Research paper A novel mutation in the TMC1 gene causes non-syndromic hearing loss in a Moroccan family. Amina Bakhchane a , Hicham Charoute a , Halima Nahili a , Rachida Roky b , Hassan Rouba a , Majida Charif a,c,d , Guy Lenaers c,d,1 , Abdelhamid Barakat a,1, ⁎ a Laboratoire de Génétique Moléculaire Humaine, Département de Recherche Scientifique, Institut Pasteur du Maroc, Casablanca, Morocco b Université Hassan II Ain Chock, Laboratoire de Physiologie et génétique moléculaire, Km 8 Route d'El Jadida, B.P. 5366 Maarif, Casablanca 20100, Morocco c Institut des Neurosciences de Montpellier, U1051 de l'INSERM, Université de Montpellier I et II, BP 74103, 34091 Montpellier cedex 05, France d PREMMi, Université d'Angers, CHU Bât IRIS/IBS, Rue des Capucins, 49933 Angers cedex 9, France abstract article info Article history: Received 12 March 2015 Received in revised form 29 June 2015 Accepted 22 July 2015 Available online xxxx Keywords: Hearing loss TMC1 Mutation Whole exome sequencing Morocco Autosomal recessive non-syndromic hearing loss (ARNSHL) is one of the most common genetic diseases in human and is subject to important genetic heterogeneity, rendering molecular diagnosis difficult. Whole- exome sequencing is thus a powerful strategy for this purpose. After excluding GJB2 mutation and other common mutations associated with hearing loss in Morocco, whole-exome sequencing was performed to study the genet- ic causes of one sibling with ARSHNL in a consanguineous Moroccan family. After filtering data and Sanger se- quencing validation, one novel pathogenic homozygous mutation c.1810C N G (p.Arg604Gly) was identified in TMC1, a gene reported to cause deafness in various populations. Thus, we identified here the first mutation in the TMC1 gene in the Moroccan population causing non-syndromic hearing loss. © 2015 Elsevier B.V. All rights reserved. 1. Introduction Hearing loss is one of the most common sensory disorder world- wide, occurring in approximately 0.2% of newborns (Hilgert et al., 2009). The majority of congenital cases are due to genetic factors and inherited across generations. Hereditary hearing loss is highly heteroge- neous, with autosomal recessive non-syndromic hearing loss (ARNSHL) being the most frequent accounting for 80% of all cases. To date, a total of 142 loci have been mapped for non syndromic deafness and 90 genes have been identified, including 31 autosomal dominant, 55 auto- somal recessive and 4 X-linked genes (http://hereditaryhearingloss. org). GJB2 mutations are responsible for almost 50% of all cases with ARNSHL in most populations (Diaz-Horta et al., 2012). Conversely, the majority of other deafness mutations are private and found in very few families, if not in a single one (Duman and Tekin, 2012). Thus screening of ARNSHL genes by standard molecular procedures is exten- sively long, expensive and time consuming (Diaz-Horta et al., 2012). Whole-exome sequencing (WES) is lately viewed as a substitute to more conventional procedures for genetic diagnosis, as it authorizes a specific enrichment and sequencing of all exons of protein-coding genes, including those involved in ARNSHL (Diaz-Horta et al., 2012). Next to the usual genes related to hearing loss published previously in Morocco, mainly GJB2 (Abidi et al., 2007; Abidi et al., 2008), LRTOMT (Charif et al., 2012) and also MT-RNR1 (Nahili et al., 2010), we here de- scribe the first mutation in the TMC1 gene in the Moroccan population affected by ARNSHL, a gene which was already identified as causative for DFNA36 and DFNB7/11 deafness presentations (Kurima et al., 2002). 2. Patients and methods 2.1. Ethics statements and DNA preparation This study was approved by the ethic committee on research of Pas- teur Institute of Morocco. We collected written informed consents from all patients before including them in the study. One unrelated Moroccan family showing prelingual, profound and bilateral sensorineural hearing loss took part to this study because of its parental consanguinity and the existence of two siblings with hearing loss issues (Fig. 1.A). Further Gene xxx (2015) xxx–xxx Abbreviations: ARNSHL, autosomal recessive non-syndromic hearing loss; DFNA, deaf- ness, autosomal dominant 36; DFNB, deafness, autosomal recessive; ESP, Exome Sequencing Project; EVS, Exome Variant Server; GJB2, gap junction protein, beta 2, 26 kDa; LRTOMT, leucine rich transmembrane and O-methyltransferase domain con- taining; MT-RNR1, Ribosomal RNA, Mitochondrial, 12S; PolyPhen, Polymorphism Phenotyping; SIFT, Sorting Intolerant From Tolerant; TMC1, transmembrane channel-like 1; WES, Whole-exome sequencing. ⁎ Corresponding author at: Laboratoire de Génétique Moléculaire et Humaine, Département de Recherche Scientifique, Institut Pasteur. 1, Place Louis Pasteur, C.P. 20360 Casablanca, Morocco. E-mail address: [email protected] (A. Barakat). 1 equal level contributors. GENE-40734; No. of pages: 6; 4C: http://dx.doi.org/10.1016/j.gene.2015.07.075 0378-1119/© 2015 Elsevier B.V. All rights reserved. Contents lists available at ScienceDirect Gene journal homepage: www.elsevier.com/locate/gene Please cite this article as: Bakhchane, A., et al., A novel mutation in the TMC1 gene causes non-syndromic hearing loss in a Moroccan family., Gene (2015), http://dx.doi.org/10.1016/j.gene.2015.07.075

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Gene xxx (2015) xxx–xxx
GENE-40734; No. of pages: 6; 4C:
Contents lists available at ScienceDirect
Gene
j ourna l homepage: www.e lsev ie r .com/ locate /gene
Research paper
A novelmutation in the TMC1 gene causes non-syndromic hearing loss ina Moroccan family.
