A Missense Mutation in the Sodium Phosphate Co-transporter Slc34a1 Impairs Phosphate Homeostasis Takayuki Iwaki,* Mayra J. Sandoval-Cooper,* Harriet S. Tenenhouse, † and Francis J. Castellino* *W.M. Keck Center for Transgene Research and the Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana; and † Department of Human Genetics, McGill University-Montreal Children’s Hospital Research Institute, Montreal, Quebec, Canada ABSTRACT The sodium phosphate co-transporters Npt2a and Npt2c play important roles in the regulation of phosphate homeostasis. Slc34a1, the gene encoding Npt2a, resides downstream of the gene encoding coagulation factor XII (f12) and was inadvertently modified while generating f12 / mice. In this report, the renal consequences of this modification are described. The combined single allelic mutant Slc34a1m contains two point mutations in exon 13: A499V is located in intracellular loop 5, and V528M is located in transmembrane domain 11. In addition to the expected coagulopathy of the f12 / phenotype, mice homozygous for the double allelic modification (f12 / /slc34a1 m/m ) displayed hypophosphatemia, hy- percalcemia, elevated levels of alkaline phosphatase, urolithiasis, and hydronephrosis. Strategic cross- breedings demonstrated that the kidney-related pathology was associated only with autosomal reces- sive transmission of the slc34a1 m gene and was not influenced by the simultaneous inactivation of f12. Npt2a[V528M] could be properly expressed in opossum kidney cells, but Npt2a[A499V] could not. These results suggest that a single amino acid substitution in Npt2a can lead to improper translocation of the protein to the cell membrane, disturbance of phosphate homeostasis, and renal calcification. Whether point mutations in the SLC34A1 gene can lead to hypophosphatemia and nephrolithiasis in humans remains unknown. J Am Soc Nephrol 19: 1753–1762, 2008. doi: 10.1681/ASN.2007121360 Three classes of sodium phosphate (Na/P i ) trans- porters have been identified. The most important of these for regulation of inorganic P i in mouse kidney are members of the type 2 family, viz, Npt2a (gene slc34a1) and Npt2c (gene slc34a3). These transport proteins are specifically expressed in the brush bor- der membranes of renal proximal tubules, where the bulk of filtered P i is reabsorbed, and mediate Na gradient– dependent transport of P i from primary urine to proximal tubular cells. 1 Regulation of Npt2a protein abundance in the apical membrane by factors such as dietary phosphate, parathyroid hormone, 2–4 and phosphatonins 5 occurs via the ac- tion of scaffolding proteins and protein kinases. 6,7 Inactivation of murine slc34a1 in mice led to a decrease in renal P i reabsorption arising from a de- fect in Na/P i co-transport. 8 The accompanying hy- pophosphatemia in slc34a1 / mice was not fully compensated by the consequent 2.8-fold increase in Npt2c protein abundance in kidney proximal tu- bules. 9 These biochemical abnormalities, as well as hypercalcemia and hypercalciuria in slc34a1 / Received December 21, 2007. Accepted April 9, 2008. Published online ahead of print. Publication date available at www.jasn.org. Correspondence: Dr. Francis J. Castellino, W.M. Keck Center for Transgene Research, 230 Raclin-Carmichael Hall, Notre Dame, IN 46556. Phone: 574-631-9152; Fax: 574-631-8017; E-mail: [email protected] Copyright 2008 by the American Society of Nephrology BASIC RESEARCH www.jasn.org J Am Soc Nephrol 19: 1753–1762, 2008 ISSN : 1046-6673/1909-1753 1753

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

A Missense Mutation in the Sodium PhosphateCo-transporter Slc34a1 Impairs PhosphateHomeostasis
Takayuki Iwaki,* Mayra J. Sandoval-Cooper,* Harriet S. Tenenhouse,† andFrancis J. Castellino*
*W.M. Keck Center for Transgene Research and the Department of Chemistry and Biochemistry, University of NotreDame, Notre Dame, Indiana; and †Department of Human Genetics, McGill University-Montreal Children’s HospitalResearch Institute, Montreal, Quebec, Canada
ABSTRACTThe sodium phosphate co-transporters Npt2a and Npt2c play important roles in the regulation ofphosphate homeostasis. Slc34a1, the gene encoding Npt2a, resides downstream of the gene encodingcoagulation factor XII (f12) and was inadvertently modified while generating f12�/� mice. In this report,the renal consequences of this modification are described. The combined single allelic mutant Slc34a1mcontains two point mutations in exon 13: A499V is located in intracellular loop 5, and V528M is locatedin transmembrane domain 11. In addition to the expected coagulopathy of the f12�/� phenotype, micehomozygous for the double allelic modification (f12�/�/slc34a1m/m) displayed hypophosphatemia, hy-percalcemia, elevated levels of alkaline phosphatase, urolithiasis, and hydronephrosis. Strategic cross-breedings demonstrated that the kidney-related pathology was associated only with autosomal reces-sive transmission of the slc34a1m gene and was not influenced by the simultaneous inactivation of f12.Npt2a[V528M] could be properly expressed in opossum kidney cells, but Npt2a[A499V] could not. Theseresults suggest that a single amino acid substitution in Npt2a can lead to improper translocation of theprotein to the cell membrane, disturbance of phosphate homeostasis, and renal calcification. Whetherpoint mutations in the SLC34A1 gene can lead to hypophosphatemia and nephrolithiasis in humansremains unknown.
J Am Soc Nephrol 19: 1753–1762, 2008. doi: 10.1681/ASN.2007121360
Three classes of sodium phosphate (Na/Pi) trans-porters have been identified. The most important ofthese for regulation of inorganic Pi in mouse kidneyare members of the type 2 family, viz, Npt2a (geneslc34a1) and Npt2c (gene slc34a3). These transportproteins are specifically expressed in the brush bor-der membranes of renal proximal tubules, wherethe bulk of filtered Pi is reabsorbed, and mediate Nagradient– dependent transport of Pi from primaryurine to proximal tubular cells.1 Regulation ofNpt2a protein abundance in the apical membraneby factors such as dietary phosphate, parathyroidhormone,2– 4 and phosphatonins5 occurs via the ac-tion of scaffolding proteins and protein kinases.6,7
Inactivation of murine slc34a1 in mice led to adecrease in renal Pi reabsorption arising from a de-
fect in Na/Pi co-transport.8 The accompanying hy-pophosphatemia in slc34a1�/� mice was not fullycompensated by the consequent 2.8-fold increase inNpt2c protein abundance in kidney proximal tu-bules.9 These biochemical abnormalities, as well ashypercalcemia and hypercalciuria in slc34a1�/�
Received December 21, 2007. Accepted April 9, 2008.
Published online ahead of print. Publication date available atwww.jasn.org.
