A), a Si-SPCPE (curve B) and the SiAt(20%)-PCPE. The SiAt(20%)-SPCPE corresponds to a carbonaste with composition: carbon powder:paraffin:modifier40%:40%:20%, w/w).i
p5
i2+ ethanolic solution for 5 min. Voltammetric conditions: pulse amplitude,0 mV; pulse width, 5.0 ms; scan rate, 20 mV s−1. (A) SPCE; (B) Si-SPCPE;C) SiAt(20%)-SPCPE.
Fig. 1 shows that both Si-SPCPE and SiAt(20%)-SPCPEre able to preconcentrate Ni2+ from ethanolic solutions whilePCPE is unable to promote Ni2+ preconcentration. The abilityf Si-SPCE to preconcentrate Ni2+ probably is due to elec-rostatic interactions between Ni2+ and Si O− groups. WheniAt(20%)-SPCPE is used the obtained peak current is approx-
mately three times higher than the obtained with Si-SPCPE.his result indicates that 2-aminothiazole organofunctional-
zed silica has higher capacity for Ni2+ preconcentrationhan non-functionalized silica. It was not observed voltam-
etric peaks without preconcentration step, indicating thatiAt(20%)-SPCPE or NH4Cl/DMG mixture do not present anyoltammetric signal in the studied potential range.
.1. Study of the operational parameters
The SiAt concentration in the carbon paste had a signifi-ant influence on the voltammetric response; this influence wasvaluated by using modifier percentages from 5 to 30%, beinghe preconcentration step performed in a 25 �mol L−1 Ni2+
thanolic solution for 5 min and the voltammograms registeredn 0.1 mol L−1 NH4Cl solution containing 2.5 mmol L−1 DMG.t was observed that maximum peak current was obtained wheniAt concentration in the paste was 20%. Higher concentrationsf SiAt significantly decreased the peak current. This is pre-umably due the reduction of conductive area (carbon particles)t the electrode surface [29]. Thus, the SiAt(20%)-SPCPE wasmployed in all subsequent experiments.
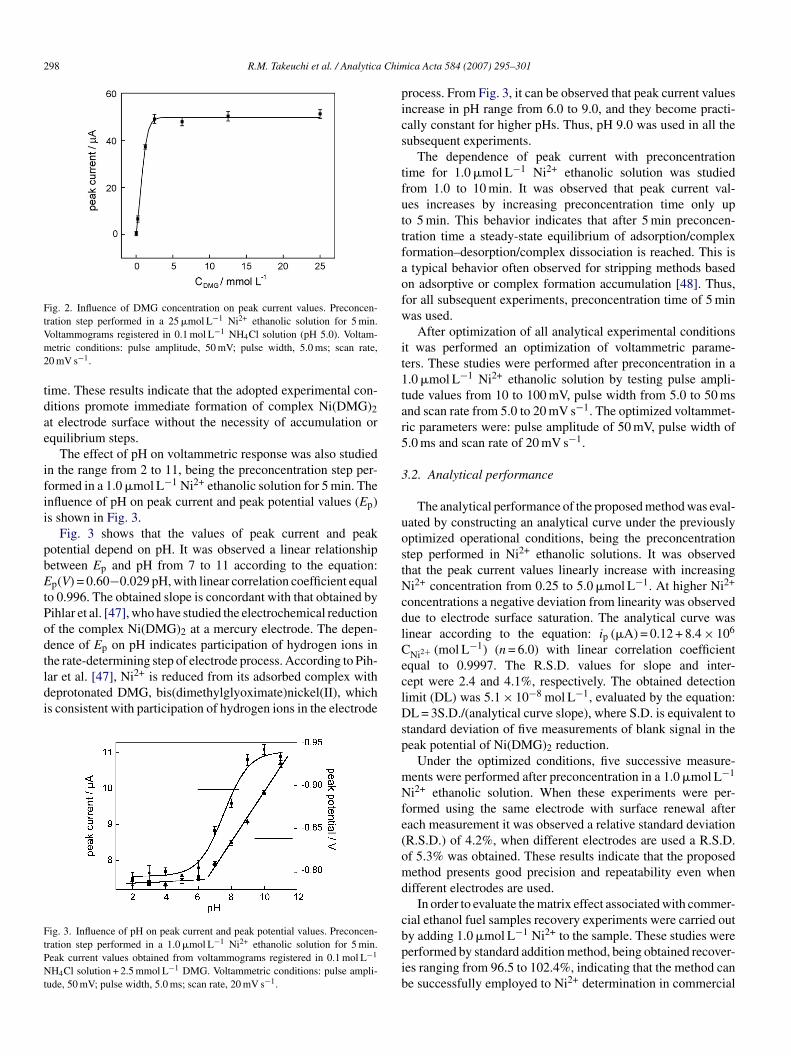
The effect of DMG concentration on the peak current valuesas also studied and the results obtained are shown in Fig. 2.As observed in Fig. 2, maximum signal is obtained for
MG concentration ranging from 2.5 to 25 mmol L−1. There-ore, aiming to obtain a maximum of signal and keep the DMGoncentration as low as possible, 2.5 mmol L−1 DMG was used
n the subsequent experiments.It was observed that the peak current values obtained afterreconcentration in 1.0 �mol L−1 Ni2+ ethanolic solution formin are independent of accumulation potential or equilibrium