Amina Bakhchane a, Hicham Charoute a, Halima Nahili a, Rachida Roky b, Hassan Rouba a, Majida Charif a,c,d,Guy Lenaers c,d,1, Abdelhamid Barakat a,1,⁎a Laboratoire de Génétique Moléculaire Humaine, Département de Recherche Scientifique, Institut Pasteur du Maroc, Casablanca, Moroccob Université Hassan II Ain Chock, Laboratoire de Physiologie et génétique moléculaire, Km 8 Route d'El Jadida, B.P. 5366 Maarif, Casablanca 20100, Moroccoc Institut des Neurosciences de Montpellier, U1051 de l'INSERM, Université de Montpellier I et II, BP 74103, 34091 Montpellier cedex 05, Franced PREMMi, Université d'Angers, CHU Bât IRIS/IBS, Rue des Capucins, 49933 Angers cedex 9, France
Abbreviations:ARNSHL, autosomal recessive non-syndness, autosomal dominant 36; DFNB, deafness, autoSequencing Project; EVS, Exome Variant Server; GJB2,26 kDa; LRTOMT, leucine rich transmembrane and O-mtaining; MT-RNR1, Ribosomal RNA, Mitochondrial, 1Phenotyping; SIFT, Sorting Intolerant From Tolerchannel-like 1; WES, Whole-exome sequencing.⁎ Corresponding author at: Laboratoire de Génétiq
Département de Recherche Scientifique, Institut Paste20360 Casablanca, Morocco.
E-mail address: [email protected] (A. Baraka1 equal level contributors.
http://dx.doi.org/10.1016/j.gene.2015.07.0750378-1119/© 2015 Elsevier B.V. All rights reserved.
Please cite this article as: Bakhchane, A., et al.(2015), http://dx.doi.org/10.1016/j.gene.201
a b s t r a c t
a r t i c l e i n f oArticle history:Received 12 March 2015Received in revised form 29 June 2015Accepted 22 July 2015Available online xxxx
Keywords:Hearing lossTMC1MutationWhole exome sequencingMorocco
Autosomal recessive non-syndromic hearing loss (ARNSHL) is one of the most common genetic diseases inhuman and is subject to important genetic heterogeneity, rendering molecular diagnosis difficult. Whole-exome sequencing is thus a powerful strategy for this purpose. After excluding GJB2mutation and other commonmutations associatedwith hearing loss inMorocco, whole-exome sequencingwas performed to study the genet-ic causes of one sibling with ARSHNL in a consanguineous Moroccan family. After filtering data and Sanger se-quencing validation, one novel pathogenic homozygous mutation c.1810C N G (p.Arg604Gly) was identified inTMC1, a gene reported to cause deafness in various populations. Thus, we identified here the first mutation inthe TMC1 gene in the Moroccan population causing non-syndromic hearing loss.
© 2015 Elsevier B.V. All rights reserved.
1. Introduction
Hearing loss is one of the most common sensory disorder world-wide, occurring in approximately 0.2% of newborns (Hilgert et al.,2009). The majority of congenital cases are due to genetic factors andinherited across generations. Hereditary hearing loss is highly heteroge-neous, with autosomal recessive non-syndromic hearing loss (ARNSHL)being the most frequent accounting for 80% of all cases. To date, a totalof 142 loci have been mapped for non syndromic deafness and 90genes have been identified, including 31 autosomal dominant, 55 auto-somal recessive and 4 X-linked genes (http://hereditaryhearingloss.org). GJB2 mutations are responsible for almost 50% of all cases withARNSHL in most populations (Diaz-Horta et al., 2012). Conversely, the
romic hearing loss; DFNA, deaf-somal recessive; ESP, Exomegap junction protein, beta 2,ethyltransferase domain con-2S; PolyPhen, Polymorphismant; TMC1, transmembrane
ue Moléculaire et Humaine,ur. 1, Place Louis Pasteur, C.P.
t).
, A novel mutation in the TMC5.07.075
majority of other deafness mutations are private and found in veryfew families, if not in a single one (Duman and Tekin, 2012). Thusscreening of ARNSHL genes by standard molecular procedures is exten-sively long, expensive and time consuming (Diaz-Horta et al., 2012).Whole-exome sequencing (WES) is lately viewed as a substitute tomore conventional procedures for genetic diagnosis, as it authorizes aspecific enrichment and sequencing of all exons of protein-codinggenes, including those involved in ARNSHL (Diaz-Horta et al., 2012).
Next to the usual genes related to hearing loss published previouslyin Morocco, mainly GJB2 (Abidi et al., 2007; Abidi et al., 2008), LRTOMT(Charif et al., 2012) and also MT-RNR1 (Nahili et al., 2010), we here de-scribe the first mutation in the TMC1 gene in the Moroccan populationaffected by ARNSHL, a gene which was already identified as causativefor DFNA36 and DFNB7/11 deafness presentations (Kurima et al., 2002).