Correspondence: Dr. Francis J. Castellino, W.M. Keck Center forTransgene Research, 230 Raclin-Carmichael Hall, Notre Dame, IN46556. Phone: 574-631-9152; Fax: 574-631-8017; E-mail:[email protected]
Copyright � 2008 by the American Society of Nephrology
BASIC RESEARCH www.jasn.org
J Am Soc Nephrol 19: 1753–1762, 2008 ISSN : 1046-6673/1909-1753 1753

mice, were associated with low body weight, mild skeletal ab-normalities,8 and urolithiasis.10
The phenotype of slc34a1�/� mice resembles that of pa-tients with hereditary hypophosphatemic rickets with hyper-calciuria (HHRH), an inherited disorder of Pi homeostasis,11
with the exception that the mice have a milder skeletal pheno-type.12,13 On this basis, it was hypothesized that mutations inthe human orthologue SLC34A1 were responsible for HHRH.However, genome-based studies on kindreds with HHRH in-dicated that mutations of NPT2a were not responsible for thisdisease,14 and subsequent studies identified mutations inSLC34A3 as possibly causative in these and other HHRH pa-tient populations.15,16 Whether point mutations in theSLC34A1 gene can lead to hypophosphatemia and nephrolithi-asis in humans is an unresolved issue. An approach to address-ing this question can be made through the generation of miceharboring known mutations in slc34a1. However, to date, nopoint mutations in Npt2a have been mechanistically linked torenal pathologies in controlled mouse studies.
The gene arrangement in the vicinity of slc34a1 is, consec-utively, 5�-[coagulation factor 12 (f12)/profilin-3 (pfn3)/slc34a1]-3�, with this latter gene arranged in reverse orienta-tion. To generate mice with a total inactivation of thecoagulation f12 gene, we first designed a targeting vector (TV)with homologous recombination 5� and 3� sites external to thef12 gene at its untranslated regions (UTR). Although we con-firmed that the resulting mice lacked f12 mRNA or FXII pro-tein, we also found unexpected kidney-related phenotypes inthese mice (unpublished observations). Since then, other linesof f12�/� mice have been generated by us and by others17,18
through exon-deletion targeting strategies, with homologousrecombination sites in the TV designed entirely within the f12gene sequence. In these mice (f12�/�-L2), f12 message andFXII protein were entirely eliminated, but kidney-related pa-
thologies were not found. Furthermore, renal disease has notbeen linked to FXII deficiencies in humans.
The unexpected phenotypes in the f12�/�-L1 mice with to-tal exon deletions were reminiscent of those in slc34a1�/�
mice,8,10 rather than the phenotypes of the f12�/�-L2 mice.17,18
Thus, in this study we examined the integrity of the slc34a1gene in f12�/�-L1 mice as a candidate mechanism for the renalpathologies that were observed. These in-depth studies al-lowed novel mechanistic revelations to be made in regard tosignificant kidney disease in a mouse line containing minimalpoint mutations in slc34a1.
RESULTS
Targeted Inactivation of the f12 Gene to Generatef12-L1 MiceOn the basis of the mouse genomic arrangement of Figure1A, a targeting vector TV (Figure 1B) was constructed andelectroporated in mouse embryonic stem cells (ESC). Afterscreening the ESC with positive (G418)/negative (5�-fluo-rocytosine) selection, approximately 400 properly targetedcells were identified. DNA samples from these cells weredigested with EcoRI, after which the DNA fragments weresubjected to Southern blotting using the external probesEP1 and EP2 (Figure 1C). From these analyses, two inde-pendent and correctly targeted homologous recombinantswere identified (Figure 2A).
Generation of f12�/�-L1 MiceBlastocyst injections of the ESC yielded five male chimericmice, which were mated with C57Bl/6J female mice. Germlinetransmission was observed with at least three pairs, and f12�/�
mice (F1, 129SvJ/C57Bl6J; 50/50) were generated. Male F1
Figure 1. Generation of f12�/�-L1 mice. (A)The top line represents the f12 gene with its 14exons (light gray boxes), the pfn3 gene with itsone exon (black box), and exons 11 through 13of the slc34a1 gene (dark gray boxes). Somekey restriction enzyme sites are indicated (RI,EcoRI; RV, EcoRV; H, HindIII; S, SalI). (B) Con-struction of the TV for f12�/�-L1 mice. An RI-Hdigestion region of the 5�-UTR (A) and an RI-Sdigestion fragment of the 3�-UTR (A) of the f12gene were used to flank the NEO cDNA, whichwas used for positive selection after homolo-gous recombination in mouse embryonic stemcells. A cytidine deaminase (CDA) cassette wasinserted downstream of the RI-S fragment andused for negative selection of recombinants.This process resulted in replacement of theentire f12 coding sequence with NEO. *One RIsite was newly generated for purposes ofscreening. EP1 and EP2 in A represent externalprobes used for the 5� and 3� flanks of the TV,respectively. (C) Expected mutated allele.
BASIC RESEARCH www.jasn.org
1754 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1753–1762, 2008

f12�/�-L1 mice were intermated with female f12�/�-L1 mice,thus providing f12�/�-L1 mice (Figure 2B).
Characterization of f12�/�-L1 mice showed the completeabsence of f12 mRNA in liver (data not shown) and ofFXII antigen in plasma (Figure 2C). Coagulation assays off12�/�-L1 plasma also demonstrated greatly elevated acti-vated partial thromboplastin times (aPTT; data not shown),similar to the activated partial thromboplastin times foundin our f12�/�-L2 mice.18
Low Body Weight, Urolithiasis, and Hydronephrosis inthe f12�/�-L1 MiceThe body sizes of the f12�/�-L1 mice were consistently smallerthan those of wild-type (WT) and of f12�/�-L1 mice at 6 wkof age (Figure 2D). Massive calcifications were observed inf12�/�-L1 kidneys (Figure 2, J through L), even as young as 6wk of age (data not shown). In 16-wk-old f12�/�-L1 mice,kidney cysts developed (Figure 2, F and G), along with severehydronephrosis at 24 wk (Figure 2H). In contrast, neither WTmice (Figure 2, E and I) nor f12�/�-L2 mice18 presented withthese kidney abnormalities, even to �1 yr of age, suggesting theabsence of functional mutations in the slc34a1 gene in thislatter mouse line. In agreement with these observations,plasma Pi levels were 9.3 � 0.31 (n � 9), 6.4 � 0.1 (n � 13),and 9.6 � 0.48 (n � 6) in WT, f12�/�-L1, and f12�/�-L2 mice,respectively, showing that hypophosphatemia is found only in
the f12�/�-L1 mice, consistent with the kidney abnormalitiesin this same line of mice.
These results were supported by transabdominal ultrasoundanalysis, which revealed high echogenic regions in f12�/�-L1 kid-neys at 6 wk of age. These data are entirely consistent with renalcalcification (Figure 2N). Similar regions were abundantly foundin the entire kidneys and were associated with low echogeniccystic regions in 16-wk-old f12�/�-L1 mice (Figure 2O). Hy-dronephrotic echo patterns were obtained from 24-wk-oldmice (Figure 2P), demonstrating significant kidney destruc-tion. In contrast, no abnormalities in were found WT mice,even at 36 wk of age (Figure 2M).
