A first principles theoretical study of vibrational spectral diffusion and hydrogen bond dynamics in aqueous ionic solutions: D2O in hydration shells of Cl− ions Bhabani S. Mallik, A. Semparithi, and Amalendu Chandra Citation: J. Chem. Phys. 129, 194512 (2008); doi: 10.1063/1.3006032 View online: http://dx.doi.org/10.1063/1.3006032 View Table of Contents: http://jcp.aip.org/resource/1/JCPSA6/v129/i19 Published by the AIP Publishing LLC. Additional information on J. Chem. Phys. Journal Homepage: http://jcp.aip.org/ Journal Information: http://jcp.aip.org/about/about_the_journal Top downloads: http://jcp.aip.org/features/most_downloaded Information for Authors: http://jcp.aip.org/authors Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

A first principles theoretical study of vibrational spectral diffusion andhydrogen bond dynamics in aqueous ionic solutions: D2O in hydrationshells of Cl− ionsBhabani S. Mallik, A. Semparithi, and Amalendu Chandra Citation: J. Chem. Phys. 129, 194512 (2008); doi: 10.1063/1.3006032 View online: http://dx.doi.org/10.1063/1.3006032 View Table of Contents: http://jcp.aip.org/resource/1/JCPSA6/v129/i19 Published by the AIP Publishing LLC. Additional information on J. Chem. Phys.Journal Homepage: http://jcp.aip.org/ Journal Information: http://jcp.aip.org/about/about_the_journal Top downloads: http://jcp.aip.org/features/most_downloaded Information for Authors: http://jcp.aip.org/authors
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

A first principles theoretical study of vibrational spectraldiffusion and hydrogen bond dynamics in aqueous ionic solutions:D2O in hydration shells of Cl− ions
Bhabani S. Mallik, A. Semparithi, and Amalendu Chandraa�
Department of Chemistry, Indian Institute of Technology, Kanpur 208016, India
�Received 19 April 2008; accepted 3 October 2008; published online 20 November 2008�
A theoretical study of vibrational spectral diffusion and hydrogen bond dynamics in aqueous ionicsolutions is presented from first principles without employing any empirical potential models. Thepresent calculations are based on ab initio molecular dynamics for trajectory generation and waveletanalysis of the simulated trajectories for time dependent frequency calculations. Results are obtainedfor two different deuterated aqueous solutions: the first one is a relatively dilute solution of a singleCl− ion and the second one is a concentrated solution of NaCl ��3M� dissolved in liquid D2O. Itis found that the frequencies of OD bonds in the anion hydration shell, i.e., those which arehydrogen bonded to the chloride ion, have a higher stretch frequency than those in the bulk water.Also, on average, the frequencies of hydration shell OD modes are found to increase with increasein the anion-water hydrogen bond distance. On the dynamical side, when the vibrational spectraldiffusion is calculated exclusively for the hydration shell water molecules in the first solution, thedynamics reveals three time scales: a short-time relaxation ��200 fs� corresponding to thedynamics of intact ion-water hydrogen bonds, a slower relaxation ��3 ps� corresponding to thelifetimes of chloride ion-water hydrogen bonds, and another longer-time constant ��20 ps�corresponding to the escape dynamics of water from the anion hydration shell. Existence of suchthree time scales for hydration shell water molecules was also reported earlier for water containinga single iodide ion using classical molecular dynamics �B. Nigro et al., J. Phys. Chem. A 110, 11237�2006��. Hence, the present study confirms the basic results of this earlier work using a differentmethodology. However, when the vibrational spectral diffusion is calculated over all the OD modes,only two time scales of �150 fs and �2.7 ps are found without the slowest component of �20 ps.This is likely because of the very small weight that the hydration shell water molecules carry to theoverall spectral diffusion in the solution containing a single ion. For the concentrated solution also,the slowest component of �20 ps is not found in the spectral diffusion of all water moleculesbecause a distinct separation between the hydration shell and bulk water in terms of their stretchfrequencies does not hold at this high concentration regime. The present first principles results arecompared with those of the available experiments and classical simulations. © 2008 AmericanInstitute of Physics. �DOI: 10.1063/1.3006032�
I. INTRODUCTION
This paper is concerned with calculations of vibrationalspectral diffusion and hydrogen bond dynamics in aqueousionic solutions from first principles without using any em-pirical model potentials. The vibrational frequencies of watermolecules in an aqueous solution fluctuate because of fluc-tuations in the local solvation environment, and studies ofthe temporal evolution of these frequency fluctuations,known as vibrational spectral diffusion, using ultrafast time-resolved infrared spectroscopy have provided a very power-ful experimental means of investigating water dynamics atmolecular level, such as the dynamics of breaking and refor-mation of hydrogen bonds in water1–10 and aqueoussolutions.11–17 For ionic solutes of negative charges such ashalide ions, hydrogen bonds are formed between the anionsand water molecules in their first hydration shells and the