2. Patients and methods
2.1. Ethics statements and DNA preparation
This study was approved by the ethic committee on research of Pas-teur Institute of Morocco.We collectedwritten informed consents fromall patients before including them in the study. One unrelatedMoroccanfamily showing prelingual, profound and bilateral sensorineural hearingloss took part to this study because of its parental consanguinity and theexistence of two siblings with hearing loss issues (Fig. 1.A). Further
1 gene causes non-syndromic hearing loss in aMoroccan family., Gene

Fig. 1. (A) Pedigree of the SF107 family, presenting the segregation of the p.Arg604Gly mutation. (B) Electrophoregram showing the heterozygous (top) and homozygous (bottom)c.1810C N G mutation.
2 A. Bakhchane et al. / Gene xxx (2015) xxx–xxx
Clinical examination of the subjects disqualified any symptom or mal-formation that could be suggestive of a syndromic form of hearingloss. Using the phenol chloroformmethod, Genomic DNAwas extractedfrom the blood from affected and unaffected family members. The pa-tients were previously tested negative for the most common connexin(GJB2, GJB6 and GJB3), mitochondrial (12sRNA) mutations and the242G N A mutation in LRTOMT gene. We also included in this study 90healthy Moroccan with normal hearing.
2.2. Exome sequencing
Whole-exome sequencing was performed on patient SF107.3 byOtogenetics Corporation (Norcross, GA, USA). After initial sample qual-ity control, fragmented genomic DNA was amplified using Illumina li-brary preparation kit (Illumina, Inc., San Diego, CA). The resultinglibraries were captured by Agilent Human exome V5 (51 Mb) capturekit, followed by paired end sequencing on a Hiseq2000 platform(Illumina, San Diego, USA), according to the manufacturer's protocol.We obtained a minimal exome coverage of 53X, which providedsufficient depth to analyze variants.
2.3. Read mapping and variant analysis
Short read alignment and variants calling were performed using theDNAnexus software package. Paired-end short reads were alignedagainst the human genome reference sequence hg19 (GRCh37). Thealigned short reads were analyzed to identify single nucleotide varia-tions (SNVs) and indels (insertions and deletions). We filtered variantsbased on dbSNP (build 135) and 1000 genome project databases. Vari-ants with a frequency less than 1% were considered as rare. Impacts ofeach variant on protein structure were predicted by SIFT (Sorting Intol-erant From Tolerant), PolyPhen-2 (Polymorphism Phenotyping) andProven softwares. The remaining variants were viewed using theDNAnexus Genome Browser.
Table 1Sequences of the primers used to validate the mutation by Sanger sequencing.
TMC1-Ex20-F TTTAAGAAGTATCTTGGGGAACTGTMC1-Ex20-R GGATCTCATTTCCACCAACC
2.4. Verification of variants (Sanger sequencing)
To ascertain the segregationwith the disease phenotype in this fam-ily, Sanger sequencing was performed to validate the mutation in the
Please cite this article as: Bakhchane, A., et al., A novel mutation in the TMC(2015), http://dx.doi.org/10.1016/j.gene.2015.07.075
candidate gene. Specific primers were designed using Primer3 (http://primer3.ut.ee/) (Table 1).
2.5. Protein structure prediction and analysis of stability
The exact three-dimensional structure of TMC1 is not available;therefore, homology modeling approach was used to predict a 3Dmodel of the protein. CPHmodels-3.2 Server built a protein 3D modelbased on the homologous protein structures available in protein databases (PDB) (Nielsen et al., 2010). The effect of the amino acid substitu-tion on TMC1 protein structurewas analyzed in silico using the I-Mutant2.0 (Capriotti et al., 2005) and SDM (Site Directed Mutator) (Worthet al., 2011)methods. The native andmutant protein structureswere vi-sualized using YASARA (Krieger and Vriend, 2014).
3. Results
3.1. Exome sequencing and SNP filtering
By whole exome sequencing, we obtained 42 279 068 reads with anaverage sequence depth of 53×. About 97% of the total short reads weremapped to human reference sequence. A total of 121,839 single nu-cleotide polymorphisms, 6205 deletions and 5404 insertions wereidentified.
These variants werefiltered to identify pathogenic variants, with thefollowing strategy: we focused on missense, nonsense and frameshiftmutations and excluded intronic, 5′UTR, 3′UTR, and synonymousvariants. We discarded common variants reported in NHLBI ExomeSequencing Project (ESP), Exome Variant Server (EVS) (Tennessenet al., 2012), and the 1000 Genomes Project, with a frequency greaterthan 1%. Deleterious SNPs predicted by SIFT, PolyPhen-2 and Provensoftwares were retained. According to the pedigree, we hypothesizedan autosomal recessive mode of inheritance. Thus, we selected
1 gene causes non-syndromic hearing loss in aMoroccan family., Gene

3A. Bakhchane et al. / Gene xxx (2015) xxx–xxx
homozygous and compoundheterozygousmutations. Based on this strat-egy, we identified a single novel deleterious mutation c.1810C N G(p.Arg604Gly) at homozygous state in the exon 20 of the TMC1 gene.This mutation (chr9: 75435804) is localized in a 4 megabases homozy-gous region located on chromosome 9q21.13. This variant has not yetbeen reported and is absent from all actual databases. Using Sanger se-quencing, we found that both parents were heterozygous, the two affect-ed offspring were homozygous for the mutation, while the healthysiblings were heterozygous or homozygous wild-type (Fig. 1.B). Thisnovel mutation was analyzed on 63 Moroccan probands from recessivemultiplex families without genetic diagnosis in order to identify thesame mutation in another family, and on 90 Moroccan controls. None ofthe patients and the controls subjects showed this novel mutation.