Sequence Analysis of pfn3 and slc34a1 in f12�/�-L1MiceBecause the genes pfn3 and slc34a1 are present in the 3�-UTRhomologous recombination region of the TV used to constructf12�/�-L1 mice, we believed that the hypophosphatemia andkidney abnormalities seen in the f12�/�-L1 mice were the re-sult of mutations in slc34a1. Thus, the sequences of both pfn3and slc34a1 were determined from f12�/�-L1 mouse genomicDNA. The portion of the slc34a1 3� sequence that was used forhomologous recombination in f12�/�-L1 mice is shown inFigure 3. Two point mutations, viz., A499V and V528M, werediscovered in exon 13 of the slc34a1 gene in the f12�/�-L1mouse genome (Figure 3), which were not detected in WT and
Figure 2. Screening and characterization off12�/�-L1 mice. (A) Southern blot analysiswith the 5�- and 3�-external probes of di-gested DNA from ESC transfected with f12.The 5� external probe (EP1 in Figure 1A) dis-tinguished the WT-f12 (11.5 kb) and null (6.7kb) alleles with RI-digested DNA, and the 3�external probe (EP2 in Figure 1A) distin-guished the WT-f12 (W; 7.8 kb) and null (N;5.2 kb) alleles with RI-digested DNA. Molec-ular weight standards (M) are shown in lane 1;control 129/SvJ DNA (C) is shown in lane 2,and individual targeted ESC samples are enu-merated in the remaining lanes. (B) PCR-based genotyping of typical WT (lane 2),f12�/�-L1 (lane 3) and f12�/�-L2 (lane 4)mouse tail-tip DNA. (C) Western blot analysisof typical WT (lane 1), f12�/�-L1 (lane 2), andf12�/�-L1 (lane 3) mouse plasmas, using apolyclonal antibody to human FXII, showingthe absence of FXII antigen in f12�/�-L1mouse plasma. (D) Body sizes of WT, f12�/�-L1, and f12�/�-L1 mice. (E) Hematoxylin II and eosin Y (H & E) stains for WT mouse kidney at 36 wk of age. (F through H) H & E stainsof f12�/�-L1 mouse kidney at 6 wk of age (F), 16 wk of age (G), and 24 wk of age (H). The arrows in F and G indicate areas of kidneywith abnormal cellularity, with widespread kidney architecture disruption in G. (I) von Kossa stains for WT mouse kidney at 36 wk of ageand of f12�/�-L1 mouse kidneys at 6 wk of age (J), 16 wk of age (K), and 24 wk of age (L). The arrows point to examples of calcificationsin J through L. Transabdominal ultrasound images for WT mouse kidney at 36 wk of age (M), f12�/�-L1 mouse kidney at 6 wk of age(N), 16 wk of age (O), and 24 wk of age (P). Red arrows in (N) and (O) indicate the high echogenic parts, which are typical indicationsof calcification. The arrow in N shows a calcification in a low echogenic cystic region of the kidney. An asterisk in (P) indicates a largearea of low echogenicity, which is a typical signature of renal hydronephritis. Magnification, �200.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1753–1762, 2008 Mutations in the Mouse Slc34a1 Gene 1755

f12�/�-L2 mice or in the human or rat counterpart slc34a1genes (Figure 3); therefore, we redefined f12�/�-L1 mice asf12�/�/slc34a1m/m to avoid any confusion with f12�/�-L2mice, the genotype of which has been experimentally deter-mined through genomic sequence analysis to be f12�/�/slc34a1�/�. Last, the pfn3 gene (Figure 3) was fully sequencedand found not to be altered in the f12�/�-L1 genome.
Additional Breedings of MiceBecause the f12�/� and slc34a1m/m genes were linked inf12�/�/slc34a1m/m mice, it was not possible to investigate theindividual contributions of each genotype in vivo with thismouse line. Thus, we created new mouse lines by multiplecross-breedings to resolve this issue. We cross-bred f12�/
�-L2 mice18 (i.e., f12�/�/slc34a1�/�) with f12�/�/slc34a1�/�8 mice to generate f12�/�/slc34a1�/� mice. Wealso cross-bred f12�/�/slc34a1m/m mice with f12�/�/slc34a1�/� mice to generate f12�/�/slc34a1m/� mice andf12�/�/slc34a1m/m mice with f12�/�/slc34a1�/� mice toprovide f12�/�/slc34a1�/m mice, each after multiple cross-breeding (Figure 4, A and B). Neither the coagulopathy norrenal abnormalities that were exhibited by singly deficientf12�/� or slc34a1�/� mice, respectively, or by the double-deficient f12�/�/slc34a1m/m mice were found in f12�/�/slc34a1�/�, fXII�/�/slc34a1�/�, or f12�/�/slc34a1�/m mice.Thus, the genetic traits were transmitted in an autosomalrecessive manner for each of the f12 and slc34a1 genes, andthe cross-breeding strategies provided us with the mice nec-essary to separate the phenotypes of the linked defects in thef12 and slc34a1 genes in the f12�/�/slc34a1m/m mice.
Serum Pi, Ca2�, and ALP Levels in MiceSerum Pi levels in f12�/�/slc34a1m/m mice were significantlylower than those in WT and f12�/�/slc34a1�/� mice (Figure4C). In contrast, serum Ca2� (Figure 4C) and ALP (Figure 4D)levels in f12�/�/slc34a1m/m mice were significantly higher thanthose in WT and f12�/�/slc34a1�/� mice. To rule out the pos-sibility that lack of FXII affected Npt2a functions, f12�/�/slc34a1�/�, f12�/�/slc34a11m/m, and f12�/�/slc34a1�/� micewere intercrossed to generate several double heterozygotecombinations depicted in Figure 4, A and B. In this series, only
Figure 3. Sequence analysis of the 3�-flanking region of f12�/�-L1. (A) Amino acid sequence comparisons of a region of theslc34a1 gene in humans and rodents with f12�/�-L1 mice, show-ing the two mutations (boxed) in the latter. TMD, transmembranedomain; ICL, intracellular loop. (B) The two point mutations (*) inexon 13 resulting from the recombination event in the 3� genomicregion of f12�/�-L1 mice are indicated and represented inslc34a1 in the bottom panel. This gene is oriented in the oppositedirection of the f12 and pfn3 genes, and only exons 11, 12, and13 (gray boxes) are shown because the 3� flank of the TV does notextend beyond this region. The relative location of the pfn3 geneis also shown with its single exon (gray box).
Figure 4. A scheme of genotype variations andplasma chemistries of f12 and slc34a1 gene alteredmice. (A) Genotypes of three different lines of f12/slc34a1 mice that were the starting points of thecross-breedings. (B) Genotypes of mice that wereproduced from the cross-breedings. (C through E)Serum Pi levels (C), serum Ca2� levels (D), and serumALP levels (E) of the various genotypic combinationsof mice obtained from the cross-breedings. *Thelevels in f12�/�/slc34a1�/� sera were significantlydifferent from those of WT, f12�/�/slc34a1�/�, f12�/�/slc34a1�/m, f12�/�/slc34a1�/m, f12�/�/slc34a1�/�,and f12�/�/slc34a1�/� sera but not significantly differ-ent from those of f12�/�/slc34a1m/m and f12�/�/slc34a1m/� sera. #The levels in f12�/�/slc34a1m/� weresignificantly different from those of WT, f12�/�/slc34a1�/�, f12�/�/slc34a1�/m, f12�/�/slc34a1�/m,f12�/�/slc34a1�/�, and f12�/�/slc34a1�/� sera, butnot significantly different from those in f12�/�/slc34a1�/� and f12�/�/slc34a1m/m sera. †The levels inf12�/�/slc34a1m/m serum were significantly different
from those in WT, f12�/�/slc34a1�/�, f12�/�/slc34a1�/m, f12�/�/slc34a1�/m, f12�/�/slc34a1�/�, and f12�/�/slc34a1�/� sera but notsignificantly different from those in f12�/�/slc34a1�/� and f12�/�/slc34a1m/� sera.
BASIC RESEARCH www.jasn.org
1756 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1753–1762, 2008

f12�/�/slc34a1m/� mice displayed similar profiles of Pi (Figure4C), Ca2� (Figure 4D), and ALP (Figure 4E) as f12�/�/slc34a1m/m and f12�/�/slc34a1�/� mice. These results clearlyindicate that the FXII deletion did not affect Npt2a functionand that one intact copy of slc34a1 with one mutated slc34a1copy was sufficient to maintain normal serum concentrationsof Pi, Ca2�, and ALP. In addition, these data ruled out domi-nant negative effects of the slc34a1m gene on WT slc34a1.
