A jet fuel surrogate formulated by real fuel properties Stephen Dooley a, * , Sang Hee Won a , Marcos Chaos a , Joshua Heyne a , Yiguang Ju a , Frederick L. Dryer a , Kamal Kumar b , Chih-Jen Sung b , Haowei Wang c , Matthew A. Oehlschlaeger c , Robert J. Santoro d , Thomas A. Litzinger d a Department of Mechanical and Aerospace Engineering, Princeton University, Princeton, NJ 08544, USA b School of Engineering, University of Connecticut, Storrs, CT, USA c Department of Mechanical, Aerospace, and Nuclear Engineering, Rensselaer Polytechnic Institute, Troy, NY, USA d Propulsion Engineering Research Center, The Pennsylvania State University, University Park, PA, USA article info Article history: Received 30 June 2010 Accepted 2 July 2010 Available online 23 July 2010 Keywords: Jet fuel Surrogate formulation Kinetic model Group additivity Fuel properties Combustion jet-A abstract An implicit methodology based on chemical group theory to formulate a jet aviation fuel surrogate by the measurements of several combustion related fuel properties is tested. The empirical formula and derived cetane number of an actual aviation fuel, POSF 4658, have been determined. A three component surrogate fuel for POSF 4658 has been formulated by constraining a mixture of n-decane, iso-octane and toluene to reproduce the hydrogen/carbon ratio and derived cetane number of the target fuel. The validity of the proposed surrogate is evaluated by experimental measurement of select combustion properties of POSF 4658, and the POSF 4658 surrogate. (1) A variable pressure flow reactor has been used to chart the chemical reactivity of stoichiometric mixtures of POSF 4658/O 2 /N 2 and POSF 4658 surrogate/O 2 /N 2 at 12.5 atm and 500–1000 K, fixing the carbon content at 0.3% for both mixtures. (2) The high temperature chemical reactivity and chemical kinetic–molecular diffusion coupling of POSF 4658 and POSF 4658 surrogate have been evaluated by measurement of the strained extinc- tion limit of diffusion flames. (3) The autoignition behavior of POSF 4658 and POSF 4658 surrogate has been measured with a shock tube at 674–1222 K and with a rapid compression machine at 645–714 K for stoichiometric mix- tures of fuel in air at pressures close to 20 atm. The flow reactor study shows that the character and extent of chemical reactivity of both fuels at low temperature (500–675 K) and high temperature (900 K+) are extremely similar. Slight differences in the transition from the end of the negative temperature coefficient regime to hot ignition are observed. The diffusion flame strained extinction limits of the fuels are observed to be indistinguishable when com- pared on a molar basis. Ignition delay measurements also show that POSF 4658 exhibits NTC behavior. Moreover, the ignition delays of both fuels are also extremely similar over the temperature range studied in both shock tube and rapid compression machine experiments. A chemical kinetic model is constructed and utilized to interpret the experimental observations and provides a rationale as to why the real fuel and surrogate fuel exhibit such similar reactivity. Ó 2010 The Combustion Institute. Published by Elsevier Inc. All rights reserved. 1. Introduction The coupling of computational fluid dynamics with detailed chemical kinetics provides the promise of optimizing engine per- formance and exploring the effect of fuel variability on pollutant formation and engine efficiency in a facile and fundamental scien- tific manner. Jet aviation fuels, diesels and gasolines are complex mixtures of hundreds of chemical components. The detailed numerical simula- tion of the combustion of real fuels continues to be out of reach when applied to any fuel which is not a pure component or a mix- ture of more than a few components. The most prevalent proposal to circumvent this problem is the use of a surrogate fuel. A surro- gate should be comprised of only a handful of components but be capable of emulating the gas phase combustion characteristics of 0010-2180/$ - see front matter Ó 2010 The Combustion Institute. Published by Elsevier Inc. All rights reserved. doi:10.1016/j.combustflame.2010.07.001 * Corresponding author. Address: Department of Mechanical and Aerospace Engineering, Princeton University, Princeton, NJ 08544, USA. E-mail address: [email protected] (S. Dooley). Combustion and Flame 157 (2010) 2333–2339 Contents lists available at ScienceDirect Combustion and Flame journal homepage: www.elsevier.com/locate/combustflame

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Combustion and Flame 157 (2010) 2333–2339
Contents lists available at ScienceDirect
Combustion and Flame
journal homepage: www.elsevier .com/locate /combustflame
A jet fuel surrogate formulated by real fuel properties
Stephen Dooley a,*, Sang Hee Won a, Marcos Chaos a, Joshua Heyne a, Yiguang Ju a, Frederick L. Dryer a,Kamal Kumar b, Chih-Jen Sung b, Haowei Wang c, Matthew A. Oehlschlaeger c, Robert J. Santoro d,Thomas A. Litzinger d
a Department of Mechanical and Aerospace Engineering, Princeton University, Princeton, NJ 08544, USAb School of Engineering, University of Connecticut, Storrs, CT, USAc Department of Mechanical, Aerospace, and Nuclear Engineering, Rensselaer Polytechnic Institute, Troy, NY, USAd Propulsion Engineering Research Center, The Pennsylvania State University, University Park, PA, USA
a r t i c l e i n f o a b s t r a c t
Article history:Received 30 June 2010Accepted 2 July 2010Available online 23 July 2010
Keywords:Jet fuelSurrogate formulationKinetic modelGroup additivityFuel propertiesCombustion jet-A
0010-2180/$ - see front matter � 2010 The Combustdoi:10.1016/j.combustflame.2010.07.001
* Corresponding author. Address: Department ofEngineering, Princeton University, Princeton, NJ 0854
E-mail address: [email protected] (S. Dooley
An implicit methodology based on chemical group theory to formulate a jet aviation fuel surrogate by themeasurements of several combustion related fuel properties is tested. The empirical formula and derivedcetane number of an actual aviation fuel, POSF 4658, have been determined. A three component surrogatefuel for POSF 4658 has been formulated by constraining a mixture of n-decane, iso-octane and toluene toreproduce the hydrogen/carbon ratio and derived cetane number of the target fuel. The validity of theproposed surrogate is evaluated by experimental measurement of select combustion properties of POSF4658, and the POSF 4658 surrogate.
(1) A variable pressure flow reactor has been used to chart the chemical reactivity of stoichiometricmixtures of POSF 4658/O2/N2 and POSF 4658 surrogate/O2/N2 at 12.5 atm and 500–1000 K, fixingthe carbon content at 0.3% for both mixtures.
(2) The high temperature chemical reactivity and chemical kinetic–molecular diffusion coupling ofPOSF 4658 and POSF 4658 surrogate have been evaluated by measurement of the strained extinc-tion limit of diffusion flames.