3.2. Modeling and stability analysis of mutant structure
Homology model building methods was used to generate three-dimensional structure of TMC1 protein, using CPHmodels-3.2 Server.Structural analyses were performed for evaluating the impact of the
Fig. 2. TMC1 gene mutation identified in family SF107 with autosomal hearing loss. A: Cytogenstructure of TMC1 protein. C: Amino acid conservation map across species demonstrating that
Please cite this article as: Bakhchane, A., et al., A novel mutation in the TMC(2015), http://dx.doi.org/10.1016/j.gene.2015.07.075
p.Arg604Gly missense mutation on the protein stability. This mutationis predicted by the I-Mutant 2.0 and SDM (Site Directed Mutator)methods to be highly destabilizing and causing conformational changeson the protein 3D structure, thus possibly leading to protein dysfunction.Indeed, the TMC1 mutation involves the change of Arginine a polar hy-drophilic residue in general found on protein surface, to Glycine a neutralhydrophobic residue, in general found inside the protein structure. Inthis respect, our prediction study revealed that in the native proteinstructure, the Arginine-604 is found on polypeptide chain surface(Fig. 3A), whereas in themutant protein, the Glycine is found in the inte-rior of the folded polypeptide (Fig. 3B). Thus, this mutation may lead tochanges in TMC1 protein folding, and consequently to its inactivationor degradation.
4. Discussion
Not only whole exome sequencing is an effective way for screeningmutations in a huge number of genes, but it is also less expensive ifwe compare this method to other classical genetic methods, especially
etic location of TMC1 gene locus on chromosome 9 and mutation position. B: SchematicArginine at position 604 is well conserved throughout vertebrates.
1 gene causes non-syndromic hearing loss in aMoroccan family., Gene

Fig. 3. Three-dimensional structuralmodeling. (A) Segments of TMC1protein highlightingposition and orientation of the native amino acid Arginine-604. (B) Same structural pre-diction as in (A) but with the Glycine-604 substitution showing a reverse orientation ofthe amino-acid residue.
Table 2Overview of all TMC1 mutations so far identified.
Origin Mutation DNA Protein Exon(
China c.589G N A p.G197R E11China c.1171C N T p.Q391X E15China c.1247 T N G p.L416RChina c.1312G N A p.A438TChina c.236 + 1G N C –China c.1107C N A p.N369KChina c.1209G N C p.W403CChina c.1253 T N A p.M418K E16India c.1960A N G p.M654V E20India c.237-6 T N G – IntronIndia c.453 + 2 T N C – IntronIndia c.628_630del p.I210del Exon 1India c.800G N A p.G267E Exon 1India c.1566 + 1G N A – IntronIran c.776 + 1G N A – E7Greece c.2350C N T p.R604X E20Morocco c.1810C N G p.Arg604Gly E20Pakistan c. IVS10-8 T N A – I10Pakistan c. IVS13 + 1G N A – I13
Pakistan c.884 + 1G N A – E13Pakistan c.830A N G p.Y277C E13Pakistan c.IVS5 + 1G N T Splice site I5Pakistan c.1534C N T p.R512X E17Pakistan c.536-8 T N A – E11Pakistan c.2004 T N G p.S668R E21Pakistan c.2035G N A p.E679K E21Pakistan c.1541C N T p.P514L E17
4 A. Bakhchane et al. / Gene xxx (2015) xxx–xxx
Please cite this article as: Bakhchane, A., et al., A novel mutation in the TMC(2015), http://dx.doi.org/10.1016/j.gene.2015.07.075
for extremely heterogeneous diseases such as ARNSHL. In this work,WES disclosed in a Moroccan family a novel missense mutationc.1810C N G (p.Arg604Gly) in exon 20 in TMC1 gene (Fig. 2.A) whichis responsible for the DFNA36 and DFNB7/11 form of deafness(Kurima et al., 2002). This mutation is predicted to be deleterious asstated by Polyphen-2 (Adzhubei et al., 2010), MutationTaster(Schwarz et al., 2010) and SIFT (http://sift.jcvi.org/). TMC1 gene encodesa transmembrane channel-like protein 1, which includes six transmem-brane domains (Fig. 2.B). This protein does not show similarity to anyother known protein and is expressed specifically in hair cells of the co-chlea and in the neurosensory epithelium of the vestibular organ, andmandatory for their normal functions (Kurima et al., 2002). The identi-fied missense mutation c.1810C N G (p.Arg604Gly) can cause structuralchanges in TMC1 structure affecting its function, and eventually leadingto the phenotype observed in the affected offspring. The pathogenic ef-fect of the p.Arg604Gly mutation on the three-dimensional structure issuggested by the prediction programs as reported in the graphicalmodel (Fig. 3) In addition, this amino-acid position is well-conservedamong species (Fig. 2.C). To date, a total of 52 different mutations havebeen detected in TMC1 gene (Table 2). In North Africa, TMC1 mutationswere identified in Algeria, Morocco (the current study) and Tunisia. InTunisia, a novel mutation (c.2260 + 2 T) was recently described bywhole exome sequencing in intron 23 of TMC1 (Riahi et al., 2014),while another mutation in TMC1 exon 7 c.100C N T (p.R34X) was veryfrequently found (5.55%) in Tunisian patients with ARSHNL, suggestinga founder effect (Ben Said et al., 2010). This mutation c.100C N T wasalso present in a family from Algeria (Ammar-Khodja et al., 2015).
In conclusion, usingwhole exome sequencing, we identified the firstTMC1mutation, c.1810C NG (p.Arg604Gly) in theMoroccan population.The identification of supplementary disease-causing mutations in thisgene should confirm further the essential role of the TMC1protein in au-ditory function.