strength and dynamics of these hydrogen bonds greatly in-fluence the energetics, dynamics, and vibrational characteris-tics of water molecules in the solutions. Thus, an understand-ing of the basic mechanism and kinetics of hydrogen bondfluctuations in aqueous solutions is a critical step towardtheoretical development and understanding of chemical reac-tion dynamics in aqueous media including many biochemicalprocesses involving charge transfer through hydrogenbonded water.
On the experimental side, Kropman and Bakker11,12 car-ried out femtosecond midinfrared spectroscopic studies ofthe dynamics of vibrational spectral diffusion in aqueous al-kali halide solutions containing Cl−, Br−, or I− as the negativeions. These authors found a rather slow long-time decay ofthe frequency shifts which was attributed to the hydrogenbond dynamics of hydration shell water molecules. For ex-ample, while a characteristic time of �1–2 ps has beenfound for the long-time component of the spectral diffusionin pure water and assigned to the hydrogen bonda�Electronic mail: [email protected].
THE JOURNAL OF CHEMICAL PHYSICS 129, 194512 �2008�
0021-9606/2008/129�19�/194512/15/$23.00 © 2008 American Institute of Physics129, 194512-1
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

dynamics,1–10 time scales of 12–25 ps were reported for thecorresponding dynamics of water in the hydration shells ofthe halide ions.11,12 A more recent experimental study13 onaqueous NaBr solutions has reported a slowing down of vi-brational spectral diffusion by about factors of 2 and 3 for3M and 6M solutions, respectively, when compared with thecorresponding dynamics for pure water. Thus, no dramaticslowing down of the type reported earlier11,12 was found inthis later study. On the theoretical side, Nigro et al.18 carriedout a detailed calculation of the spectral diffusion of hydra-tion shell water molecules in a dilute aqueous I− solutionusing a combination of classical molecular dynamics andperturbative method. In particular, these authors calculatedthe fluctuating frequencies of water molecules as they escapefrom the I− hydration shell and argued that the long-timecomponent of OH spectral diffusion in Refs. 11 and 12 cor-responds to the escape dynamics of water from the anionhydration shell. The relation between rotational motion ofwater in an ion hydration shell and escape dynamics has alsobeen looked at in a subsequent study.19
There are also theoretical studies in literature which havelooked at the hydrogen bond and residence dynamics of wa-ter molecules in ion hydration shells through properly con-structed population correlation functions but without any cal-culations of associated frequency fluctuations. For example,Chowdhuri and Chandra20 carried out classical molecular dy-namics studies of halide ion–water systems and found asomewhat slower dynamics of the anion-water hydrogenbonds than that of water-water hydrogen bonds for chlorideand bromide ions. For iodide ion, the anion-water hydrogenbond dynamics was, in fact, found to be faster than the cor-responding water-water dynamics.20 We note that in classicalmolecular dynamics, one uses predefined empirical interac-tion potentials and the use of this empirical potential can beavoided in the so-called ab initio molecular dynamics simu-lations where the forces on the nuclei are obtained directlyfrom quantum electronic structure calculations performed“on the fly” via an adiabatic dynamics principle.21,22 Re-cently, Raugei and Klein23 and Heuft and Meijer24 carriedout such ab initio molecular dynamics studies of halide ionsdissolved in water. Although these authors did not considerthe relaxation of hydrogen bonds or vibrational spectral dif-fusion directly, the residence time of a water molecule in theinner solvation shell of a halide ion was calculated and it wasfound to be in the range of 10–20 ps and this value wascompared with the experimentally measured slow time scaleof vibrational spectral diffusion in alkali halide solutionscontaining chloride, bromide, or iodide ions.11,12 We againnote in this context the work in Ref. 18, where a direct rela-tion between the long-time component of the spectral diffu-sion and escape dynamics of hydration shell water was es-tablished. However, these studies were carried out at verylow ion concentration where the hydration shell and bulkwater molecules can be clearly distinguished. For high con-centrations, such a clear-cut separation between hydrationshell and bulk water may not exist because of the overlap ofhydration shells and, in such situations, interpretation of anexperimentally observed time scale in terms of escape dy-namics may not be very meaningful. Clearly, there is a need
to carry out a detailed theoretical study, preferably from firstprinciples without involving any empirical potential models,of vibrational spectral diffusion in concentrated aqueousionic solutions and its relations to ion-water hydrogen bondand escape dynamics. Such a study is presented here for thefirst time.