(3) The autoignition behavior of POSF 4658 and POSF 4658 surrogate has been measured with a shocktube at 674–1222 K and with a rapid compression machine at 645–714 K for stoichiometric mix-tures of fuel in air at pressures close to 20 atm.
The flow reactor study shows that the character and extent of chemical reactivity of both fuels at lowtemperature (500–675 K) and high temperature (900 K+) are extremely similar. Slight differences in thetransition from the end of the negative temperature coefficient regime to hot ignition are observed. Thediffusion flame strained extinction limits of the fuels are observed to be indistinguishable when com-pared on a molar basis. Ignition delay measurements also show that POSF 4658 exhibits NTC behavior.Moreover, the ignition delays of both fuels are also extremely similar over the temperature range studiedin both shock tube and rapid compression machine experiments. A chemical kinetic model is constructedand utilized to interpret the experimental observations and provides a rationale as to why the real fueland surrogate fuel exhibit such similar reactivity.
� 2010 The Combustion Institute. Published by Elsevier Inc. All rights reserved.
1. Introduction
The coupling of computational fluid dynamics with detailedchemical kinetics provides the promise of optimizing engine per-formance and exploring the effect of fuel variability on pollutant
ion Institute. Published by Elsevier
Mechanical and Aerospace4, USA.).
formation and engine efficiency in a facile and fundamental scien-tific manner.
Jet aviation fuels, diesels and gasolines are complex mixtures ofhundreds of chemical components. The detailed numerical simula-tion of the combustion of real fuels continues to be out of reachwhen applied to any fuel which is not a pure component or a mix-ture of more than a few components. The most prevalent proposalto circumvent this problem is the use of a surrogate fuel. A surro-gate should be comprised of only a handful of components but becapable of emulating the gas phase combustion characteristics of
Inc. All rights reserved.
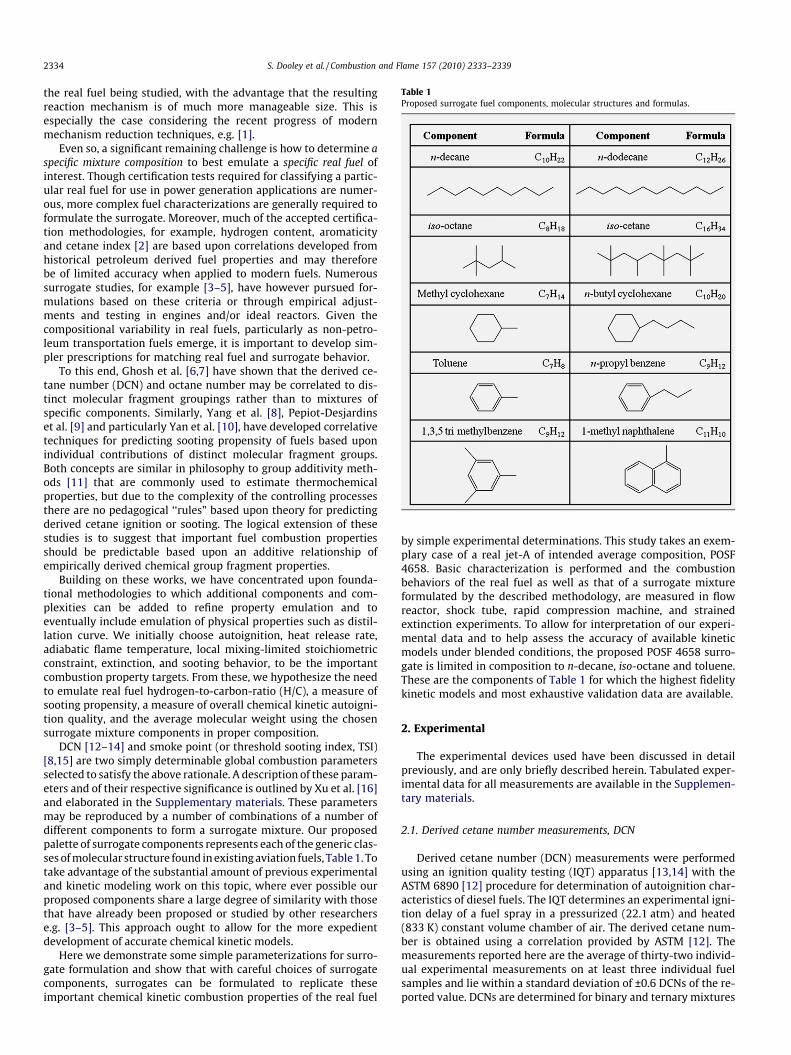
Table 1Proposed surrogate fuel components, molecular structures and formulas.
2334 S. Dooley et al. / Combustion and Flame 157 (2010) 2333–2339
the real fuel being studied, with the advantage that the resultingreaction mechanism is of much more manageable size. This isespecially the case considering the recent progress of modernmechanism reduction techniques, e.g. [1].
Even so, a significant remaining challenge is how to determine aspecific mixture composition to best emulate a specific real fuel ofinterest. Though certification tests required for classifying a partic-ular real fuel for use in power generation applications are numer-ous, more complex fuel characterizations are generally required toformulate the surrogate. Moreover, much of the accepted certifica-tion methodologies, for example, hydrogen content, aromaticityand cetane index [2] are based upon correlations developed fromhistorical petroleum derived fuel properties and may thereforebe of limited accuracy when applied to modern fuels. Numeroussurrogate studies, for example [3–5], have however pursued for-mulations based on these criteria or through empirical adjust-ments and testing in engines and/or ideal reactors. Given thecompositional variability in real fuels, particularly as non-petro-leum transportation fuels emerge, it is important to develop sim-pler prescriptions for matching real fuel and surrogate behavior.
To this end, Ghosh et al. [6,7] have shown that the derived ce-tane number (DCN) and octane number may be correlated to dis-tinct molecular fragment groupings rather than to mixtures ofspecific components. Similarly, Yang et al. [8], Pepiot-Desjardinset al. [9] and particularly Yan et al. [10], have developed correlativetechniques for predicting sooting propensity of fuels based uponindividual contributions of distinct molecular fragment groups.Both concepts are similar in philosophy to group additivity meth-ods [11] that are commonly used to estimate thermochemicalproperties, but due to the complexity of the controlling processesthere are no pedagogical ‘‘rules” based upon theory for predictingderived cetane ignition or sooting. The logical extension of thesestudies is to suggest that important fuel combustion propertiesshould be predictable based upon an additive relationship ofempirically derived chemical group fragment properties.