Disclosure statements
The authors declare not having any conflict of interest.
E)/Intron(I) Type of variant Domain Reference
Missense IC1 Gao et al. (2013)Nonsense EC2 Gao et al. (2013)Missense EC2 Chen et al. (2015)Missense EC2 Chen et al. (2015)Splicing site – Yang et al. (2013)Missense TM3 Yang et al. (2013)Missense EC2 Yang et al. (2013)Missense EC2 Zhao et al. (2014)Missense TM5 Kurima et al. (2002)
7 Splice site regulation – Ganapathy et al. (2014)9 Splice site regulation – Ganapathy et al. (2014)1 Deletion TM1 Ganapathy et al. (2014)3 Missense EC1 Ganapathy et al. (2014)17 Splice site regulation – Ganapathy et al. (2014)
Splice site mutation – Hilgert et al. (2008)Nonsense EC3 Hilgert et al. (2008)Missense IC3 The present studySplice site mutation – Kurima et al. (2002)Splice site mutation – Kurima et al. (2002)
Santos et al. (2005)Splice site mutation – Kurima et al. (2002)Missense TM2 Santos et al. (2005)Splice site mutation – Kitajiri et al. (2007b)Nonsense IC3 Kurima et al. (2002)Splice site mutation – Santos et al. (2005)Missense EC3 Santos et al. (2005)Missense EC3 Santos et al. (2005)Missense IC3 Kitajiri et al. (2007b)
1 gene causes non-syndromic hearing loss in aMoroccan family., Gene
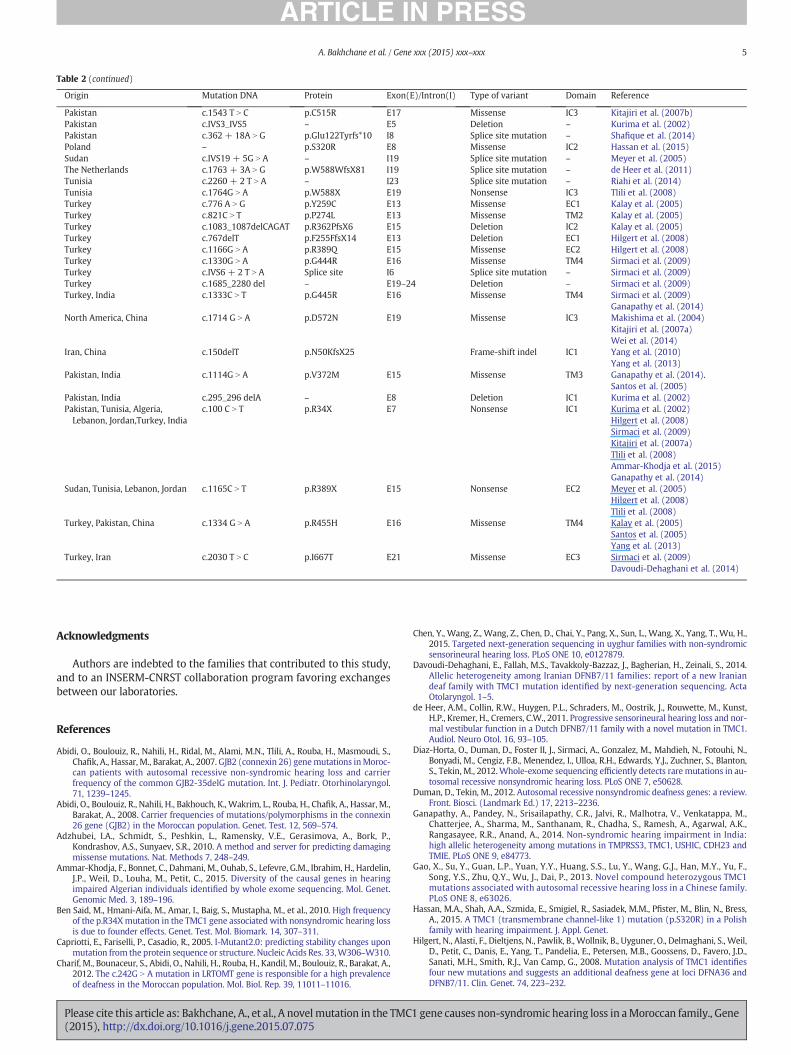
Table 2 (continued)
Origin Mutation DNA Protein Exon(E)/Intron(I) Type of variant Domain Reference
Pakistan c.1543 T N C p.C515R E17 Missense IC3 Kitajiri et al. (2007b)Pakistan c.IVS3_IVS5 – E5 Deletion – Kurima et al. (2002)Pakistan c.362 + 18A N G p.Glu122Tyrfs*10 I8 Splice site mutation – Shafique et al. (2014)Poland – p.S320R E8 Missense IC2 Hassan et al. (2015)Sudan c.IVS19 + 5G N A – I19 Splice site mutation – Meyer et al. (2005)The Netherlands c.1763 + 3A N G p.W588WfsX81 I19 Splice site mutation – de Heer et al. (2011)Tunisia c.2260 + 2 T N A – I23 Splice site mutation – Riahi et al. (2014)Tunisia c.1764G N A p.W588X E19 Nonsense IC3 Tlili et al. (2008)Turkey c.776 A N G p.Y259C E13 Missense EC1 Kalay et al. (2005)Turkey c.821C N T p.P274L E13 Missense TM2 Kalay et al. (2005)Turkey c.1083_1087delCAGAT p.R362PfsX6 E15 Deletion IC2 Kalay et al. (2005)Turkey c.767delT p.F255FfsX14 E13 Deletion EC1 Hilgert et al. (2008)Turkey c.1166G N A p.R389Q E15 Missense EC2 Hilgert et al. (2008)Turkey c.1330G N A p.G444R E16 Missense TM4 Sirmaci et al. (2009)Turkey c.IVS6 + 2 T N A Splice site I6 Splice site mutation – Sirmaci et al. (2009)Turkey c.1685_2280 del – E19–24 Deletion – Sirmaci et al. (2009)Turkey, India c.1333C N T p.G445R E16 Missense TM4 Sirmaci et al. (2009)
Ganapathy et al. (2014)North America, China c.1714 G N A p.D572N E19 Missense IC3 Makishima et al. (2004)
Kitajiri et al. (2007a)Wei et al. (2014)
Iran, China c.150delT p.N50KfsX25 Frame-shift indel IC1 Yang et al. (2010)Yang et al. (2013)
Pakistan, India c.1114G N A p.V372M E15 Missense TM3 Ganapathy et al. (2014).Santos et al. (2005)
Pakistan, India c.295_296 delA – E8 Deletion IC1 Kurima et al. (2002)Pakistan, Tunisia, Algeria,Lebanon, Jordan,Turkey, India
c.100 C N T p.R34X E7 Nonsense IC1 Kurima et al. (2002)Hilgert et al. (2008)Sirmaci et al. (2009)Kitajiri et al. (2007a)Tlili et al. (2008)Ammar-Khodja et al. (2015)Ganapathy et al. (2014)