Transcript Levels of slc34a1 in KidneysKidney levels of slc34a1 transcript were similar in WT mice(1.9 � 0.4 � 108 copies/100 ng total RNA [n � 6]), andf12�/�/slc34a1m/m mice (1.9 � 0.5 � 108 copies/100 ng totalRNA [n � 6]). Testicular transcript levels of pfn3 in WT and inf12�/�/slc34a1m/m mice were 6.1 � 1.2 � 106 copies/100 ngtotal RNA (n � 6) and 11.0 � 2.3 � 106 100 ng total RNA (n �6), respectively. These data show that transcription of the twoimmediate downstream genes is not affected by the mutationin the f12 gene.
Immunohistochemistry of Npt2a in KidneyAnti-Npt2a immunostainings of kidney sections from themice of Figure 4B were positive in the epithelia of proximaltubules from WT mice (Figure 5A). As expected, Npt2a immu-nostaining was negative in f12�/�/slc34a1�/� mice (Figure5B). Whereas f12�/�/slc34a1�/� mice also expressed Npt2a(Figure 5C), immunostaining for Npt2a was not evident inf12�/�/slc34a1m/m mice (Figure 5D), despite that its transcriptlevels were similar to those in WT mice. Furthermore, f12�/�/slc34a1�/m mice expressed Npt2a (Figure 5E). For furtherprobing of the reason for interference in Npt2a expression in
f12�/�/slc34a1m/m mice, f12�/�/slc34a1�/�, f12�/�/slc34a1m/�,and f12�/�/slc34a1�/m mice were generated. As clearly illustrated,f12�/�/slc34a1�/� (Figure 5F) and f12�/�/slc34a1m/� (Figure5H) mice stained positively for Npt2a. Conversely, f12�/�/slc34a1m/� mice did not express Npt2a antigen (Figure 5G). Thus,there was no interference in Npt2a expression from the absence ofFXII, indicating that the double-mutated alleles in the slc34a1gene were the cause of the abnormal renal phenotype in f12�/�/slc34a1m/m mice.
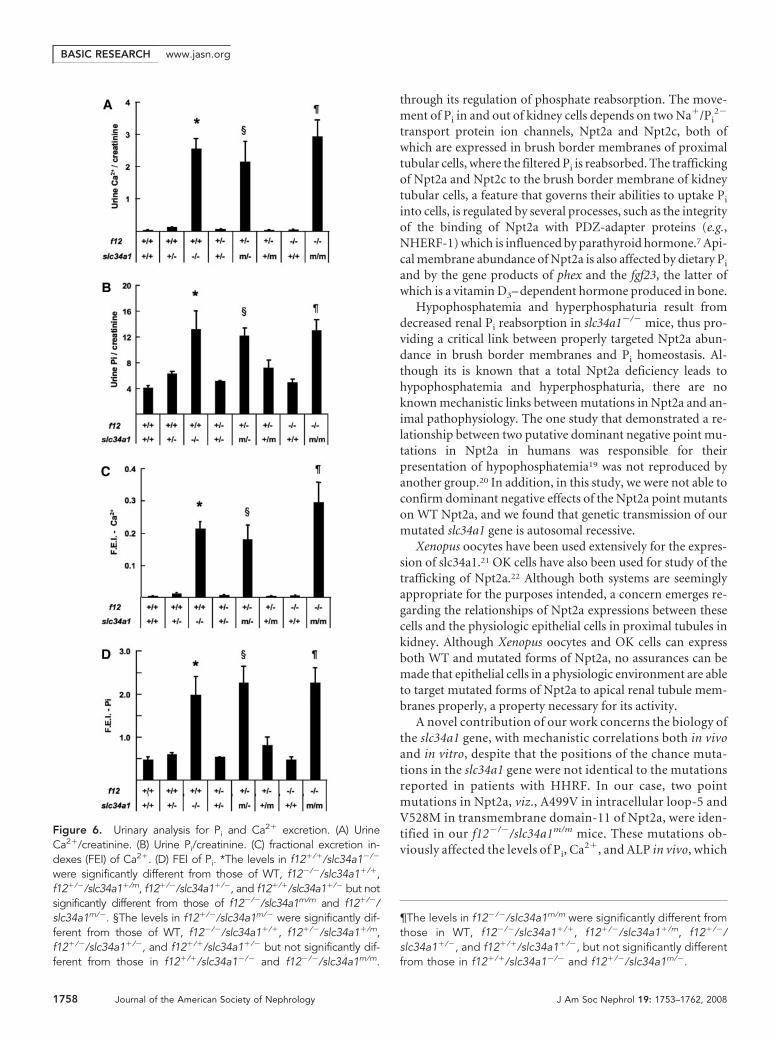
Urinary Analyses for Ca2� and Pi ExcretionUrinary analyses clearly indicated that creatinine-normalizedurine levels of Pi and Ca2� were increased in f12�/�/slc34a1�/�
and further elevated in f12�/�/slc34a1�/� mice (Figure 6, Aand B). These alterations also corresponded to the fractionalexcretion of them (Figure 6, C and D). These values in f12�/�/slc34a1�/� were similar to those in WT mice. These values inf12�/�/slc34a1�/m and f12�/�/slc34a1�/� were close to thosein f12�/�/slc34a1�/� mice, and those in f12�/�/slc34a1m/� andf12�/�/slc34a1m/m were close to those in f12�/�/slc34a1�/�
mice (Figure 6).
Fluorescence Microscopy of Transfected OpossumKidney CellsFluorescence microscopic analysis showed that there were noendogenous positive recombinant green fluorescence protein(GFP) signals in opossum kidney (OK) cells transfected withpCS2-empty plasmid (Figure 7A) or in OK cells transfectedwith pCS2-containing cDNA that express WT-Npt2a,Npt2a[A499V], Npt2a[V528M], or Npt2a[A499V/V528M](data not shown). Conversely, as expected, strongly positiveGFP signals were found in OK cells transfected with pCS2-GFP(Figure 7B). Alexa Fluora 488 –positive stainings were found inOK cells expressing recombinant WT-Npt2a orNpt2a[V528M] (Figure 7, C and E, respectively), showing ex-pression of Npt2a. In contrast, Alexa Fluora 488 –positivestainings were not found in OK cells expressing heterologousNpt2a[A499V] or Npt2a[A499V/V528M] (Figure 7, D and F,respectively). These results show that there was no expressionof Npt2a[A499V] or Npt2a[A499V/V528M] and/or that themutant proteins were conformationally altered and not recog-nized by the antibody to WT-Npt2a. Five separate transienttransfections confirmed that neither Npt2a[A499V] norNpt2a[A499V/V528M] antigen was observed in the cells.Higher magnified and cross-sectional images demonstratedthat the positive GFP signals were diffusely observed in thecytoplasm of OK cells expressing GFP (Figure 7, G and g).Apical anti-Npt2a stains were seen in OK cells expressing re-combinant WT-Npt2a (Figure 7, H and h) or Npt2a[V528M](Figure 7, I and i).