Building on these works, we have concentrated upon founda-tional methodologies to which additional components and com-plexities can be added to refine property emulation and toeventually include emulation of physical properties such as distil-lation curve. We initially choose autoignition, heat release rate,adiabatic flame temperature, local mixing-limited stoichiometricconstraint, extinction, and sooting behavior, to be the importantcombustion property targets. From these, we hypothesize the needto emulate real fuel hydrogen-to-carbon-ratio (H/C), a measure ofsooting propensity, a measure of overall chemical kinetic autoigni-tion quality, and the average molecular weight using the chosensurrogate mixture components in proper composition.
DCN [12–14] and smoke point (or threshold sooting index, TSI)[8,15] are two simply determinable global combustion parametersselected to satisfy the above rationale. A description of these param-eters and of their respective significance is outlined by Xu et al. [16]and elaborated in the Supplementary materials. These parametersmay be reproduced by a number of combinations of a number ofdifferent components to form a surrogate mixture. Our proposedpalette of surrogate components represents each of the generic clas-ses of molecular structure found in existing aviation fuels, Table 1. Totake advantage of the substantial amount of previous experimentaland kinetic modeling work on this topic, where ever possible ourproposed components share a large degree of similarity with thosethat have already been proposed or studied by other researcherse.g. [3–5]. This approach ought to allow for the more expedientdevelopment of accurate chemical kinetic models.
Here we demonstrate some simple parameterizations for surro-gate formulation and show that with careful choices of surrogatecomponents, surrogates can be formulated to replicate theseimportant chemical kinetic combustion properties of the real fuel
by simple experimental determinations. This study takes an exem-plary case of a real jet-A of intended average composition, POSF4658. Basic characterization is performed and the combustionbehaviors of the real fuel as well as that of a surrogate mixtureformulated by the described methodology, are measured in flowreactor, shock tube, rapid compression machine, and strainedextinction experiments. To allow for interpretation of our experi-mental data and to help assess the accuracy of available kineticmodels under blended conditions, the proposed POSF 4658 surro-gate is limited in composition to n-decane, iso-octane and toluene.These are the components of Table 1 for which the highest fidelitykinetic models and most exhaustive validation data are available.
2. Experimental
The experimental devices used have been discussed in detailpreviously, and are only briefly described herein. Tabulated exper-imental data for all measurements are available in the Supplemen-tary materials.
2.1. Derived cetane number measurements, DCN
Derived cetane number (DCN) measurements were performedusing an ignition quality testing (IQT) apparatus [13,14] with theASTM 6890 [12] procedure for determination of autoignition char-acteristics of diesel fuels. The IQT determines an experimental igni-tion delay of a fuel spray in a pressurized (22.1 atm) and heated(833 K) constant volume chamber of air. The derived cetane num-ber is obtained using a correlation provided by ASTM [12]. Themeasurements reported here are the average of thirty-two individ-ual experimental measurements on at least three individual fuelsamples and lie within a standard deviation of ±0.6 DCNs of the re-ported value. DCNs are determined for binary and ternary mixtures
S. Dooley et al. / Combustion and Flame 157 (2010) 2333–2339 2335
of n-decane, iso-octane and toluene. In all, fourteen different mix-tures were investigated, Table S1, Supplementary materials.
2.2. Surrogate formulation
The empirical formula of POSF 4658 was determined experi-mentally (Perkin Elmer 2400 Series II CHN analyzer) employingASTM method D5291 to be C10.17H19.91 (H/C = 1.957). The DCN ofPOSF 4658 determined using the same IQT testing method de-scribed above is 47.1 ± 0.3, and the TSI was determined using thesmoke point technique [8] to be 21.4 (personal communication,Santoro, R.J.). These data provide four constraints which withinthe present methodology a surrogate should emulate, DCN, H/C,TSI and an average ‘molecular’ mass of 142 ± 5 g/mol.
The data presented in Table S1, have been delineated by an ex-treme vertices matrix to parameterize the DCN, H/C, MW and TSIas a function of n-decane, iso-octane and toluene mole fraction.Available TSI data for pure components [17] were utilized with alinear relationship between TSI and mixture fraction for binaryand ternary mixtures. Though the four targets can be approxi-mated with the mixtures of the surrogate components listed inTable 1, indeed, this has been shown for real fuel TSI [10], it isnot possible to compute a mixture formula of n-decane/iso-oc-tane/toluene in any proportion that satisfies the H/C ratio and TSIconstraints simultaneously. Furthermore, mixtures of the threecomponents investigated here cannot match the molecular weightconstraint. The experimental validations investigated herein arepredominately chemical kinetic in nature and therefore morestrongly influenced by the H/C ratio than by the TSI constraint.Hence in the context of the present study H/C ratio is allowed pre-cedence over TSI. A three component n-decane/iso-octane/toluenemixture of 42.67/33.02/24.31 mol.% is predicted to have a DCN of47.1 and a TSI of 14, is of H/C 2.01 and average ‘molecular’ massof 120.67 g mol�1. This mixture was prepared on a mass basisand we label it as the POSF 4658 surrogate. The DCN of the POSF4658 surrogate was measured as 47.4 ± 0.3, indicating the applica-bility of our DCN correlation.