Sudan, Tunisia, Lebanon, Jordan c.1165C N T p.R389X E15 Nonsense EC2 Meyer et al. (2005)Hilgert et al. (2008)Tlili et al. (2008)
Turkey, Pakistan, China c.1334 G N A p.R455H E16 Missense TM4 Kalay et al. (2005)Santos et al. (2005)Yang et al. (2013)
Turkey, Iran c.2030 T N C p.I667T E21 Missense EC3 Sirmaci et al. (2009)Davoudi-Dehaghani et al. (2014)
5A. Bakhchane et al. / Gene xxx (2015) xxx–xxx
Acknowledgments
Authors are indebted to the families that contributed to this study,and to an INSERM-CNRST collaboration program favoring exchangesbetween our laboratories.
References
Abidi, O., Boulouiz, R., Nahili, H., Ridal, M., Alami, M.N., Tlili, A., Rouba, H., Masmoudi, S.,Chafik, A., Hassar, M., Barakat, A., 2007. GJB2 (connexin 26) genemutations inMoroc-can patients with autosomal recessive non-syndromic hearing loss and carrierfrequency of the common GJB2-35delG mutation. Int. J. Pediatr. Otorhinolaryngol.71, 1239–1245.
Abidi, O., Boulouiz, R., Nahili, H., Bakhouch, K., Wakrim, L., Rouba, H., Chafik, A., Hassar, M.,Barakat, A., 2008. Carrier frequencies of mutations/polymorphisms in the connexin26 gene (GJB2) in the Moroccan population. Genet. Test. 12, 569–574.
Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P.,Kondrashov, A.S., Sunyaev, S.R., 2010. A method and server for predicting damagingmissense mutations. Nat. Methods 7, 248–249.
Ammar-Khodja, F., Bonnet, C., Dahmani, M., Ouhab, S., Lefevre, G.M., Ibrahim, H., Hardelin,J.P., Weil, D., Louha, M., Petit, C., 2015. Diversity of the causal genes in hearingimpaired Algerian individuals identified by whole exome sequencing. Mol. Genet.Genomic Med. 3, 189–196.
Ben Said, M., Hmani-Aifa, M., Amar, I., Baig, S., Mustapha, M., et al., 2010. High frequencyof the p.R34Xmutation in the TMC1 gene associated with nonsyndromic hearing lossis due to founder effects. Genet. Test. Mol. Biomark. 14, 307–311.
Capriotti, E., Fariselli, P., Casadio, R., 2005. I-Mutant2.0: predicting stability changes uponmutation from the protein sequence or structure. Nucleic Acids Res. 33,W306–W310.
Charif, M., Bounaceur, S., Abidi, O., Nahili, H., Rouba, H., Kandil, M., Boulouiz, R., Barakat, A.,2012. The c.242G N A mutation in LRTOMT gene is responsible for a high prevalenceof deafness in the Moroccan population. Mol. Biol. Rep. 39, 11011–11016.
Please cite this article as: Bakhchane, A., et al., A novel mutation in the TMC(2015), http://dx.doi.org/10.1016/j.gene.2015.07.075
Chen, Y., Wang, Z., Wang, Z., Chen, D., Chai, Y., Pang, X., Sun, L., Wang, X., Yang, T., Wu, H.,2015. Targeted next-generation sequencing in uyghur families with non-syndromicsensorineural hearing loss. PLoS ONE 10, e0127879.
Davoudi-Dehaghani, E., Fallah, M.S., Tavakkoly-Bazzaz, J., Bagherian, H., Zeinali, S., 2014.Allelic heterogeneity among Iranian DFNB7/11 families: report of a new Iraniandeaf family with TMC1 mutation identified by next-generation sequencing. ActaOtolaryngol. 1–5.
de Heer, A.M., Collin, R.W., Huygen, P.L., Schraders, M., Oostrik, J., Rouwette, M., Kunst,H.P., Kremer, H., Cremers, C.W., 2011. Progressive sensorineural hearing loss and nor-mal vestibular function in a Dutch DFNB7/11 family with a novel mutation in TMC1.Audiol. Neuro Otol. 16, 93–105.