In the present work, we have carried out a first principlestheoretical study of vibrational spectral diffusion and hydro-gen bond dynamics in aqueous ionic solutions of two differ-ent concentrations by employing the method of ab initio mo-lecular dynamics21,22 and wavelet analysis25–28 withoutinvolving any empirical potential models. In particular, welooked at the dynamics of hydration shell water around achloride ion as compared to that of pure water and also theeffects of ion concentration on the dynamics of spectral dif-fusion and hydrogen bonds. We first investigated the equilib-rium aspects of frequency-structure correlations of water inthe anionic hydration shell such as the relations between thestretch frequencies of hydration shell OD modes and the ion-water hydrogen bond distance, the distribution of hydrationshell OD frequencies as compared to that of bulk water, andalso the influence of hydrogen bond angle on the OD stretchfrequencies. Subsequently, we calculated the hydrogen bondand residence dynamics of hydration shell and bulk watermolecules by means of population time correlation functionapproach and the dynamics of vibrational spectral diffusionthrough frequency time correlation and hole dynamics calcu-lations. The observed dynamics of the spectral diffusion ofhydration shell water molecules at low ion concentration areanalyzed in terms of the dynamics of intact ion-water hydro-gen bonds, lifetimes of ion-water hydrogen bonds, and alsothe residence time of water in the anionic hydration shell. Athigh ion concentration, where the distinction between thehydration shell and bulk water becomes blurred in terms oftheir OD stretch frequencies, the calculated spectral diffusionfor all the water molecules reveal only the first two dynami-cal processes without any noticeable component of slow es-cape dynamics. Also, the time scales of the dynamics of ion-water intact hydrogen bonds and their lifetimes are found tobe longer than those of pure water and also these time scalesbecome longer with an increase in the ion concentration, inagreement with the observations of recent experiments.13
We have organized the rest of the paper as follows. InSec. II, we present the details of ab initio molecular dynam-ics simulations and calculations of fluctuating frequencies.The results of frequency distributions and frequency-structure correlations are discussed in Sec. III for hydrationshell and bulk water molecules. In Sec. IV, we present ourresults of the relaxation of chloride ion-water hydrogenbonds and also of the escape dynamics of water moleculesfrom the hydration shell of the anion. The results of vibra-tional spectral diffusion of hydration shell water in the solu-tion containing a single ion are discussed in Sec. V, and theeffects of ion concentration on the dynamics of spectral dif-fusion and hydrogen bonds are discussed in Sec. VI. Ourconclusions are briefly summarized in Sec. VII.