DISCUSSION
The kidney is primarily responsible for phosphate homeostasisand, in general, is a determinant for blood phosphate levels
Figure 5. (A through H) Immunostains of Npt2a in WT (A),f12�/�/slc34a1�/� (B), f12�/�/slc34a1�/� (C), f12�/�/slc34a1m/m
(D), f12�/�/slc34a1�/m (E), f12�/�/slc34a1�/� (F), f12�/�/slc34a1m/�, (G) and f12�/�/slc34a1�/m (H) kidneys. Magnifica-tion, �400.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1753–1762, 2008 Mutations in the Mouse Slc34a1 Gene 1757
through its regulation of phosphate reabsorption. The move-ment of Pi in and out of kidney cells depends on two Na�/Pi
2�
transport protein ion channels, Npt2a and Npt2c, both ofwhich are expressed in brush border membranes of proximaltubular cells, where the filtered Pi is reabsorbed. The traffickingof Npt2a and Npt2c to the brush border membrane of kidneytubular cells, a feature that governs their abilities to uptake Pi
into cells, is regulated by several processes, such as the integrityof the binding of Npt2a with PDZ-adapter proteins (e.g.,NHERF-1) which is influenced by parathyroid hormone.7 Api-cal membrane abundance of Npt2a is also affected by dietary Pi
and by the gene products of phex and the fgf23, the latter ofwhich is a vitamin D3– dependent hormone produced in bone.
Hypophosphatemia and hyperphosphaturia result fromdecreased renal Pi reabsorption in slc34a1�/� mice, thus pro-viding a critical link between properly targeted Npt2a abun-dance in brush border membranes and Pi homeostasis. Al-though its is known that a total Npt2a deficiency leads tohypophosphatemia and hyperphosphaturia, there are noknown mechanistic links between mutations in Npt2a and an-imal pathophysiology. The one study that demonstrated a re-lationship between two putative dominant negative point mu-tations in Npt2a in humans was responsible for theirpresentation of hypophosphatemia19 was not reproduced byanother group.20 In addition, in this study, we were not able toconfirm dominant negative effects of the Npt2a point mutantson WT Npt2a, and we found that genetic transmission of ourmutated slc34a1 gene is autosomal recessive.
Xenopus oocytes have been used extensively for the expres-sion of slc34a1.21 OK cells have also been used for study of thetrafficking of Npt2a.22 Although both systems are seeminglyappropriate for the purposes intended, a concern emerges re-garding the relationships of Npt2a expressions between thesecells and the physiologic epithelial cells in proximal tubules inkidney. Although Xenopus oocytes and OK cells can expressboth WT and mutated forms of Npt2a, no assurances can bemade that epithelial cells in a physiologic environment are ableto target mutated forms of Npt2a to apical renal tubule mem-branes properly, a property necessary for its activity.
A novel contribution of our work concerns the biology ofthe slc34a1 gene, with mechanistic correlations both in vivoand in vitro, despite that the positions of the chance muta-tions in the slc34a1 gene were not identical to the mutationsreported in patients with HHRF. In our case, two pointmutations in Npt2a, viz., A499V in intracellular loop-5 andV528M in transmembrane domain-11 of Npt2a, were iden-tified in our f12�/�/slc34a1m/m mice. These mutations ob-viously affected the levels of Pi, Ca2�, and ALP in vivo, which
Figure 6. Urinary analysis for Pi and Ca2� excretion. (A) UrineCa2�/creatinine. (B) Urine Pi/creatinine. (C) fractional excretion in-dexes (FEI) of Ca2�. (D) FEI of Pi. *The levels in f12�/�/slc34a1�/�
were significantly different from those of WT, f12�/�/slc34a1�/�,f12�/�/slc34a1�/m, f12�/�/slc34a1�/�, and f12�/�/slc34a1�/� but notsignificantly different from those of f12�/�/slc34a1m/m and f12�/�/slc34a1m/�. §The levels in f12�/�/slc34a1m/� were significantly dif-ferent from those of WT, f12�/�/slc34a1�/�, f12�/�/slc34a1�/m,f12�/�/slc34a1�/�, and f12�/�/slc34a1�/� but not significantly dif-ferent from those in f12�/�/slc34a1�/� and f12�/�/slc34a1m/m.
¶The levels in f12�/�/slc34a1m/m were significantly different fromthose in WT, f12�/�/slc34a1�/�, f12�/�/slc34a1�/m, f12�/�/slc34a1�/�, and f12�/�/slc34a1�/�, but not significantly differentfrom those in f12�/�/slc34a1�/� and f12�/�/slc34a1m/�.
BASIC RESEARCH www.jasn.org
1758 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1753–1762, 2008

led to hypophosphatemia in these mice. Only mice homozy-gous for the double mutation in Npt2a displayed renal pa-thologies, consistent with altered Na�/Pi
2� transport. Fur-thermore, the presence of this protein in kidney wasdiminished in both f12�/�/slc34a1m/m and f12�/�/slc34a1m/� mice. This diminution of protein expressionmight be a result of mutation-based misfolding, because weshow nearly normal levels of slc34a1 mRNA in kidneys off12�/�/slc34a1m/m mice.
In vitro expression experiments demonstrated that renalproximal tubular OK cells that were transfected with slc34a1 orwith the mutated slc34a1 gene that led to Npt2a[V528M] cor-rectly expressed Npt2a; however, OK cells transfected with
genes mutated to produce Npt2a[A499V] orNpt2a[A499V/V528M] did not express Npt2a. Themechanisms required for successful expression ofNpt2a on apical membranes of polarized cells, invitro, depend on the cell types used and on aminoacid residues in the C-terminal intracellular tailsof this protein.23 Our results suggested that Npt2aprotein folding is important for its correct local-ized expression and is likely mediated by specificchaperones and by interactions with scaffoldingproteins, which are present in the brush bordermembrane of renal proximal tubules; therefore,Npt2a[A499V] protein likely is not recognized bythese chaperones and/or scaffolds, and this could re-sult in its premature degradation in the cells.
Additional examples of point mutations in trans-porter proteins that lead to severe pathologies areknown (e.g., cystic fibrosis [CF], a disease caused bymutations in the gene that encodes the CF trans-membrane conductance regulator [CFTR]). Manyreports of point mutations in this protein that resultin its malfunction are known, and the most commonof these CF-causing mutations (�F508) reaches theplasma membrane in reduced amounts as a result ofprotein misfolding.24 Other single amino acid substi-tutions in the CFTR gene also result in protein mis-folding and lead to reduced expression on the cellsurface.25,26 Thus, it seems that point mutations inthe Npt2a co-transporter may have a similar impacton protein processing. In conclusion, this work hasshown that abnormal kidney pathologies can mech-anistically result from simple point mutations inNpt2a.