Diaz-Horta, O., Duman, D., Foster II, J., Sirmaci, A., Gonzalez, M., Mahdieh, N., Fotouhi, N.,Bonyadi, M., Cengiz, F.B., Menendez, I., Ulloa, R.H., Edwards, Y.J., Zuchner, S., Blanton,S., Tekin, M., 2012.Whole-exome sequencing efficiently detects raremutations in au-tosomal recessive nonsyndromic hearing loss. PLoS ONE 7, e50628.
Duman, D., Tekin, M., 2012. Autosomal recessive nonsyndromic deafness genes: a review.Front. Biosci. (Landmark Ed.) 17, 2213–2236.
Ganapathy, A., Pandey, N., Srisailapathy, C.R., Jalvi, R., Malhotra, V., Venkatappa, M.,Chatterjee, A., Sharma, M., Santhanam, R., Chadha, S., Ramesh, A., Agarwal, A.K.,Rangasayee, R.R., Anand, A., 2014. Non-syndromic hearing impairment in India:high allelic heterogeneity among mutations in TMPRSS3, TMC1, USHIC, CDH23 andTMIE. PLoS ONE 9, e84773.
Gao, X., Su, Y., Guan, L.P., Yuan, Y.Y., Huang, S.S., Lu, Y., Wang, G.J., Han, M.Y., Yu, F.,Song, Y.S., Zhu, Q.Y., Wu, J., Dai, P., 2013. Novel compound heterozygous TMC1mutations associated with autosomal recessive hearing loss in a Chinese family.PLoS ONE 8, e63026.
Hassan, M.A., Shah, A.A., Szmida, E., Smigiel, R., Sasiadek, M.M., Pfister, M., Blin, N., Bress,A., 2015. A TMC1 (transmembrane channel-like 1) mutation (p.S320R) in a Polishfamily with hearing impairment. J. Appl. Genet.
Hilgert, N., Alasti, F., Dieltjens, N., Pawlik, B., Wollnik, B., Uyguner, O., Delmaghani, S., Weil,D., Petit, C., Danis, E., Yang, T., Pandelia, E., Petersen, M.B., Goossens, D., Favero, J.D.,Sanati, M.H., Smith, R.J., Van Camp, G., 2008. Mutation analysis of TMC1 identifiesfour new mutations and suggests an additional deafness gene at loci DFNA36 andDFNB7/11. Clin. Genet. 74, 223–232.
1 gene causes non-syndromic hearing loss in aMoroccan family., Gene

6 A. Bakhchane et al. / Gene xxx (2015) xxx–xxx
Hilgert, N., Smith, R.J., Van Camp, G., 2009. Function and expression pattern of nonsyn-dromic deafness genes. Curr. Mol. Med. 9, 546–564.
Kalay, E., Karaguzel, A., Caylan, R., Heister, A., Cremers, F.P., Cremers, C.W., Brunner, H.G.,de Brouwer, A.P., Kremer, H., 2005. Four novel TMC1 (DFNB7/DFNB11) mutations inTurkish patients with congenital autosomal recessive nonsyndromic hearing loss.Hum. Mutat. 26, 591.
Kitajiri, S., Makishima, T., Friedman, T.B., Griffith, A.J., 2007a. A novel mutation at theDFNA36 hearing loss locus reveals a critical function and potential genotype–phenotype correlation for amino acid-572 of TMC1. Clin. Genet. 71, 148–152.
Kitajiri, S.I., McNamara, R., Makishima, T., Husnain, T., Zafar, A.U., Kittles, R.A., Ahmed,Z.M., Friedman, T.B., Riazuddin, S., Griffith, A.J., 2007b. Identities, frequencies and or-igins of TMC1 mutations causing DFNB7/B11 deafness in Pakistan. Clin. Genet. 72,546–550.
Krieger, E., Vriend, G., 2014. YASARA View — molecular graphics for all devices — fromsmartphones to workstations. Bioinformatics 30, 2981–2982.
Kurima, K., Peters, L.M., Yang, Y., Riazuddin, S., Ahmed, Z.M., Naz, S., Arnaud, D., Drury, S.,Mo, J., Makishima, T., Ghosh, M., Menon, P.S., Deshmukh, D., Oddoux, C., Ostrer, H.,Khan, S., Riazuddin, S., Deininger, P.L., Hampton, L.L., Sullivan, S.L., Battey Jr., J.F.,Keats, B.J., Wilcox, E.R., Friedman, T.B., Griffith, A.J., 2002. Dominant and recessivedeafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cellfunction. Nat. Genet. 30, 277–284.
Makishima, T., Kurima, K., Brewer, C.C., Griffith, A.J., 2004. Early onset and rapid progres-sion of dominant nonsyndromic DFNA36 hearing loss. Otol. Neurotol. 25, 714–719.
Meyer, C.G., Gasmelseed, N.M., Mergani, A., Magzoub, M.M., Muntau, B., Thye, T.,Horstmann, R.D., 2005. Novel TMC1 structural and splice variants associated withcongenital nonsyndromic deafness in a Sudanese pedigree. Hum. Mutat. 25, 100.
Nahili, H., Charif, M., Boulouiz, R., Bounaceur, S., Benrahma, H., Abidi, O., Chafik, A., Rouba,H., Kandil, M., Barakat, A., 2010. Prevalence of themitochondrial A 1555Gmutation inMoroccan patients with non-syndromic hearing loss. Int. J. Pediatr. Otorhinolaryngol.74, 1071–1074.