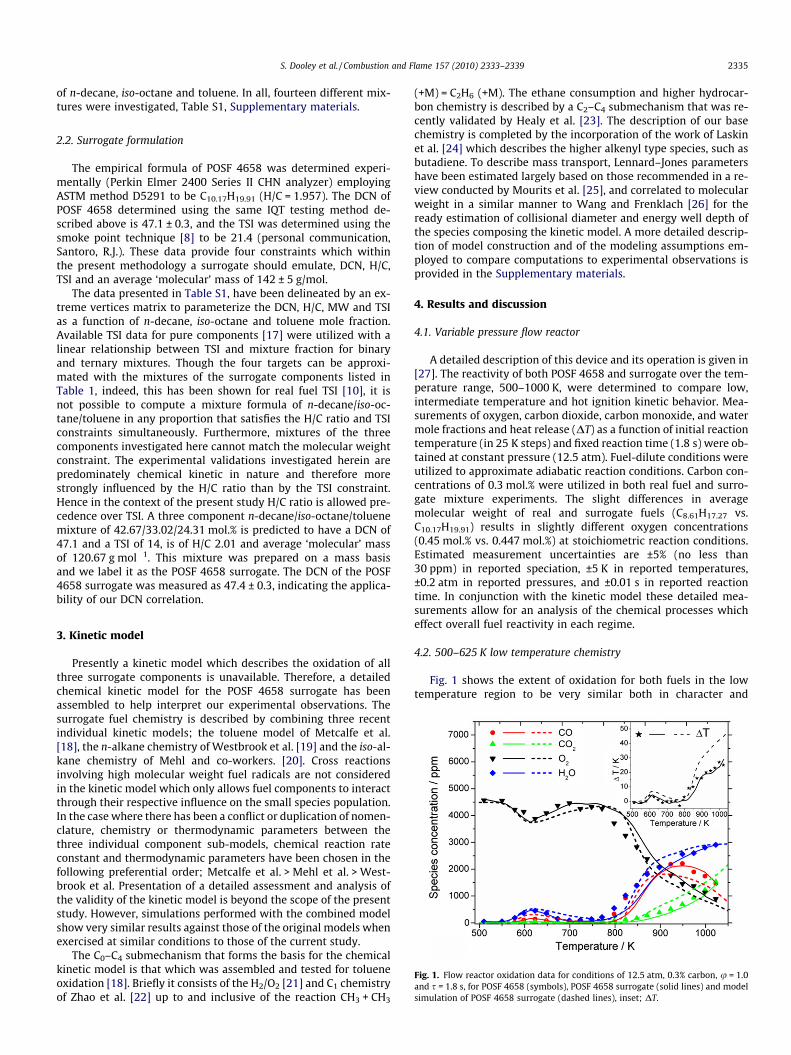
Fig. 1. Flow reactor oxidation data for conditions of 12.5 atm, 0.3% carbon, u = 1.0and s = 1.8 s, for POSF 4658 (symbols), POSF 4658 surrogate (solid lines) and modelsimulation of POSF 4658 surrogate (dashed lines), inset; DT.
3. Kinetic model
Presently a kinetic model which describes the oxidation of allthree surrogate components is unavailable. Therefore, a detailedchemical kinetic model for the POSF 4658 surrogate has beenassembled to help interpret our experimental observations. Thesurrogate fuel chemistry is described by combining three recentindividual kinetic models; the toluene model of Metcalfe et al.[18], the n-alkane chemistry of Westbrook et al. [19] and the iso-al-kane chemistry of Mehl and co-workers. [20]. Cross reactionsinvolving high molecular weight fuel radicals are not consideredin the kinetic model which only allows fuel components to interactthrough their respective influence on the small species population.In the case where there has been a conflict or duplication of nomen-clature, chemistry or thermodynamic parameters between thethree individual component sub-models, chemical reaction rateconstant and thermodynamic parameters have been chosen in thefollowing preferential order; Metcalfe et al. > Mehl et al. > West-brook et al. Presentation of a detailed assessment and analysis ofthe validity of the kinetic model is beyond the scope of the presentstudy. However, simulations performed with the combined modelshow very similar results against those of the original models whenexercised at similar conditions to those of the current study.
The C0–C4 submechanism that forms the basis for the chemicalkinetic model is that which was assembled and tested for tolueneoxidation [18]. Briefly it consists of the H2/O2 [21] and C1 chemistryof Zhao et al. [22] up to and inclusive of the reaction CH3 + CH3
(+M) = C2H6 (+M). The ethane consumption and higher hydrocar-bon chemistry is described by a C2–C4 submechanism that was re-cently validated by Healy et al. [23]. The description of our basechemistry is completed by the incorporation of the work of Laskinet al. [24] which describes the higher alkenyl type species, such asbutadiene. To describe mass transport, Lennard–Jones parametershave been estimated largely based on those recommended in a re-view conducted by Mourits et al. [25], and correlated to molecularweight in a similar manner to Wang and Frenklach [26] for theready estimation of collisional diameter and energy well depth ofthe species composing the kinetic model. A more detailed descrip-tion of model construction and of the modeling assumptions em-ployed to compare computations to experimental observations isprovided in the Supplementary materials.
4. Results and discussion
4.1. Variable pressure flow reactor
A detailed description of this device and its operation is given in[27]. The reactivity of both POSF 4658 and surrogate over the tem-perature range, 500–1000 K, were determined to compare low,intermediate temperature and hot ignition kinetic behavior. Mea-surements of oxygen, carbon dioxide, carbon monoxide, and watermole fractions and heat release (DT) as a function of initial reactiontemperature (in 25 K steps) and fixed reaction time (1.8 s) were ob-tained at constant pressure (12.5 atm). Fuel-dilute conditions wereutilized to approximate adiabatic reaction conditions. Carbon con-centrations of 0.3 mol.% were utilized in both real fuel and surro-gate mixture experiments. The slight differences in averagemolecular weight of real and surrogate fuels (C8.61H17.27 vs.C10.17H19.91) results in slightly different oxygen concentrations(0.45 mol.% vs. 0.447 mol.%) at stoichiometric reaction conditions.Estimated measurement uncertainties are ±5% (no less than30 ppm) in reported speciation, ±5 K in reported temperatures,±0.2 atm in reported pressures, and ±0.01 s in reported reactiontime. In conjunction with the kinetic model these detailed mea-surements allow for an analysis of the chemical processes whicheffect overall fuel reactivity in each regime.
4.2. 500–625 K low temperature chemistry
Fig. 1 shows the extent of oxidation for both fuels in the lowtemperature region to be very similar both in character and

2336 S. Dooley et al. / Combustion and Flame 157 (2010) 2333–2339
magnitude; the oxidation of POSF 4658 is onset at 550–578 K, therate of oxidation increases until 623–648 K, where the fuel exhibitsnegative temperature coefficient (NTC) behavior. The POSF 4658surrogate shows the onset of reactivity at 564–589 K and exhibitsNTC behavior at 613–636 K. The extent of reactivity is reflected bythe amount of O2 consumed and of CO2, CO and H2O formed. Giventhe estimated experimental uncertainties, both fuels exhibit verysimilar reactivity (all measurements agree to ±15%) and certainlysatisfactory emulation of reactivity in this temperature regime un-der the conditions studied.
The kinetic model reproduces the qualitative features of thePOSF 4658 surrogate oxidation very well, although the extent ofreactivity in the NTC regime is characteristically over-predicted.Both model and surrogate show the point of highest reactivity inthe low temperature regime at approximately 600 K. It has previ-ously been established under very similar conditions to those ofthe present study, that iso-octane e.g. [28] and toluene e.g. [18]are not capable of sufficient radical-chain branching activity tosupport oxidation below approximately 800 and 900 K, respec-tively. Analysis of the model simulations at 600 K support this con-clusion, showing at s = 1.8 s only 2.2% of the initial mole fraction ofn-decane remains, compared to 37.3% and 60.2% of the initial molefractions of iso-octane and toluene respectively. At the much latertime of 3.6 s, model predictions show iso-octane and toluene molefractions remain largely unchanged (35.4% and 59.0%, respec-tively); at these low temperatures, aromatic and iso-alkane compo-nents are oxidized by the radical pool generated by the n-alkane,and in turn, generate few active radicals themselves.