CONCISE METHODS
Screening of �-Phage LibrariesOligonucleotide primer set X1UP (5�-GGTCTCTGCTG-
ATGAGTCTG, cDNA position 43 to 62) and X2RP (5�-
GTGCGTCCTTAAATTTCTTG, cDNA position 98 to
117), or X13UP (5�-GGCTGAAGAATACTCCACCT,
cDNA position 1499 to 1518) and X14RP (5�-CACTAGCTTCACT-
GTGACCC, cDNA position 1880 to 1899), based on the murine f12
cDNA sequence in the NCBI server (NM 021489), was used to obtain
the fragment of the f12 gene that contained partial exon 1/intron
A/partial exon 2 (F1) or partial exon 13/intron M/partial exon 14
(F2), respectively, from tail genomic DNA of 129/SvJ mice (Jackson
Laboratories, Bar Harbor, ME) using PCR. PCR products resulting
from amplifications with these 418-bp F1 or 486-bp F2 PCR probes
were used to isolate genomic clones containing the f12 gene from a
129/SvJ �-FIXII genomic library (Stratagene, La Jolla, CA), and sev-
eral appropriate DNA clones were obtained. The resulting genomic
clones spanned approximately 9 kb of the 5�-UTR, followed by the
entire f12 gene (14 exons and 13 introns) and approximately 1 kb of
Figure 7. Fluorescence and laser scanning confocal microscopy for trans-fected OK cells. (A through F) Immunostains of Npt2a in OK cells transfectedwith pCS2-empty plasmid (A), pCS2-GFP (B), pCS2-slc34a1 (C), pCS2-slc34a1[A499V] (D), pCS2-slc34a1[V528M] (E), and pCS2-slc34a1[A499/V528M] (F). (G through I) Immunostains of Npt2a in OK cells transfected withpCS2-GFP (G), pCS2-slc34a1 (H), and pCS2-slc34a1[V528M] (I) using an oilimmersion objective. (g through i) Cross-sections along the planes from Gthrough I represented by white lines, respectively. Clipping planes wereplaced at the regions of interest, which are indicated as white lines in Gthrough I, and used to expose the GFP-positive (g) or NPT2 Alexa Fluor488–positive (h and i) labeling and its cellular localization. The images wererendered to 120� perspective projection and captured at a zoom factor of0.857 pixel/voxel. XY and XZ orthogonal images were captured. Magnifica-tions: �20 in A through F; �60 in G through I.
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1753–1762, 2008 Mutations in the Mouse Slc34a1 Gene 1759
the 3�-UTR. Despite many rescreening attempts to obtain a longer
3�-UTR for more effective genomic recombination, no additional
useful clones were obtained. Previous results27 suggested that use of
�-libraries containing genomic sequences led to rearrangements of
sequences between the 3�-UTR of the f12 and slc34a1 genes, and cor-
rect sequences were not obtained using this approach. The strategy for
screening was then changed to vectorette and suppression long and
accurate PCR (VS-LA-PCR), and 6 kb of the 3�-UTR was obtained.
This allowed a TV to be constructed with ample flanking sequences of
the 5�- and 3�-UTR regions of mouse f12.
Extended 3�-UTR Sequences of the f12 GeneGenome walking with VS-LA-PCR was performed as described pre-
viously.28 Genomic DNA (50 mg) from 129/SvJ mice, prepared as in
the previous section, was digested by the restriction endonucleases
DraI, EcoICR1, EcoRV, PvuII, ScaI, SspI, and StuI (Promega, Madi-
son, WI), individually. After the digestion, each fragment was ligated
to an adaptor consisting of two oligonucleotides, 5�-pACCAGCCC-
NH2 and 5�-GTAATACGACTCACTATAGGGCACGCGTGGTCG-
ACGGCCCGGGCTGGT. VS-LA-PCR was performed several times
with a gene-specific primer and the adaptor-specific primer to obtain
sufficient sequence lengths.
The TV was constructed similar to those that we have previously
described29 from a basic plasmid containing the neomycin-resistant
gene (NEO) for positive selection along with 5�- and 3�-flanking se-
quences from the murine f12 gene. For the latter, a 5.2-kb EcoRV/
HindIII fragment and a 4.8-kb EcoRI/SalI fragment of the f12 gene
were used as the 5� and 3� flanks of f12, respectively, of the NEO gene
in the TV. The cytosine deaminase (CDA) cassette was inserted down-
stream of the 3�-flanking region of the TV for f12 for negative selec-
tion against random chromosomal integrants.
Homologous Recombination in ESCThe TV was used to replace, by homologous recombination, a 9.0-kb
fragment of the f12 gene with the NEO gene. This process eliminated
the entire f12 coding sequence from the mouse genome. The genera-
tion of targeted ESC and subsequent blastocyst injections were per-
formed.
Southern blot analyses with external probes on ESC DNA, after
digestion with EcoRI, were carried out to assess the integrity of the
homologous recombination at both the 5� and 3� terminal regions.
The 5� external probe (EP1; 421 bp, spanning bp 5846 to 5446 up-
stream of the ATG initiation codon of the f12 gene) was obtained by
PCR using primers 5�-CTCTCAGGAGAAAAAGTATACC and 5�-
CAGACCACATTCTCTTCATCC. The 3� external probe (EP2; 471
bp, spanning bp 5453 to 5923 from the TAA stop codon of the f12
gene) was also obtained by PCR using the primers 5�-GTCTACAT-
AGTTCTAACCAG and 5�-GGGTCCATGTGTTACCTTGTC.
GenotypingGenomic DNA was obtained by ear-punch biopsy. PCR was per-
formed with oligonucleotide primer sets: A common forward primer
(5�-TCTTCAGTCCGCTACTCCCAC, spanning bp 200 to 180 up-
stream of the ATG initiation codon of the f12 gene), and a reverse
primer in exon 1 of the f12 gene (5�-AGATCCAGACTCATCAGCAG,
spanning bp 48 to 67 of the f12 cDNA) to detect the WT allele (247
bp), or the aforementioned common forward primer along with a
reverse primer residing within NEO (5�-ACAAGCAAAACCAAAT-
TAAGGGCCA) to detect the null allele (466 bp).
Histochemistry and ImmunohistochemistryKidneys were obtained from mice at 6 wk of age, fixed with periodate-
lysine-paraformaldehyde, embedded in paraffin, and sectioned at a
thickness of 4 �m. The slides were stained with hematoxylin II and
eosin Y (Richard Allen Scientific, Kalamazoo, MI) for morphologic
analysis and von Kossa stain to detect calcification.
For immunostaining, tissue sections were deparaffinized and
placed in avidin and biotin blocking solutions (Zymed Laboratories,
South San Francisco, CA), followed by Peroxo-block (Zymed) to in-
hibit endogenous peroxidase activity. Anti-Npt2a immunostains
were conducted using the ImmPress Kit (Vector Laboratories, Bur-
lingame, CA). The sections were incubated with rabbit anti-human
primary antibodies (Alpha Diagnostics, San Antonio, TX), followed
by horseradish peroxidase– conjugated goat anti-rabbit IgG. The
slides were developed with 3-amino-9-ethylcarbazole and counter-
stained.
Transabdominal Ultrasound AnalysisMice were sedated by inhalation of 1.5% isoflurane sufficient to main-
tain their heart rates at approximately 500 bpm. The abdomens of the
mice were cleaned with a chemical hair remover (Nair) to minimize
ultrasound attenuation. Transabdominal ultrasound analysis was ac-
complished with a Vevo 770 system (VisualSonics, Toronto, ON,
Canada).
Sequence Analysis of slc34a1 cDNATotal RNA was obtained from kidneys using Trizol (Invitrogen,
Carlsbad, CA). First-strand cDNA was synthesized using Superscript-
Table 1. Primers and probes used for sequence analysis
Gene Type Sequence Position Size (bp)
pfn3 Forward 5�-CTAATGCAAGCACAAGTTGC 20 to 39 506Reverse 5�-GTTCACGGTTTATTCTGGTC 506 to 525
slc34a1 Forward 5�-GAGCTGAGCCACAGTCAAGG 14 to 33 864Fragment-1 Reverse 5�-GTGAAGGGCTCTGTGATGAC 858 to 877slc34a1 Forward 5�-CTTTTGCAGGGGCGACCGTG 700 to 719 735Fragment-2 Reverse 5�-GGCTGGCAACGGCTGCCAGG 1415 to 1434slc34a1 Forward 5�-GTGTGATCAGCATTGAGCGG 1345 to 1364 780Fragment-3 Reverse 5�-CTTAATATGCACAGGTACC 2106 to 2124
BASIC RESEARCH www.jasn.org
1760 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1753–1762, 2008

III reverse transcriptase (Invitrogen). Reverse transcriptase–PCR was
performed with the primers in Table 1 to obtain the designated frag-
ments for sequence analysis. After purification of PCR products, they
were directly sequenced using the same primers at the DNA Core
Sequencing Facility at the University of Illinois at Urbana-Cham-
paign.