298 R.M. Takeuchi et al. / Analytica Chim
Fig. 2. Influence of DMG concentration on peak current values. Preconcen-tration step performed in a 25 �mol L−1 Ni2+ ethanolic solution for 5 min.Vm2
tdae
ifii
pbEtPodtldi
FtPNt
pics
tfuttfaofw
it1tar5
3
uostNcdlC
oltammograms registered in 0.1 mol L−1 NH4Cl solution (pH 5.0). Voltam-etric conditions: pulse amplitude, 50 mV; pulse width, 5.0 ms; scan rate,
0 mV s−1.
ime. These results indicate that the adopted experimental con-itions promote immediate formation of complex Ni(DMG)2t electrode surface without the necessity of accumulation orquilibrium steps.
The effect of pH on voltammetric response was also studiedn the range from 2 to 11, being the preconcentration step per-ormed in a 1.0 �mol L−1 Ni2+ ethanolic solution for 5 min. Thenfluence of pH on peak current and peak potential values (Ep)s shown in Fig. 3.
Fig. 3 shows that the values of peak current and peakotential depend on pH. It was observed a linear relationshipetween Ep and pH from 7 to 11 according to the equation:p(V) = 0.60−0.029 pH, with linear correlation coefficient equal
o 0.996. The obtained slope is concordant with that obtained byihlar et al. [47], who have studied the electrochemical reductionf the complex Ni(DMG)2 at a mercury electrode. The depen-ence of Ep on pH indicates participation of hydrogen ions in
he rate-determining step of electrode process. According to Pih-ar et al. [47], Ni2+ is reduced from its adsorbed complex witheprotonated DMG, bis(dimethylglyoximate)nickel(II), whichs consistent with participation of hydrogen ions in the electrodeig. 3. Influence of pH on peak current and peak potential values. Preconcen-ration step performed in a 1.0 �mol L−1 Ni2+ ethanolic solution for 5 min.eak current values obtained from voltammograms registered in 0.1 mol L−1
H4Cl solution + 2.5 mmol L−1 DMG. Voltammetric conditions: pulse ampli-ude, 50 mV; pulse width, 5.0 ms; scan rate, 20 mV s−1.
eclDsp
mNfe(omd
cbpib
ica Acta 584 (2007) 295–301
rocess. From Fig. 3, it can be observed that peak current valuesncrease in pH range from 6.0 to 9.0, and they become practi-ally constant for higher pHs. Thus, pH 9.0 was used in all theubsequent experiments.