The similarity in reactivity exhibited by model, surrogate andPOSF 4658 at this condition is extremely encouraging. It appearsthat the n-alkane component of the surrogate is behaving in a verysimilar way to the more complex mixture of n-alkyl componentspresent in POSF 4658, Fig. 1. Therefore it is strongly suggestive thatthe prescribed quantity of n-decane is accurately emulating thechemical processes of the total n-alkyl components in the real fuel.Moreover, it appears that the aromatic and iso-alkane componentsare also present in appropriate quantities to provide the intendedradical termination processes sufficiently so as to replicate the glo-bal reactivity of the real fuel.
4.3. 625–825 K negative temperature coefficient
POSF 4658 exhibits NTC behavior in the temperature range623–648 to 723 K, Fig. 1. At 723 K little or no conversion of oxygenor formation of products are observed, indicating that the reactiv-ity of the system has halted almost entirely under these conditions.Subsequently, 723–771 K, the systems shows very modest reactiv-ity as exhibited by the O2 and H2O profiles, before showing a muchlarger increase in reactivity at 771–798 K (hot ignition condition).The POSF 4658 surrogate emulates the temperature dependence ofthe target fuel very well with regard to the NTC regime. Howeverthe fuels deviate in reactivity between 723 and 771 K, with the sur-rogate remaining un-reactive until 787 K, i.e. a temperatureapproximately 54 K higher than the real fuel.
At 800 K, the model again shows the surrogate fuel to be con-sumed from radicals that are largely generated through the activityof the n-alkane component, only 25% of which remains at this reac-tion time (vs. 71% iso-octane and 85% toluene). By virtue of thisdeviation in reactivity, we may surmise that the much richermolecular structural palette contained in the real fuel allows POSF4658 to become oxidized at the lower temperatures of the inter-mediate temperature regime. The functional temperature depen-dence of chemical reactivity results from a complex switching ofthe fate of alkyl, alkylperoxy and hydroperoxy alkyl radicals whichis dependent in a detailed manner on molecular structure. Recogn-ising this, the modest divergence in reactivity between real fuel
and such a simple surrogate is not discouraging. In fact, given themuch simpler molecular structure of the surrogate fuel it is extre-mely encouraging that it should emulate the more diverse real fuelcomposition so closely in regard to the complex temperaturedependence of combustion oxidation kinetics, particularly so inreference to the exit of the NTC region.
It is likely that in our provisional surrogate the iso-alkane com-ponent, is in fact too highly branched to emulate very precisely thereactivity of the much lesser branched alkanes that are present inPOSF 4658 [2] at these temperatures of transitioning regimes ofchemistry. The other contributing factor involved at these condi-tions is in the selection of toluene as representative of the genericaromatic class of compounds. The longer alkyl chain(s) of the aro-matics found in real jet aviation fuels [2] ought to allow these moi-eties to become oxidized at much lower temperatures thantoluene. We view these as relatively minor deficiencies especiallyas our ‘‘first generation” surrogate components allow a much morethorough understanding via analyses provided by more reliable ki-netic models than those that exist for alternative components. Theincorporation of one or more n-alkyl aromatics or perhaps a simi-larly reactive, better studied, compound such as a xylene, would al-low the surrogate to emulate the reactivity of the real fuel withimproved fidelity.
4.4. 825–1050 K intermediate to high temperature chemistry
Fig. 1 shows that the reactivity of the POSF 4658 surrogate re-mains diminished relative to POSF 4658 until temperatures greaterthan approximately 900 K. Although the kinetic model reproducesthese measurements quite well, it is noted that at temperaturesabove 900 K carbon monoxide is under-predicted and carbon diox-ide over-predicted relative to experiment. Analysis of the chemicalflux at 950 K shows that both alkane components are consumed al-most completely by typical high temperature hydrogen abstractionalkyl radical beta-scission processes, with iso-octane showing asmall (5%) level of unimolecular decomposition. This chemistry isin contrast to the rather more complex competition betweenbeta-scission and addition of O2 to alkyl radicals that are formedbelow 900 K. This may be a significant observation as the reactivityof the ternary system at these higher temperatures depends largelyon the identity and quantity of beta-scission products formed.Since POSF 4658 and surrogate share similar species evolutions,it is likely that the net reactivity (radical producing/consumingabilities) of the intermediates formed by both fuels culminatingin the crucial formation and decomposition of H2O2 are approxi-mately equal in this temperature range.
4.5. Diffusion flame extinction limits
A counterflow burner was utilized to measure the extinctionstrain rate as a function of nitrogen dilution for POSF 4658 andthe proposed POSF 4658 surrogate for diffusion flames. The tem-perature of the fuel side was maintained at 500 ± 5 K, and that ofthe oxidizer (air) side at 298 K ± 2 K, in an identical setup to Wonet al. [29]. In addition to being an important property to under-stand the stabilization mechanism of a turbulent flame, extinctionlimits of diffusion flames are commanding in practical combustionsystems, and thus necessary for a surrogate to satisfy.