Nielsen, M., Lundegaard, C., Lund, O., Petersen, T.N., 2010. CPHmodels-3.0—remote ho-mology modeling using structure—guided sequence profiles. Nucleic Acids Res. 38,W576–W581.
Riahi, Z., Bonnet, C., Zainine, R., Louha, M., Bouyacoub, Y., Laroussi, N., Chargui, M., Kefi, R.,Jonard, L., Dorboz, I., Hardelin, J.P., Salah, S.B., Levilliers, J., Weil, D., McElreavey, K.,Boespflug, O.T., Besbes, G., Abdelhak, S., Petit, C., 2014. Whole exome sequencingidentifies new causative mutations in Tunisian families with non-syndromic deaf-ness. PLoS ONE 9, e99797.
Santos, R.L., Wajid, M., Khan, M.N., McArthur, N., Pham, T.L., Bhatti, A., Lee, K., Irshad, S.,Mir, A., Yan, K., Chahrour, M.H., Ansar, M., Ahmad, W., Leal, S.M., 2005. Novel
Please cite this article as: Bakhchane, A., et al., A novel mutation in the TMC(2015), http://dx.doi.org/10.1016/j.gene.2015.07.075
sequence variants in the TMC1 gene in Pakistani families with autosomal recessivehearing impairment. Hum. Mutat. 26, 396.
Schwarz, J.M., Rodelsperger, C., Schuelke, M., Seelow, D., 2010. MutationTaster evaluatesdisease-causing potential of sequence alterations. Nat. Methods 7, 575–576.
Shafique, S., Siddiqi, S., Schraders, M., Oostrik, J., Ayub, H., Bilal, A., Ajmal, M., Seco, C.Z.,Strom, T.M., Mansoor, A., Mazhar, K., Shah, S.T., Hussain, A., Azam, M., Kremer, H.,Qamar, R., 2014. Genetic spectrum of autosomal recessive non-syndromic hearingloss in Pakistani families. PLoS ONE 9, e100146.
Sirmaci, A., Duman, D., Ozturkmen-Akay, H., Erbek, S., Incesulu, A., Ozturk-Hismi, B., Arici,Z.S., Yuksel-Konuk, E.B., Tasir-Yilmaz, S., Tokgoz-Yilmaz, S., Cengiz, F.B., Aslan, I.,Yildirim, M., Hasanefendioglu-Bayrak, A., Aycicek, A., Yilmaz, I., Fitoz, S., Altin, F.,Ozdag, H., Tekin, M., 2009. Mutations in TMC1 contribute significantly to nonsyn-dromic autosomal recessive sensorineural hearing loss: a report of five novel muta-tions. Int. J. Pediatr. Otorhinolaryngol. 73, 699–705.
Tennessen, J.A., Bigham, A.W., O'Connor, T.D., Fu, W., Kenny, E.E., Gravel, S., McGee, S., Do,R., Liu, X., Jun, G., Kang, H.M., Jordan, D., Leal, S.M., Gabriel, S., Rieder, M.J., Abecasis, G.,Altshuler, D., Nickerson, D.A., Boerwinkle, E., Sunyaev, S., Bustamante, C.D., Bamshad,M.J., Akey, J.M., Broad, G.O., Seattle, G.O., Project, N.E.S., 2012. Evolution and function-al impact of rare coding variation from deep sequencing of human exomes. Science337, 64–69.
Tlili, A., Rebeh, I.B., Aifa-Hmani, M., Dhouib, H., Moalla, J., Tlili-Chouchene, J., Said, M.B.,Lahmar, I., Benzina, Z., Charfedine, I., Driss, N., Ghorbel, A., Ayadi, H., Masmoudi, S.,2008. TMC1 but not TMC2 is responsible for autosomal recessive nonsyndromic hear-ing impairment in Tunisian families. Audiol. Neuro Otol. 13, 213–218.
Wei, Q., Zhu, H., Qian, X., Chen, Z., Yao, J., Lu, Y., Cao, X., Xing, G., 2014. Targeted genomiccapture and massively parallel sequencing to identify novel variants causing Chinesehereditary hearing loss. J. Transl. Med. 12, 311.
Worth, C.L., Preissner, R., Blundell, T.L., 2011. SDM—a server for predicting effects of mu-tations on protein stability and malfunction. Nucleic Acids Res. 39, W215–W222.
Yang, T., Kahrizi, K., Bazazzadeghan, N., Meyer, N., Najmabadi, H., Smith, R.J., 2010. A novelmutation adjacent to the Bthmousemutation in the TMC1 genemakes this mouse anexcellent model of human deafness at the DFNA36 locus. Clin. Genet. 77, 395–398.
Yang, T., Wei, X., Chai, Y., Li, L., Wu, H., 2013. Genetic etiology study of the non-syndromicdeafness in Chinese Hans by targeted next-generation sequencing. Orphanet J. RareDis. 8, 85.
Zhao, Y., Wang, D., Zong, L., Zhao, F., Guan, L., Zhang, P., Shi, W., Lan, L., Wang, H., Li, Q.,Han, B., Yang, L., Jin, X., Wang, J., Wang, J., Wang, Q., 2014. A novel DFNA36 mutationin TMC1 orthologous to the Beethoven (Bth) mouse associated with autosomal dom-inant hearing loss in a Chinese family. PLoS ONE 9, e97064.
1 gene causes non-syndromic hearing loss in aMoroccan family., Gene
Related Documents