Other MiceSlc34a1�/� mice were obtained from Dr. Beate Lanske (Harvard
School of Medicine, Boston, MA). The generation and characteriza-
tion of other mice used in this study, viz., f12�/�/slc34a1�/�8 and
f12�/�/slc34a1�/�,18 have been described.
Measurements of Serum and Urine Levels of Pi, Ca2�,Creatinine, and ALPSerum and urine levels of Pi, Ca2�, creatinine, and ALP were mea-
sured using the VetTest chemistry analyzer (IDEXX Laboratories,
Westbrook, ME). The fractional excretion indexes Pi or Ca2� were
calculated as follows: Urine Pi or Ca2�/(urine creatinine � serum Pi
or Ca2�).
Npt2a ConstructsAn expression vector backbone, pCS2, that contains the cytomegalo-
virus immediate early promoter to drive downstream inserted genes,
was obtained from Dr. Paul Huber (University of Notre Dame, Notre
Dame, Indiana) and first digested with BamHI and XbaI. A fragment
of recombinant GFP (Stratagene, La Jolla, CA) was amplified with the
forward primer hrGFP.F (sequences provided next), which contained
a BglII site (italic) and Kozak sequence (underlined), followed by the
initiation codon (bold), along with the reverse primer phrGFP.R,
which contained XbaI site (italic) just downstream of the termination
codon (bold) of phrGFP.R. All amplifications were performed using
PrimeStar DNA polymerase (TakaraBio, Madison, WI). The PCR
amplicon was digested by BglII and XbaI. The BglII- and XbaI-di-
gested fragment was ligated to pCS2 that was digested with BamHI
and XbaI. The resulting plasmid was designated pCS2-hrGFP. A frag-
ment of murine slc34a1 was amplified with the forward primer
Slc34a1.F, which contained a BglII site (italics) and the Kozak se-
quence (underlined), followed by the initiation codon (bold), and the
reverse primer Sc34a1.R, which contained a XbaI site (italics), just
downstream of the termination codon (bold) using an NIH Mamma-
lian Gene Collection (MGC) clone (clone 4223484; Invitrogen) as the
template. The PCR amplicon was digested by BglII and XbaI, and the
fragment obtained was ligated to pCS2 and digested by BamHI and
XbaI. The plasmid was designated pCS2-slc34a1.
hrGFP.F: 5�-GAAGATCTGCCGCCACCATGGTGAGCAAGCA-
GATCCT
hrGFP.R: 5�-GTTCTAGATTACACCCACTCGTGCAGGC
Slc34a1.F: 5�-GAAGATCTGCCGCCACCATGATGTCCTACAG-
CGAGAGATTG
Slc34a1.R: 5�-GTTCTAGACTAGAGACGGGTGGCATTGTG
Site-Directed MutagenesisThe single Npt2a mutations, viz., A499V and V528M, and the double
mutation, Npt2a[A499V/V528M], were introduced into pCS2-
slc34a1 by site-directed mutagenesis to construct a vector to express
Npt2a[A499V], Npt2a[V528M], and Npt2a[A499V/V528M]. For
this, an inverse PCR reaction was performed with forward primer
A499V.F, which contained the underlined mutation (next), along
with reverse primer, A499V.R, using pCS2-slc34a1 as the template to
construct an expression plasmid for Npt2a[A499V]. Also, two inverse
PCR reactions were performed with forward primer, V528M.F, which
contains the underlined mutation, in combination with reverse
primer, V528M.R, using pCS2-slc34a1 as the template to construct a
plasmid expressing Npt2a[V528M]. This same strategy, with pCS2-
slc34a1[A499V] as the template, was used to construct the plasmid
expressing Npt2a[A499V/V528M]. After the PCR reactions, these
amplicons were treated by TaKaRa BKL Kit (TakaraBio) for self-liga-
tion.
A499V.F: 5�-TCAAGGCCCTGGGCAAACGCAC
A499V.R: 5�-CCATGCGTATGGGCAGGCGTG
V528M.F: 5�-ATGTTTGGCATTTCCATGGCAGGCTGG
V528M.R: 5�-CAGCGAGGGCAAGAGCAGGAAGC
Cell Culture, Transfection, and FluorescenceMicroscopyOK cells were purchased from ATCC (Manassas, VA) and maintained
in DMEM/Ham’s F-12 medium (1:1), containing 10% FBS and 1�
antibiotic solution (Mediatech, Herndon, VA). Cells were seeded into
each chamber of a four-chamber culture slide (BD Biosciences, Bed-
ford, MA), and 50% confluent cultures were transiently transfected
with pCS2-empty plasmid, pCS2-GFP, and pCS2 plasmids containing
cDNA expressing WT-Npt2a, Npt2a[A499V], Npt2a[V528M], and
Npt2a[A499V/V528M]. Transfections were performed using 1 �g of
plasmid DNA and 3 �l of FuGENE6 (Roche Applied Science, India-
napolis, IN) per well. After the transfections, the cells were fixed with
periodate-lysine-paraformaldehyde and stained with rabbit anti-hu-
man Npt2a, followed by an Alexa Fluora 488 –labeled goat-anti-rabbit
IgG (Invitrogen). These cells were further stained with DAPI (Invitro-
gen). Laser Scanning Confocal Microscopy was conducted with a Ni-
kon Eclipse C1si confocal system on a Nikon TE2000-E microscope
with Spectral Imaging. An EZ-C1 imaging software (Nikon, Melville,
NY) was used for image acquisition. For gaining three-dimensional
visualization control of the regions of interest, images were processed
with Imaris (Bitplane AG, Zurich, Switzerland). The DAPI-labeled
nuclei were pseudocolored (red) for enhanced contrast against the
GFP-transfected cells or the Alexa Fluor 488 NPT2 positively labeled
cells (green). The Surpass mode was used to create a volume recon-
struction of the data set (Z-stack). The 20� data sets’ volume prop-
erties were rendered to a maximum intensity projection (MIP) mode
of all layers along the viewing direction (black background). A
Shadow Projection mode was used for the 60� data sets with a trans-
parency adjustment of the blend opacity to 33.6% for the green chan-
nel. The nuclei’s (red) blend opacity was maintained at 100%.
ACKNOWLEDGMENTS
This work was supported in part by grant HL073750 (to F.J.C.).
We thank Diana Cruz-Topete for assistance of histologic analysis,
BASIC RESEARCHwww.jasn.org
J Am Soc Nephrol 19: 1753–1762, 2008 Mutations in the Mouse Slc34a1 Gene 1761

Kyle Kisiel at Nikon Imaging Center at Northwestern University for
fluorescence and confocal imaging for transfected OK cells, and the
veterinary staff of the Freimann Life Sciences Facility for providing
husbandry of the mice.
DISCLOSURESNone.