Fig. 2 shows that the extinction behavior exhibited by POSF4658 is very closely emulated by the surrogate when evaluatedon a mole fraction basis. Also shown are computations performedby a reduced kinetic model produced by the path flux analysestechnique [30]. The model reproduces the extinction strain ratequantitatively very well, though predictions at higher mole/massfractions under predict experiment by up to 14%. It has been dem-onstrated that the extinction limit of diffusion flames have a linear
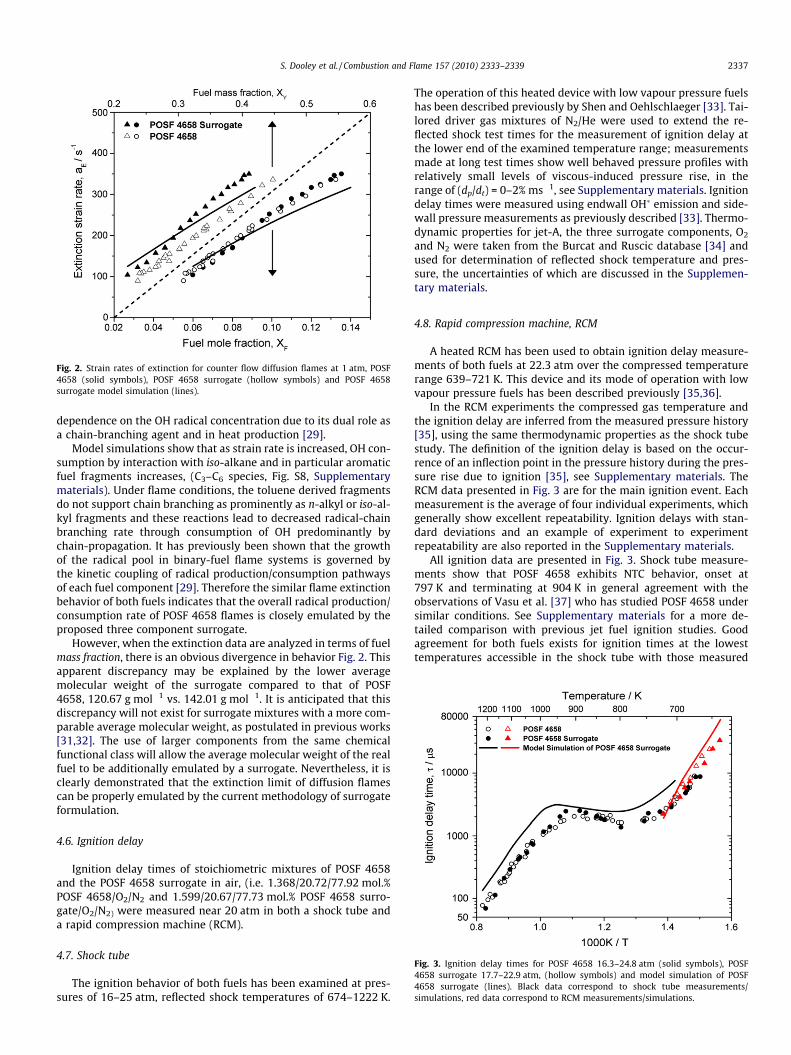
Fig. 2. Strain rates of extinction for counter flow diffusion flames at 1 atm, POSF4658 (solid symbols), POSF 4658 surrogate (hollow symbols) and POSF 4658surrogate model simulation (lines).
Fig. 3. Ignition delay times for POSF 4658 16.3–24.8 atm (solid symbols), POSF4658 surrogate 17.7–22.9 atm, (hollow symbols) and model simulation of POSF4658 surrogate (lines). Black data correspond to shock tube measurements/simulations, red data correspond to RCM measurements/simulations.
S. Dooley et al. / Combustion and Flame 157 (2010) 2333–2339 2337
dependence on the OH radical concentration due to its dual role asa chain-branching agent and in heat production [29].
Model simulations show that as strain rate is increased, OH con-sumption by interaction with iso-alkane and in particular aromaticfuel fragments increases, (C3–C6 species, Fig. S8, Supplementarymaterials). Under flame conditions, the toluene derived fragmentsdo not support chain branching as prominently as n-alkyl or iso-al-kyl fragments and these reactions lead to decreased radical-chainbranching rate through consumption of OH predominantly bychain-propagation. It has previously been shown that the growthof the radical pool in binary-fuel flame systems is governed bythe kinetic coupling of radical production/consumption pathwaysof each fuel component [29]. Therefore the similar flame extinctionbehavior of both fuels indicates that the overall radical production/consumption rate of POSF 4658 flames is closely emulated by theproposed three component surrogate.
However, when the extinction data are analyzed in terms of fuelmass fraction, there is an obvious divergence in behavior Fig. 2. Thisapparent discrepancy may be explained by the lower averagemolecular weight of the surrogate compared to that of POSF4658, 120.67 g mol�1 vs. 142.01 g mol�1. It is anticipated that thisdiscrepancy will not exist for surrogate mixtures with a more com-parable average molecular weight, as postulated in previous works[31,32]. The use of larger components from the same chemicalfunctional class will allow the average molecular weight of the realfuel to be additionally emulated by a surrogate. Nevertheless, it isclearly demonstrated that the extinction limit of diffusion flamescan be properly emulated by the current methodology of surrogateformulation.
4.6. Ignition delay
Ignition delay times of stoichiometric mixtures of POSF 4658and the POSF 4658 surrogate in air, (i.e. 1.368/20.72/77.92 mol.%POSF 4658/O2/N2 and 1.599/20.67/77.73 mol.% POSF 4658 surro-gate/O2/N2) were measured near 20 atm in both a shock tube anda rapid compression machine (RCM).
4.7. Shock tube
The ignition behavior of both fuels has been examined at pres-sures of 16–25 atm, reflected shock temperatures of 674–1222 K.
The operation of this heated device with low vapour pressure fuelshas been described previously by Shen and Oehlschlaeger [33]. Tai-lored driver gas mixtures of N2/He were used to extend the re-flected shock test times for the measurement of ignition delay atthe lower end of the examined temperature range; measurementsmade at long test times show well behaved pressure profiles withrelatively small levels of viscous-induced pressure rise, in therange of (dp/dt) = 0–2% ms�1, see Supplementary materials. Ignitiondelay times were measured using endwall OH� emission and side-wall pressure measurements as previously described [33]. Thermo-dynamic properties for jet-A, the three surrogate components, O2
and N2 were taken from the Burcat and Ruscic database [34] andused for determination of reflected shock temperature and pres-sure, the uncertainties of which are discussed in the Supplemen-tary materials.
4.8. Rapid compression machine, RCM
A heated RCM has been used to obtain ignition delay measure-ments of both fuels at 22.3 atm over the compressed temperaturerange 639–721 K. This device and its mode of operation with lowvapour pressure fuels has been described previously [35,36].
In the RCM experiments the compressed gas temperature andthe ignition delay are inferred from the measured pressure history[35], using the same thermodynamic properties as the shock tubestudy. The definition of the ignition delay is based on the occur-rence of an inflection point in the pressure history during the pres-sure rise due to ignition [35], see Supplementary materials. TheRCM data presented in Fig. 3 are for the main ignition event. Eachmeasurement is the average of four individual experiments, whichgenerally show excellent repeatability. Ignition delays with stan-dard deviations and an example of experiment to experimentrepeatability are also reported in the Supplementary materials.
All ignition data are presented in Fig. 3. Shock tube measure-ments show that POSF 4658 exhibits NTC behavior, onset at797 K and terminating at 904 K in general agreement with theobservations of Vasu et al. [37] who has studied POSF 4658 undersimilar conditions. See Supplementary materials for a more de-tailed comparison with previous jet fuel ignition studies. Goodagreement for both fuels exists for ignition times at the lowesttemperatures accessible in the shock tube with those measured

2338 S. Dooley et al. / Combustion and Flame 157 (2010) 2333–2339
in the RCM. The authors recognize that shocktube and RCM dataare not strictly comparable and this is clearly discernable frommodel simulations also plotted in Fig. 3. Shock tube ignition timesare 10–30% shorter than those observed in the RCM. This is attrib-uted to differences in the pre-ignition pressure behavior character-istic of the two experiments, particularly to post compression heatlosses in the RCM, leading to slightly longer ignition times.