194512-2 Mallik, Semparithi, and Chandra J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

II. DETAILS OF SIMULATIONS AND FREQUENCYCALCULATIONS
The ab initio molecular dynamics simulations have beencarried out by employing the Car–Parrinello method21,22 andthe CPMD code.29 We have considered two systems of differ-ent ion concentrations: system 1 contains a single Cl− dis-solved in a solvent of 31 D2O and system 2 is a concentratedsolution ��3.3M NaCl� with 2 Cl−, 2 Na+, and 28 D2O. Bothsystems were kept in cubic boxes with their lengths deter-mined from the experimental number densities of NaCl inH2O at 300 K.30,31 For system 1, which contains a singlechloride ion giving an ion concentration of �1.5M, we cal-culated the box length assuming a 1.5M NaCl solution con-sisting of 1 Cl−, 1 Na+, and 30 water, and then replaced theNa+ by a water molecule. Periodic boundary conditions wereapplied in all three dimensions and the electronic structure ofthe extended systems was represented by the Kohn–Sham�KS� formulation32 of density functional theory within aplane wave basis. The core electrons were treated via theatomic pseudopotentials of Troullier–Martins33 and the planewave expansion of the KS orbitals was truncated at a kineticenergy of 80 Ry. Although system 1 of the present studycontains only a single chloride ion, we do not expect itssolvation properties to be significantly altered by the pres-ence of a counterion. For example, simulations of a singleBr− and of HBr dissolved in 31 water showed, within errorbars, essentially the same structure and dynamics of the Br−
hydration shell.23 Also, regarding the overall small size ofthe present simulation systems, we note that majority of theexisting ab initio simulations of water and aqueous solutionhave dealt with similar sized systems because of a ratherlarge computational cost of these simulations.34 Also, a veryrecent ab initio molecular dynamics study35 considered liq-uid water systems of 32 and 64 water molecules and thecalculated structure and mean square displacements of thewater molecules were found to be not greatly different forthe two system sizes.
A fictitious mass of �=800 a.u. was assigned to theelectronic degrees of freedom and the coupled equations ofmotion describing the system dynamics was integrated byusing a time step of 5 a.u., which is equal to about 0.12 fs.All hydrogen atoms were assigned the mass of deuterium toreduce the influence of quantum effects on the dynamicalproperties. Also, our choice of D2O in place of H2O ensuresthat electronic adiabaticity and energy conservation aremaintained throughout the simulations for the chosen valuesof the fictitious electronic mass parameter and time step. Thechoice of a proper value of the electronic mass parameter isan important issue in performing Car–Parrinello simulations.While a value of 800 a.u. is likely not reliable for H2O �Ref.36� for maintaining electronic adiabaticity, it is found to beacceptable for the present D2O systems as no significant driftof the electronic kinetic energy was observed during the en-tire simulations. Also, in the context of earlier reports on thedependence of water properties on the fictitious electronicmass parameter,37 we note the more recent work in Refs. 36and 38, where it has been shown that Car–Parrinello simula-tions with electronic adiabaticity and energy conservationsatisfied throughout the runs produce results that are essen-
tially the same as those produced by other ab initio simula-tion methods such as Born–Oppenheimer dynamics and abinitio Monte Carlo simulations for the same density func-tional.
The present ab initio molecular dynamics simulationswere performed using the BLYP �Ref. 39� functional. Thisfunctional has been used in many ab initio molecular dynam-ics simulations of hydrogen bonded systems such as liquidand supercritical water,40–46 aqueous solutions,47–54
methanol,55–57 ammonia,58,59 mixed systems such as water-methanol mixtures60 and also air-water interfaces.61 Al-though this functional, like many other functionals withingeneralized gradient approximation �GGA� such as PBE,62
PW91,63 and TPSS,64 leads to a somewhat overstructure anda slower rotational and translational dynamics of water,35,41
other functionals such as OLYP �Ref. 65� or HCTH407 �Ref.66� have been found to exhibit too little structure to comparewell with experiments35 for liquid water. One of the reasonsof these quantitative discrepancies could be the fact that theGGA functionals do not describe the dispersive interactionssatisfactorily and also different simulations may not be ex-actly at the same temperature, especially when they are rununder microcanonical conditions. The BLYP functional is be-ing continually used for liquid representations not only undernonreactive situations but also for simulating chemical reac-tions in aqueous media.67 For proton transfer processes inaqueous solutions, it has been shown recently that BLYPperforms reliably, while some of the other functionals pro-duced results which were inconsistent with experiments.68
Thus, in spite of some quantitative shortcomings, this func-tional is widely used because it captures the right overallphysical and chemical picture of water over a wide range ofchemical and thermodynamic states where empirical modelsstruggle and also because of the fact that ab initio moleculardynamics is intrinsically a more powerful method to study adynamical system than its classical analog. Besides, it hasbeen shown very recently that the performance of BLYP canbe improved if simulations are carried out at experimentalpressures rather than at experimental densities.69 Thus, froman overall perspective, BLYP continues to remain the mostcommonly used functional for exploring hydrogen bondedsystems through ab initio simulations.