The dependence of peak current with preconcentrationime for 1.0 �mol L−1 Ni2+ ethanolic solution was studiedrom 1.0 to 10 min. It was observed that peak current val-es increases by increasing preconcentration time only upo 5 min. This behavior indicates that after 5 min preconcen-ration time a steady-state equilibrium of adsorption/complexormation–desorption/complex dissociation is reached. This istypical behavior often observed for stripping methods based
n adsorptive or complex formation accumulation [48]. Thus,or all subsequent experiments, preconcentration time of 5 minas used.After optimization of all analytical experimental conditions
t was performed an optimization of voltammetric parame-ers. These studies were performed after preconcentration in a.0 �mol L−1 Ni2+ ethanolic solution by testing pulse ampli-ude values from 10 to 100 mV, pulse width from 5.0 to 50 msnd scan rate from 5.0 to 20 mV s−1. The optimized voltammet-ic parameters were: pulse amplitude of 50 mV, pulse width of.0 ms and scan rate of 20 mV s−1.
.2. Analytical performance
The analytical performance of the proposed method was eval-ated by constructing an analytical curve under the previouslyptimized operational conditions, being the preconcentrationtep performed in Ni2+ ethanolic solutions. It was observedhat the peak current values linearly increase with increasingi2+ concentration from 0.25 to 5.0 �mol L−1. At higher Ni2+
oncentrations a negative deviation from linearity was observedue to electrode surface saturation. The analytical curve wasinear according to the equation: ip (�A) = 0.12 + 8.4 × 106
Ni2+ (mol L−1) (n = 6.0) with linear correlation coefficientqual to 0.9997. The R.S.D. values for slope and inter-ept were 2.4 and 4.1%, respectively. The obtained detectionimit (DL) was 5.1 × 10−8 mol L−1, evaluated by the equation:L = 3S.D./(analytical curve slope), where S.D. is equivalent to
tandard deviation of five measurements of blank signal in theeak potential of Ni(DMG)2 reduction.
Under the optimized conditions, five successive measure-ents were performed after preconcentration in a 1.0 �mol L−1
i2+ ethanolic solution. When these experiments were per-ormed using the same electrode with surface renewal afterach measurement it was observed a relative standard deviationR.S.D.) of 4.2%, when different electrodes are used a R.S.D.f 5.3% was obtained. These results indicate that the proposedethod presents good precision and repeatability even when