At comparable conditions, the maximum difference in delaytime between POSF 4658 and surrogate is 30%, indicative of the su-perb capability of this particular surrogate for emulating real fuelbehavior under practical propulsion engine conditions. These datafurther demonstrate the validity of the proposed surrogate formu-lation technique and of the applicability of the underlying princi-ples of chemical group additivity.
In reference to kinetic model simulations of shock tube data, itis important to recognize that the model should reproduce the flowreactor observations quite well yet consistently predict slowerignition delay times by approximately a-factor-of-two relative toignition experiments. More precisely, for an example surrogateexperiment at 962 K, Fig. 3 shows the surrogate to have exitedthe NTC region yet the model predicts ignition at 962 K to be insidethe NTC regime. Simulations accounting for the viscous-inducedpressure rises of 0.4 atm ms�1 do not significantly alter the com-puted ignition delays. Given the evident disagreement betweenmodel and experiment we interpret the model results with cau-tion. Notwithstanding this, the simulated ignition delay time forthis condition is 3200 ls. At a time of 2800 ls, the model showsthat the temperature has risen to 1044 K and that 15.5%, 53.3%,77.1% of the respective initial concentrations of n-decane, iso-oc-tane and toluene remain. Consistent with model analysis of theother environments, the n-alkyl component is largely responsiblefor radical production during surrogate oxidation, and by extensionalso in POSF 4658.
Much more fuel is present in the ignition simulations comparedto the flow reactor simulations, 6851/5303/3903 vs. 149/115/85 ppm n-decane/iso-octane/toluene, respectively. It is possiblethat any in-accurately assigned reaction rate constants describingcrucial branching ratios might propagate an error, and manifestin larger deviations from experiment when more fuel is present.For example the elongated NTC behavior shown by the modelmay be due to an under-emphasis on beta-scission of n-decyl/alkylradicals leading to exaggerated low temperature chemistry pro-cesses to the detriment of high temperature chain branching. Thisconclusion is supported by analysis of the model simulation at thiscondition which shows a strong competition for n-decyl alkyl rad-icals between beta-scission (approximately 40%) and formation ofRO2 (approximately 60%), with almost all of the RO2 formed en-gaged in radical chain-propagation processes. Given this analysis,it appears again that the most important facet in the accuratenumerical simulation of a jet fuel surrogate is in the accuratedescription of n-alkyl chemistries, not only confined to alkanesthemselves but also to any alkyl aromatics or lightly branchedalkanes.
The RCM ignition experiments are noteworthy in that while theoverall autoignition delay times of the real fuel and its surrogateare very similar, the quality of ignition differs markedly. The keydistinguishing feature is the existence of a rather strong pseudo-first-stage ignition activity (s1) for POSF 4658, while the surrogatedoes not show first-stage ignition, Fig. S3. The other distinguishingfeature is the higher post ignition rate of pressure rise for the sur-rogate mixture, consistent with a more sudden ignition event. Ki-netic model computations account for heat losses by theconsideration of the full pressure history of non reactive mixturesof identical Cp/Cv (c) as described by Mittal and Sung [35] andreproduce the quality of RCM data very well, Figs. 3 and S5. How-ever, consistent with shock tube simulations, the model predicts
the main ignition event to occur at longer times than experiment,by approximately a-factor-of-two. The emulation of two-stagePOSF 4658 ignition would appear to be extremely challengingand will likely require a surrogate of more structurally diversechemical fragments than those generated by a simple mixture ofn-decane, iso-octane and toluene.
5. Conclusions
This study outlines a method for the formulation of a fuel spe-cific jet fuel surrogate via the empirical correlation of relatively eas-ily measurable fuel properties. In so doing we predict the surrogatefuel to mimic wide ranging combustion properties of a target realfuel. This philosophy is tested by the formulation of a surrogate forthe real life jet-A, POSF 4658 by the emulation of DCN and H/Conly. Although demonstrated here for a jet aviation fuel, the de-scribed methodology of chemical group additivity ought to beapplicable to the study of fuels elsewhere on the distillation curve,including gasolines and diesels as well as to emerging syntheticfuels. The proviso qualification being that the surrogate componentpalette supports the formation of the distinct chemical functional-ities formed by the oxidation of the target fuel.
The presented experiments show that the POSF 4658 surrogateclosely emulates the chemical kinetic related behavior of POSF4658 beyond expectation, supporting considerable optimism thatsurrogates composed of somewhat larger molecular weight al-kanes and alkyl aromatics can lead to further improvements in sur-rogate performance. It is anticipated the addition of TSI as a furtherconstraint will allow emulation of the sooting propensity of thereal fuel and also improve and extend the ability for a surrogateto emulate more global combustion parameters with improvedprecision. Moreover, the ignition and diffusion flame strainedextinction studies comparison in particular, lends strong credenceto the proposed surrogate formulation method for application tothe study of practical combustion systems.
It is likely that simple surrogate fuels, such as the first genera-tion test-case presented here will be deficient in emulating high-detailed observations of real fuels such as detailed speciation andtwo-stage ignition character. Emulation of common small molecu-lar weight intermediate species such as ethylene and formalde-hyde may be much more achievable given the intrinsic roles ofthese species in regulating chemical reactivity. Indeed, althoughthe tested surrogate emulates overall ignition times, it fails toemulate the two-stage ignition behavior exhibited by POSF 4658in the RCM study. This is not surprising as the surrogate was notformulated to do so. Such targets may be achievable if an increaseddiversity of molecular structure is present in the surrogate compo-nent palette or/and if additional constraints can be added for sur-rogate formulation. One such additional constraint might bethrough modifications of the IQT testing procedures.
A kinetic model for the presented n-decane/iso-octane/toluenePOSF 4658 surrogate has been constructed and its computationscompared with experimental observations to elucidate similaritiesin the reactivity of surrogate and real fuel. Analysis shows that thechemical reactivity of the surrogate is strongly dependent on theprocesses of its n-alkane component. At lower temperatures, thedependence on n-alkyl radical kinetics is so strong that the fateof iso-alkyl and aromatic radicals formed in competing abstractionreactions are in comparison of little consequence. By this virtue,the accurate numerical modeling of any multi component surro-gate fuel depends crucially on the accurate description of n-alkylchemistry. Finally, although the present first generation surrogatedoes not allow simultaneous emulation of H/C, DCN, and TSI, theuse of a higher carbon number alkane such as n-dodecane canaccommodate sufficient addition of aromatic components to

S. Dooley et al. / Combustion and Flame 157 (2010) 2333–2339 2339
emulate the entire spectrum of fuel properties that are known forvarious real gas turbine fuels [16]. Further demonstrations of uti-lizing these concepts to emulate real fuel combustion kinetic prop-erties are underway.