In the present study, the initial configuration of watermolecules and ions were generated by carrying out a classi-cal molecular dynamics simulation using the empirical mul-tisite interaction potentials.70–72 Then, for each of the ab ini-tio molecular dynamics simulations, we equilibrated thesystem for 8 ps using rescaling of velocities at 300 K and,thereafter, we continued the run in NVE ensemble for an-other 50 and 40 ps for systems 1 and 2, respectively. We notethat these run periods of 50 and 40 ps, although seem quiteshort compared to typical lengths of classical simulations,are rather long in the context of ab initio molecular dynam-ics. To our knowledge, all of the existing ab initio simula-tions of aqueous ionic solutions23,24,49–53 involved runlengths that were shorter than those of the present simula-tions.
The vibrational frequency of an OD bond in the solu-tions fluctuates due to fluctuations in its interactions with the
194512-3 Vibrational spectral diffusion in aqueous solutions J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
environment caused by continual motion of the surroundingmolecules. A quantitative calculation of the time dependentvibrational frequencies of OD bonds can be carried outthrough a time series analysis of the ab initio molecular dy-namics trajectories using the wavelet method.25 Here we out-line the key features of this method, and the reader is referredto Refs. 26–28 for further details. In the wavelet method oftime series analysis, a time dependent function f�t� is ex-pressed in terms of basis functions which are constructed astranslations and dilations of a mother wavelet �
�a,b�t� = a−1/2�� t − b
a� , �1�
and the coefficients of the expansion are given by the wave-let transform of f�t�, which is defined as
L�f�a,b� = a−1/2�−�
�
f�t��� t − b
a�dt �2�
for a�0 and b real. The mother wavelet has to have compactsupport for it to be useful, i.e., it should decay to zero rapidlyfor t→ ��. Following previous work,26–28 we have used theMorlet–Grossman wavelet73 as the mother wavelet
��t� =1
�2�e2�ite−t2/2�2
, �3�
with =1, �=2, and i represents the imaginary number. Thewavelet transform of Eq. �2� produces a complex surface as afunction of the variables a and b. The inverse of the scalefactor a is proportional to the frequency and thus the wavelettransform at each b gives the frequency content of f�t� over atime window about b. Following our recent work on purewater,28 the time dependent function f�t� for a given ODbond is constructed to be a complex function
f�t� = r�t� + ip�t� , �4�
with its real and imaginary parts, r�t� and p�t�, correspondingto the instantaneous fluctuations in OD distance and the cor-responding momentum along the OD bond at time t. Thestretch frequency of this bond at a given time t=b is thendetermined from the scale a that maximizes the modulus ofthe corresponding wavelet transform at b. The process isthen repeated for the entire trajectories and for all the ODbonds that are there in the two solutions considered in thiswork.