ifferent electrodes are used.In order to evaluate the matrix effect associated with commer-
ial ethanol fuel samples recovery experiments were carried out
y adding 1.0 �mol L−1 Ni2+ to the sample. These studies wereerformed by standard addition method, being obtained recover-es ranging from 96.5 to 102.4%, indicating that the method cane successfully employed to Ni2+ determination in commercial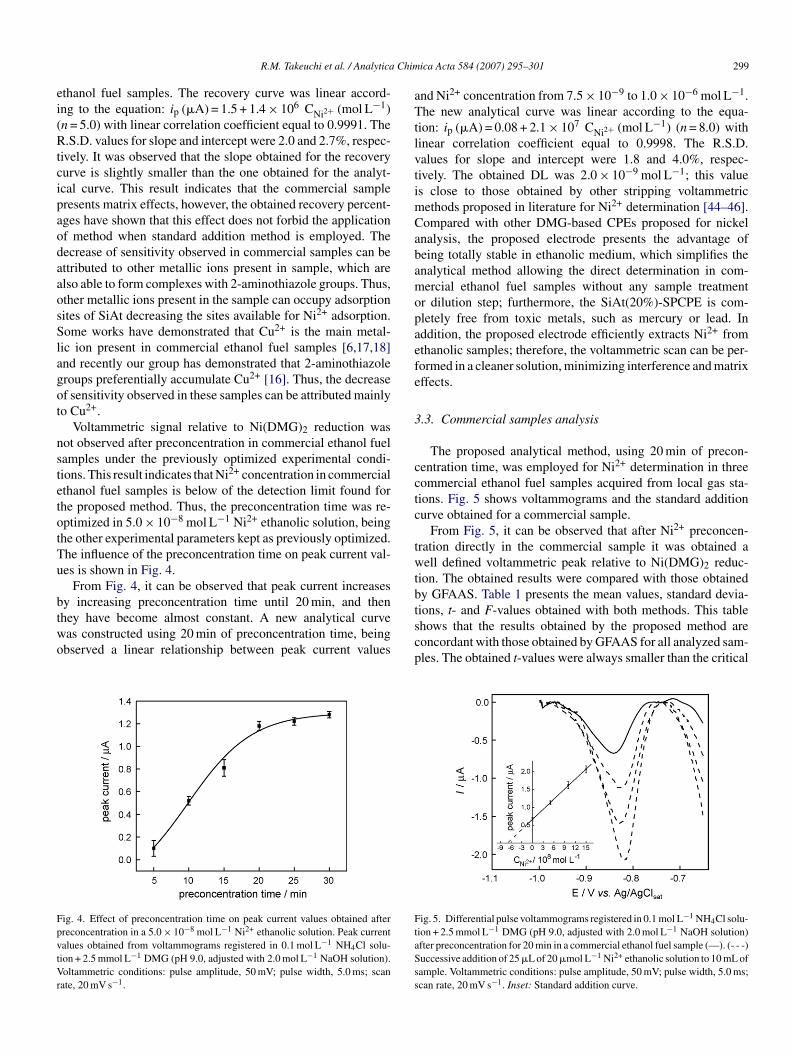
Chim
ei(RtcipaodaaosSlagot
nstetotTu
btwo
FpvtVr
aTtlvtimCabamopaefe
3
cctc
twtb
R.M. Takeuchi et al. / Analytica
thanol fuel samples. The recovery curve was linear accord-ng to the equation: ip (�A) = 1.5 + 1.4 × 106 CNi2+ (mol L−1)n = 5.0) with linear correlation coefficient equal to 0.9991. The.S.D. values for slope and intercept were 2.0 and 2.7%, respec-
ively. It was observed that the slope obtained for the recoveryurve is slightly smaller than the one obtained for the analyt-cal curve. This result indicates that the commercial sampleresents matrix effects, however, the obtained recovery percent-ges have shown that this effect does not forbid the applicationf method when standard addition method is employed. Theecrease of sensitivity observed in commercial samples can bettributed to other metallic ions present in sample, which arelso able to form complexes with 2-aminothiazole groups. Thus,ther metallic ions present in the sample can occupy adsorptionites of SiAt decreasing the sites available for Ni2+ adsorption.ome works have demonstrated that Cu2+ is the main metal-
ic ion present in commercial ethanol fuel samples [6,17,18]nd recently our group has demonstrated that 2-aminothiazoleroups preferentially accumulate Cu2+ [16]. Thus, the decreasef sensitivity observed in these samples can be attributed mainlyo Cu2+.
Voltammetric signal relative to Ni(DMG)2 reduction wasot observed after preconcentration in commercial ethanol fuelamples under the previously optimized experimental condi-ions. This result indicates that Ni2+ concentration in commercialthanol fuel samples is below of the detection limit found forhe proposed method. Thus, the preconcentration time was re-ptimized in 5.0 × 10−8 mol L−1 Ni2+ ethanolic solution, beinghe other experimental parameters kept as previously optimized.he influence of the preconcentration time on peak current val-es is shown in Fig. 4.