Acknowledgments
Work at PU, PSU and UC is part of MURI collaboration supportedby the Air Force Office of Scientific Research under Grant NumberFA9550-07-1-0515. Work at RPI is supported by AFOSR GrantNumber FA9550-07-1-0114, both programs are monitored by Dr.Julian Tishkoff. We are grateful for collaborative discussions withProf. Kenneth Brezinsky, University of Chicago. We are also gratefulto Dr. Tim Edwards, for the provision of POSF 4658, to Dr. MarcoMehl, Lawrence Livermore National Laboratory and his co-workersfor making their latest iso-alkane kinetic model available to usprior to publication. Mr. Wenting Sun, Princeton University isappreciated for discussions on model reduction.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.combustflame.2010.07.001.
References
[1] Tianfeng Lu, Chung K. Law, Prog. Energy Combust. Sci. 35 (2009) 192–215.[2] Defense Energy Support Centre, Petroleum Quality Information Systems,
Annual Report (PQIS2008), 2008. <https://www.desc.dla.mil/DCM/Files/2004PQISreportsmall.pdf>.
[3] A. Violi, S. Yan, E.G. Eddings, A.F. Sarofim, S. Granata, T. Faravelli, E. Ranzi,Combust. Sci. Technol. 174 (2002) 399–417.
[4] P. Dagaut, A.E. Bakali, A. Ristori, Fuel 85 (2006) 944–956.[5] A. Agosta, N.P. Cernansky, D.L. Miller, T. Favarelli, E. Ranzi, Exp. Therm. Fluid
Sci. 28 (2004) 701–708.[6] P. Ghosh, S.B. Jaffe, Ind. Eng. Chem. Res. 45 (2006) 346–351.[7] P. Ghosh, K.J. Hickey, S.B. Jaffe, Ind. Eng. Chem. Res. 45 (2006) 337–345.
[8] Y. Yang, A.L. Boehman, R.J. Santoro, Combust. Flame 149 (2007) 191–205.[9] P. Pepiot-Desjardins, H. Pitsch, R. Malhotra, S.R. Kirby, A.L. Boehman, Combust.
Flame, 2009.[10] S. Yan, E.G. Eddings, A.B. Palotas, R.J. Pugmire, A.F. Sarofim, Energy Fuels 19
(2005) 2408–2415.[11] S.W. Benson, Thermochemical Kinetics, John Wiley and Sons Inc., New York,
1976.[12] ASTM D 6890, ASTM International, West Conshohocken, PA, 2007.[13] L.N. Allard, N.J. Hole, G.D. Webster, T.W.I. Ryan, D. Ott, A. Beregszazy, C.W.
Fairbridge, J. Cooley, K. Mitchell, E.K. Richardson, N.G. Elliot, D.J. Rickeard, SAETech. Pap. (1997) 971636.
[14] L.N. Allard, G.D. Webster, T.W.I. Ryan, A. Matheaus, G. Baker, A. Beregszazy, H.Read, K. Mortimer, G.J. Jones, SAE Tech. Pap. (2001) 2001013527.
[15] H.F. Calcote, D.M. Mano, Combust. Flame 49 (1983) 289–304.[16] H. Xu, Z. Yang, M. Chaos, F.L. Dryer, in: JANNAF 42nd Combustion Joint Sub-
Committee Meeting, 2008.[17] A. Mensch, R.J. Santoro, T.A. Litzinger, S.-Y. Lee, Combust. Flame 157 (2010)
1097–1105.[18] W.K. Metcalfe, S. Dooley, F.L. Dryer, in: Proc. Combust. Inst. WIPP, 2010.[19] C.K. Westbrook, W.J. Pitz, O. Herbinet, H.J. Curran, E.J. Silke, Combust. Flame
156 (2009) 181–199.[20] M. Mehl, H.J. Curran, W.J. Pitz, C.K. Westbrook, in: Proc. Combust. Inst., 2010.[21] J. Li, Z. Zhao, A. Kazakov, M. Chaos, F.L. Dryer, J.J. Scire Jr., Int. J. Chem. Kinet. 39
(2007) 109–136.[22] Z. Zhao, M. Chaos, A. Kazakov, F.L. Dryer, Int. J. Chem. Kinet. 10 (2008) 1–18.[23] D. Healy, H.J. Curran, J.M. Simmie, D.M. Kalitan, C.M. Zinner, A.B. Barrett, E.L.
Petersen, G. Bourque, Combust. Flame 155 (2008) 441–448.[24] A. Laskin, H. Wang, C.K. Law, Int. J. Chem. Kinet. 32 (2000) 589–614.[25] F.M. Mourits, F.H.A. Rummens, Can. J. Chem. 55 (1977) 3007–3020.[26] H. Wang, M. Frenklach, Combust. Flame 96 (1994) 163–170.[27] T.J. Held, F.L. Dryer, Int. J. Chem. Kinet. 30 (1998) 805–830.[28] H.J. Curran, P. Gaffuri, W.J. Pitz, C.K. Westbrook, Combust. Flame 129 (2002)
253–280.[29] S.H. Won, W. Sun, Y. Ju, Combust. Flame 157 (2010) 411–420.[30] W. Sun, Z. Chen, X. Gou, Y. Ju, Combust. Flame 157 (2010) 1298–1307.[31] S. Honnet, K. Seshadri, U. Niemann, N. Peters, Proc. Combust. Inst. 32 (2009)
485–492.[32] S. Humer, A. Frassoldati, S. Granata, T. Faravelli, E. Ranzi, R. Seiser, K. Seshadri,
Proc. Combust. Inst. 31 (2007) 393–400.[33] H.-P.S. Shen, M.A. Oehlschlaeger, Combust. Flame 156 (2009) 1053–1062.[34] A. Burcat, B. Ruscic, Ideal Gas Thermochemical Database. <http://
garfield.chem.elte.edu/burcat/burcat.html>.[35] G. Mittal, C.J. Sung, Combust. Sci. Technol. 179 (2007) 497–530.[36] K. Kumar, G. Mittal, C.J. Sung, Combust. Flame 156 (2009) 1278–1288.[37] S.S. Vasu, D.F. Davidson, R.K. Hanson, Combust. Flame 152 (2008) 125–143.
Related Documents