III. VIBRATIONAL FREQUENCIES OF WATER IN ANDOUTSIDE THE HYDRATION SHELL OF Cl−
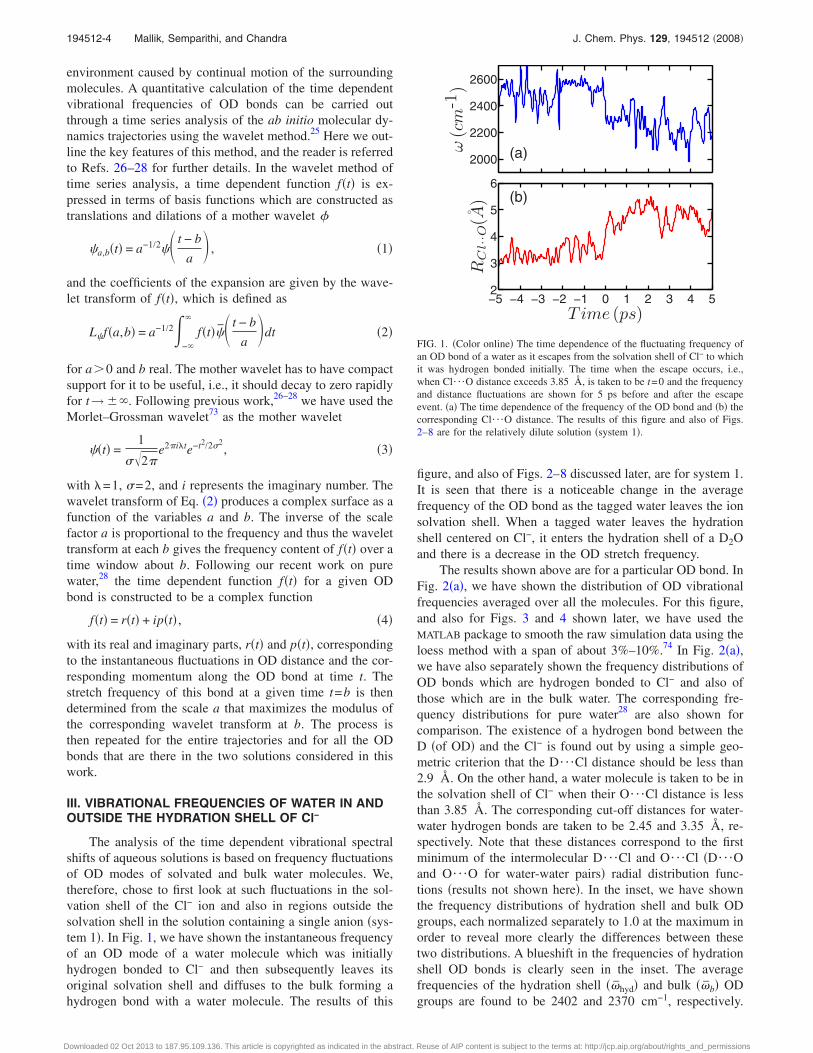
The analysis of the time dependent vibrational spectralshifts of aqueous solutions is based on frequency fluctuationsof OD modes of solvated and bulk water molecules. We,therefore, chose to first look at such fluctuations in the sol-vation shell of the Cl− ion and also in regions outside thesolvation shell in the solution containing a single anion �sys-tem 1�. In Fig. 1, we have shown the instantaneous frequencyof an OD mode of a water molecule which was initiallyhydrogen bonded to Cl− and then subsequently leaves itsoriginal solvation shell and diffuses to the bulk forming ahydrogen bond with a water molecule. The results of this
figure, and also of Figs. 2–8 discussed later, are for system 1.It is seen that there is a noticeable change in the averagefrequency of the OD bond as the tagged water leaves the ionsolvation shell. When a tagged water leaves the hydrationshell centered on Cl−, it enters the hydration shell of a D2Oand there is a decrease in the OD stretch frequency.
The results shown above are for a particular OD bond. InFig. 2�a�, we have shown the distribution of OD vibrationalfrequencies averaged over all the molecules. For this figure,and also for Figs. 3 and 4 shown later, we have used theMATLAB package to smooth the raw simulation data using theloess method with a span of about 3%–10%.74 In Fig. 2�a�,we have also separately shown the frequency distributions ofOD bonds which are hydrogen bonded to Cl− and also ofthose which are in the bulk water. The corresponding fre-quency distributions for pure water28 are also shown forcomparison. The existence of a hydrogen bond between theD �of OD� and the Cl− is found out by using a simple geo-metric criterion that the D¯Cl distance should be less than2.9 Å. On the other hand, a water molecule is taken to be inthe solvation shell of Cl− when their O¯Cl distance is lessthan 3.85 Å. The corresponding cut-off distances for water-water hydrogen bonds are taken to be 2.45 and 3.35 Å, re-spectively. Note that these distances correspond to the firstminimum of the intermolecular D¯Cl and O¯Cl �D¯Oand O¯O for water-water pairs� radial distribution func-tions �results not shown here�. In the inset, we have shownthe frequency distributions of hydration shell and bulk ODgroups, each normalized separately to 1.0 at the maximum inorder to reveal more clearly the differences between thesetwo distributions. A blueshift in the frequencies of hydrationshell OD bonds is clearly seen in the inset. The averagefrequencies of the hydration shell �hyd� and bulk �b� ODgroups are found to be 2402 and 2370 cm−1, respectively.