From Fig. 4, it can be observed that peak current increasesy increasing preconcentration time until 20 min, and then
hey have become almost constant. A new analytical curveas constructed using 20 min of preconcentration time, beingbserved a linear relationship between peak current valuesig. 4. Effect of preconcentration time on peak current values obtained afterreconcentration in a 5.0 × 10−8 mol L−1 Ni2+ ethanolic solution. Peak currentalues obtained from voltammograms registered in 0.1 mol L−1 NH4Cl solu-ion + 2.5 mmol L−1 DMG (pH 9.0, adjusted with 2.0 mol L−1 NaOH solution).oltammetric conditions: pulse amplitude, 50 mV; pulse width, 5.0 ms; scan
ate, 20 mV s−1.
tscp
FtaSss
ica Acta 584 (2007) 295–301 299
nd Ni2+ concentration from 7.5 × 10−9 to 1.0 × 10−6 mol L−1.he new analytical curve was linear according to the equa-
ion: ip (�A) = 0.08 + 2.1 × 107 CNi2+ (mol L−1) (n = 8.0) withinear correlation coefficient equal to 0.9998. The R.S.D.alues for slope and intercept were 1.8 and 4.0%, respec-ively. The obtained DL was 2.0 × 10−9 mol L−1; this values close to those obtained by other stripping voltammetric
ethods proposed in literature for Ni2+ determination [44–46].ompared with other DMG-based CPEs proposed for nickelnalysis, the proposed electrode presents the advantage ofeing totally stable in ethanolic medium, which simplifies thenalytical method allowing the direct determination in com-ercial ethanol fuel samples without any sample treatment
r dilution step; furthermore, the SiAt(20%)-SPCPE is com-letely free from toxic metals, such as mercury or lead. Inddition, the proposed electrode efficiently extracts Ni2+ fromthanolic samples; therefore, the voltammetric scan can be per-ormed in a cleaner solution, minimizing interference and matrixffects.
.3. Commercial samples analysis
The proposed analytical method, using 20 min of precon-entration time, was employed for Ni2+ determination in threeommercial ethanol fuel samples acquired from local gas sta-ions. Fig. 5 shows voltammograms and the standard additionurve obtained for a commercial sample.
From Fig. 5, it can be observed that after Ni2+ preconcen-ration directly in the commercial sample it was obtained aell defined voltammetric peak relative to Ni(DMG)2 reduc-
ion. The obtained results were compared with those obtainedy GFAAS. Table 1 presents the mean values, standard devia-
ions, t- and F-values obtained with both methods. This tablehows that the results obtained by the proposed method areoncordant with those obtained by GFAAS for all analyzed sam-les. The obtained t-values were always smaller than the criticalig. 5. Differential pulse voltammograms registered in 0.1 mol L−1 NH4Cl solu-ion + 2.5 mmol L−1 DMG (pH 9.0, adjusted with 2.0 mol L−1 NaOH solution)fter preconcentration for 20 min in a commercial ethanol fuel sample (—). (- - -)uccessive addition of 25 �L of 20 �mol L−1 Ni2+ ethanolic solution to 10 mL ofample. Voltammetric conditions: pulse amplitude, 50 mV; pulse width, 5.0 ms;can rate, 20 mV s−1. Inset: Standard addition curve.

300 R.M. Takeuchi et al. / Analytica Chimica Acta 584 (2007) 295–301
Table 1Comparison between the Ni2+ concentration determined in commercial ethanol fuel samples by the proposed method and by GFAAS
Sample Proposed methoda (10−8 mol L−1) GFAASa (10−8 mol L−1) tb Fc
1 7.2 ± 0.3 6.9 ± 0.4 1.37 2.82 2.6 ± 0.4 2.3 ± 0.2 1.41 1.83 2.0 ± 0.5 1.6 ± 0.2 1.30 6.3
a n = 3.
vtnpir
4
emsfiptwc2cetl1mlcs
A
(fa
R
[
[
[
[[
[
[
[
[[
[
[
[[[[
[
[
[[[
[[
[
[
[[[[
b tcritical = 2.78 (P = 0.05 with four freedom level).c F2/2critical = 19.0 (P = 0.05).
alue, indicating that there is not statistical difference betweenhe results obtained with both methods. Obtained F-values haveot exceeded the critical value, indicating that both methodsresent similar precisions. Finally, the Ni2+ contents determinedn commercial samples are in good agreement with those valueseported in literature [12,49].