REFERENCES
1. Forster IC, Kohler K, Biber J, Murer H: Forging the link betweenstructure and function of electrogenic cotransporters: The renal typeIIa Na�/Pi cotransporter as a case study. Prog Biophys Mol Biol 80:69–108, 2002
2. Murer H, Forster I, Biber J: The sodium phosphate cotransporterfamily SLC34. Pflugers Arch 447: 763–767, 2004
3. Forster IC, Hernando N, Biber J, Murer H: Proximal tubular handling ofphosphate: A molecular perspective. Kidney Int 70: 1548–1559, 2006
4. Tenenhouse HS: Phosphate transport: Molecular basis, regulation andpathophysiology. J Steroid Biochem Mol Biol 103: 572–577, 2007
5. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y,Fujita T, Nakahara K, Fukumoto S, Yamashita T: FGF-23 is a potentregulator of vitamin D metabolism and phosphate homeostasis.J Bone Miner Res 19: 429–435, 2004
6. Khadeer MA, Tang Z, Tenenhouse HS, Eiden MV, Murer H, HernandoN, Weinman EJ, Chellaiah MA, Gupta A: Na�-dependent phosphatetransporters in the murine osteoclast: Cellular distribution and proteininteractions. Am J Physiol Cell Physiol 284: C1633–C1644, 2003
7. Weinman EJ, Biswas RS, Peng Q, Shen L, Turner CL, Steplock D,Shenolikar S, Cunningham R: Parathyroid hormone inhibits renal phos-phate transport by phosphorylation of serine 77 of sodium-hydrogenexchanger regulatory factor-1. J Clin Invest 117: 3412–3420, 2007
8. Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, TenenhouseHS: Targeted inactivation of Npt2 in mice leads to severe renal phos-phate wasting, hypercalciuria, and skeletal abnormalities. Proc NatlAcad Sci U S A 95: 5372–5377, 1998
9. Tenenhouse HS, Martel J, Gauthier C, Segawa H, Miyamoto K: Dif-ferential effects of Npt2a gene ablation and X-linked Hyp mutation onrenal expression of Npt2c. Am J Physiol Renal Physiol 285: F1281–F1278, 2003
10. Chau H, El-Maadawy S, McKee MD, Tenenhouse HS: Renal calcifica-tion in mice homozygous for the disrupted type IIa Na/Pi cotrans-porter gene Npt2. J Bone Miner Res 18: 6446–6457, 2003
11. Tenenhouse HS, Sabbagh Y: Novel phosphate-regulating genes in thepathogenesis of renal phosphate wasting disorders. Pflugers Arch444: 317–326, 2002
12. Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D,Liberman UA: Hereditary hypophosphatemic rickets with hypercalci-uria. N Engl J Med 312: 611–617, 1985
13. Tieder M, Modai D, Shaked U, Samuel R, Arie R, Halabe A, Maor J,Weissgarten J, Averbukh Z, Cohen N: “Idiopathic” hypercalciuria andhereditary hypophosphatemic rickets: Two phenotypical expressionsof a common genetic defect. N Engl J Med 316: 125–129, 1987
14. Jones A, Tzenova J, Frappier D, Crumley M, Roslin N, Kos C, Tieder M,Langman C, Proesmans W, Carpenter T, Rice A, Anderson D, MorganK, Fujiwara T, Tenenhouse H: Hereditary hypophosphatemic ricketswith hypercalciuria is not caused by mutations in the Na/Pi cotrans-porter NPT2 gene. J Am Soc Nephrol 12: 507–514, 2001
15. Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabe-dian M, Sermet I, Fujiwara TM, Morgan K, Tenenhouse HS, Juppner H:SLC34A3 mutations in patients with hereditary hypophosphatemicrickets with hypercalciuria predict a key role for the sodium-phosphatecotransporter NaPi-IIc in maintaining phosphate homeostasis. Am JHum Genet 78: 179–192, 2006
16. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-RakoverY, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P,Schnabel D, Hochberg Z, Strom TM: Hereditary hypophosphatemicrickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet 78: 193–201, 2006
17. Pauer HU, Burfeind P, Kostering H, Emons G, Hinney B: Factor XIIdeficiency is strongly associated with primary recurrent abortions.Fertil Steril 80: 590–594, 2003
18. Iwaki T, Castellino FJ: Plasma levels of bradykinin are suppressed infactor XII-deficient mice. Thromb Haemost 95: 1003–1010, 2006
19. Prie D, Huart V, Bakouh N, Planelles G, Dellis O, Gerard B, Hulin P,Benque-Blanchet F, Silve C, Grandchamp B, Friedlander G: Nephro-lithiasis and osteoporosis associated with hypophosphatemia causedby mutations in the type 2a sodium-phosphate cotransporter. N EnglJ Med 347: 983–991, 2002
20. Virkki LV, Forster IC, Hernando N, Biber J, Murer H: Functional char-acterization of two naturally occurring mutations in the human sodium-phosphate cotransporter type IIa. J Bone Miner Res 18: 2135–2141,2003
21. Werner A, Biber J, Forgo J, Palacin M, Murer H: Expression of renaltransport systems for inorganic phosphate and sulfate in Xenopuslaevis oocytes. J Biol Chem 265: 12331–12336, 1990
22. Sorribas V, Markovich D, Hayes G, Stange G, Forgo J, Biber J, MurerH: Cloning of a Na/Pi cotransporter from opossum kidney cells. J BiolChem 269: 6615–6621, 1994
23. Hernando N, Sheikh S, Karim-Jimenez Z, Galliker H, Forgo J, Biber J,Murer H: Asymmetrical targeting of type II Na-P(i) cotransporters inrenal and intestinal epithelial cell lines. Am J Physiol Renal Physiol 278:F361–F368, 2000
24. Thomas PJ, Ko YH, Pedersen PL: Altered protein folding may be themolecular basis of most cases of cystic fibrosis. FEBS Lett 312: 7–9,1992
25. Ostedgaard LS, Zeiher B, Welsh MJ: Processing of CFTR bearing theP574H mutation differs from wild-type and deltaF508-CFTR. J Cell Sci112: 2091–2098, 1999
26. Mendes F, Roxo-Rosa M, Dragomir A, Farinha CM, Roomans GM,Amaral MD, Penque D: Unusually common cystic fibrosis mutation inPortugal encodes a misprocessed protein. Biochem Biophys Res Com-mun 311: 665–671, 2003
27. Braun A, Aszodi A, Hellebrand H, Berna A, Fassler R, Brandau O:Genomic organization of profilin-III and evidence for a transcript ex-pressed exclusively in testis. Gene 283: 219–225, 2002
28. Siebert PD, Chenchik A, Kellogg DE, Lukyanov KA, Lukyanov SA: Animproved PCR method for walking in uncloned genomic DNA. NucleicAcids Res 23: 1087–1088, 1995
29. Rosen E, Chan JC, Idusogie E, Clotman F, Vlasuk G, Luther T, JalbertL, Albrecht S, Zhong L, Lissens A, Schoonjans L, Moons L, Collen D,Castellino FJ, Carmeliet P: Mice lacking Factor VII develop normallybut suffer fatal perinatal bleeding. Nature 390: 290–294, 1997
See related editorial, “Of Mice and Men: Who Is in Control of Renal PhosphateReabsorption?” on pages 1625–1626.
BASIC RESEARCH www.jasn.org
1762 Journal of the American Society of Nephrology J Am Soc Nephrol 19: 1753–1762, 2008
Related Documents