2000
2200
2400
2600
(a)ω(c
m- 1
)
−5 −4 −3 −2 −1 0 1 2 3 4 52
3
4
5
6(b)
T ime (ps)
RC
l··O(A
)
FIG. 1. �Color online� The time dependence of the fluctuating frequency ofan OD bond of a water as it escapes from the solvation shell of Cl− to whichit was hydrogen bonded initially. The time when the escape occurs, i.e.,when Cl¯O distance exceeds 3.85 Å, is taken to be t=0 and the frequencyand distance fluctuations are shown for 5 ps before and after the escapeevent. �a� The time dependence of the frequency of the OD bond and �b� thecorresponding Cl¯O distance. The results of this figure and also of Figs.2–8 are for the relatively dilute solution �system 1�.
194512-4 Mallik, Semparithi, and Chandra J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
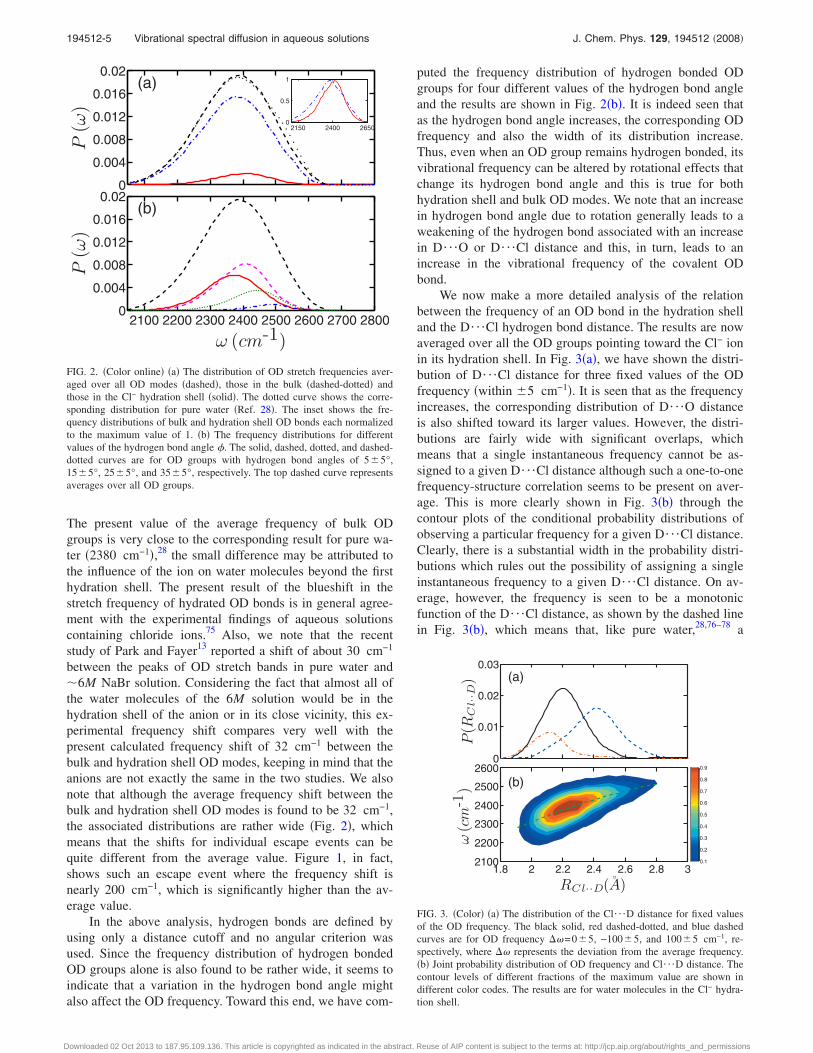
The present value of the average frequency of bulk ODgroups is very close to the corresponding result for pure wa-ter �2380 cm−1�,28 the small difference may be attributed tothe influence of the ion on water molecules beyond the firsthydration shell. The present result of the blueshift in thestretch frequency of hydrated OD bonds is in general agree-ment with the experimental findings of aqueous solutionscontaining chloride ions.75 Also, we note that the recentstudy of Park and Fayer13 reported a shift of about 30 cm−1
between the peaks of OD stretch bands in pure water and�6M NaBr solution. Considering the fact that almost all ofthe water molecules of the 6M solution would be in thehydration shell of the anion or in its close vicinity, this ex-perimental frequency shift compares very well with thepresent calculated frequency shift of 32 cm−1 between thebulk and hydration shell OD modes, keeping in mind that theanions are not exactly the same in the two studies. We alsonote that although the average frequency shift between thebulk and hydration shell OD modes is found to be 32 cm−1,the associated distributions are rather wide �Fig. 2�, whichmeans that the shifts for individual escape events can bequite different from the average value. Figure 1, in fact,shows such an escape event where the frequency shift isnearly 200 cm−1, which is significantly higher than the av-erage value.
In the above analysis, hydrogen bonds are defined byusing only a distance cutoff and no angular criterion wasused. Since the frequency distribution of hydrogen bondedOD groups alone is also found to be rather wide, it seems toindicate that a variation in the hydrogen bond angle mightalso affect the OD frequency. Toward this end, we have com-