. Conclusions
This work demonstrates that SiAt-SPCPE can be successfullymployed for reliable Ni2+ determination directly in com-ercial ethanol fuel samples without pretreatment or dilution
teps. The use of paraffin as binder agent and the modi-er chemically attached on silica have completely overcomeroblems associated with binder agent or modifier dissolu-ion by ethanol; as a consequence, the developed electrodeas totally stable in ethanolic solutions. The proposed method
ombines the high Ni2+ adsorption capacity presented by-aminothiazole organofunctionalized silica with the electro-hemical proprieties of Ni(DMG)2 complex, which was a morefficient strategy to Ni2+ determination in ethanol sampleshan using a DMG-based conventional CPE whose stability isimited to water:ethanol mixtures containing in the maximum0% of ethanol. The obtained results were in good agree-ent with those obtained with GFAAS and those reported in
iterature, indicating that the developed electrode can be suc-essfully used for Ni2+ determination in commercial ethanol fuelamples.
cknowledgements
The authors are grateful to FAPESP (contract no. 03/09334-7RMT) and no. 03/05567-7 (ALS)), CTPetro/FINEP and ANPor financial support. We also thank Dr. Maria Jose Medeirosnd her son Joao Medeiros for English corrections.
eferences
[1] A.E. Wheals, L.C. Basso, D.M.C. Alves, H.V. Amorin, Trends Biotechnol.17 (1999) 482.
[2] I.C. Macedo, Biomass Bioenergy 14 (1998) 77.[3] F. Rosillo-Calle, L.A.B. Cortez, Biomass Bioenergy 14 (1998) 115.
[4] K. Sandelim, R. Backman, Environ. Sci. Technol. 33 (1999) 4508.[5] D.B. Taylor, R.E. Synovec, Talanta 40 (1993) 495.[6] T.D. Saint’Pierre, V.L.A. Frescura, A.J. Curtius, Talanta 68 (2006)957.[7] I.M.R.A. Bruning, E.B. Malm, Bol. Tec. Petrobras 25 (1982) 217.
[
[[
[8] A.P. Oliveira, M. Morais, J.A.G. Neto, E.C. Lima, At. Spectrosc. 23 (2000)39.
[9] A.P. Oliveira, M. Morais, J.A.G. Neto, E.C. Lima, At. Spectrosc. 23 (2000)190.
10] L.A.M. Gomes, P.M. Padilha, J.C. Moreira, N.L. Dias-Filho, Y. Gushiken,J. Braz. Chem. Soc. 9 (1998) 494.
11] P.M. Padilha, C.C.F. Padilha, J.C. Rocha, Chim. Anal. 18 (1999)299.
12] P.S. Roldan, I.L. Alcantara, G.R. Castro, J.C. Rocha, C.C.F. Padilha, P.M.Padilha, Anal. Bional. Chem. 375 (2003) 574.
13] X. Xie, D. Stueben, Z. Berner, Anal. Lett. 38 (2005) 2281.14] M.T. Lam, J. Murimboh, N.M. Hassan, C.L. Chakrabarti, Anal. Chim. Acta
402 (1999) 195.15] M.F. Bergamini, S.I. Vital, A.L. Santos, N.R. Stradiotto, Eclet. Quim. 31
(2006) 45.16] R.M. Takeuchi, A.L. Santos, P.M. Padilha, N.R. Stradiotto, Talanta 71
(2007) 771.17] M.F. Oliveira, A.A. Saczk, L.L. Okumura, A.P. Fernandes, M. Moraes,
N.R. Stradiotto, Anal. Bioanal. Chem. 380 (2004) 135.18] R.A.A. Munoz, L. Angnes, Microchem. J. 77 (2004) 157.19] R.S.A. Machado-Junior, M.G. Fonseca, L.N.H. Arakaki, J.G.P. Espınola,
S.F. Oliveira, Talanta 63 (2004) 317.20] N.L. Dias-Filho, Y. Gushikem, W.L. Polito, J.C. Moreira, E.O. Ehirim,
Talanta 42 (1995) 1625.21] P. Lessi, N.L. Dias-Filho, J.C. Moreira, J.T.S. Campos, Anal. Chim. Acta
327 (1996) 183.22] A.G.S. Prado, C. Airoldi, Anal. Chim. Acta 432 (2001) 201.23] A. Walcarius, Electroanalysis 10 (1998) 1214.24] A. Walcarius, Electroanalysis 13 (2001) 701.25] L.M. Aleixo, M.F.B. Souza, O.E.S. Godinho, G. Oliveira-Neto, Y.
Gushikem, Anal. Chim. Acta 271 (1993) 143.26] A. Walcarius, N. Luthi, J.-L. Blin, B.-L. Su, L. Lamberts, Electrochim.
Acta 44 (1999) 4601.27] W. Yantasee, Y. Lin, G.E. Fryxell, B.J. Beusche, Anal. Chim. Acta 502
(2004) 207.28] I. Jureviciute, A. Malinauskas, Chem. Anal. (Warsaw) 49 (2004) 339.29] A. Meyer, S. Hoffler, K. Fischer, Anal. Chim. Acta 571 (2006) 105.30] I. Svancara, K. Vytras, J. Barek, Z. Zima, Crit. Rev. Anal. Chem. 31 (2001)
311.31] M.F.S. Teixeira, A.Z. Pinto, O. Fatibello-Filho, Talanta 45 (1997) 249.32] L. Moreno-Baron, A. Merkoci, S. Alegret, Electrochim. Acta 48 (2003)
2599.33] Y. Ballesteros, M.J.G. Huebra, M.C. Quintana, P. Hernandez, L. Hernandez,
Microchem. J. 74 (2003) 193.34] A.G.-V. Prada, N. Pena, C. Parrado, A.J. Reviejo, J.M. Pingarron, Talanta
62 (2004) 896.35] R.K. Mendes, S. Claro-Neto, E.T.G. Cavalheiro, Talanta 57 (2002) 909.36] C. Petit, J.-M. Kauffmann, Anal. Proc. 32 (1995) 11.37] C. Petit, A. Gonzalez-Cortes, J.-M. Kauffmann, Talanta 42 (1995) 1783.38] P. Gonzalez, V.A. Cortınez, C.A. Fontan, Talanta 58 (2002) 679.39] R.B. Baldwin, J.K. Cristensen, L. Kryger, Anal. Chem. 58 (1986)
1790.40] Z.-Q. Zhang, H. Liu, H. Zhang, Y.-F. Li, Anal. Chim. Acta 333 (1996) 119.41] F.O. Tartarotti, M.F. Oliveira, V.R. Balbo, N.R. Stradiotto, Microchim. Acta
155 (2006) 397.

Chim
[[
[
[
[
R.M. Takeuchi et al. / Analytica
42] M. Korolczuk, Talanta 53 (2000) 679.
43] D. Sancho, L. Deban, I. Campos, R. Pardo, M. Vega, Food Chem. 71 (2000)139.44] E.A. Hutton, B. Ogorevc, S.B. Hocevar, M.R. Smyth, Anal. Chim. Acta
557 (2006) 57.45] S. Legeai, S. Bois, O. Vittori, J. Electroanal. Chem. 591 (2006) 93.
[[[
ica Acta 584 (2007) 295–301 301
46] M. Korolczuk, K. Tyszczuk, M. Grabarczyk, Electrochem. Commun. 7
(2005) 1185.47] B. Pihlar, P. Valenta, H.W. Nurnberg, J. Electroanal. Chem. 214 (1986) 157.48] H. Alemu, B.S. Chandravanshi, Anal. Chim. Acta 368 (1998) 165.49] T.D. Saint’Pierre, T.A. Maranhao, V.L.A. Frescura, A.J. Curtius, Spec-
trochim. Acta B 60 (2005) 605.
Related Documents