puted the frequency distribution of hydrogen bonded ODgroups for four different values of the hydrogen bond angleand the results are shown in Fig. 2�b�. It is indeed seen thatas the hydrogen bond angle increases, the corresponding ODfrequency and also the width of its distribution increase.Thus, even when an OD group remains hydrogen bonded, itsvibrational frequency can be altered by rotational effects thatchange its hydrogen bond angle and this is true for bothhydration shell and bulk OD modes. We note that an increasein hydrogen bond angle due to rotation generally leads to aweakening of the hydrogen bond associated with an increasein D¯O or D¯Cl distance and this, in turn, leads to anincrease in the vibrational frequency of the covalent ODbond.
We now make a more detailed analysis of the relationbetween the frequency of an OD bond in the hydration shelland the D¯Cl hydrogen bond distance. The results are nowaveraged over all the OD groups pointing toward the Cl− ionin its hydration shell. In Fig. 3�a�, we have shown the distri-bution of D¯Cl distance for three fixed values of the ODfrequency �within �5 cm−1�. It is seen that as the frequencyincreases, the corresponding distribution of D¯O distanceis also shifted toward its larger values. However, the distri-butions are fairly wide with significant overlaps, whichmeans that a single instantaneous frequency cannot be as-signed to a given D¯Cl distance although such a one-to-onefrequency-structure correlation seems to be present on aver-age. This is more clearly shown in Fig. 3�b� through thecontour plots of the conditional probability distributions ofobserving a particular frequency for a given D¯Cl distance.Clearly, there is a substantial width in the probability distri-butions which rules out the possibility of assigning a singleinstantaneous frequency to a given D¯Cl distance. On av-erage, however, the frequency is seen to be a monotonicfunction of the D¯Cl distance, as shown by the dashed linein Fig. 3�b�, which means that, like pure water,28,76–78 a
0
0.004
0.008
0.012
0.016
0.02P
(ω)
(a)
2150 2400 26500
0.5
1
2100 2200 2300 2400 2500 2600 2700 28000
0.004
0.008
0.012
0.016
0.02
ω (cm-1)
P(ω
)
(b)
FIG. 2. �Color online� �a� The distribution of OD stretch frequencies aver-aged over all OD modes �dashed�, those in the bulk �dashed-dotted� andthose in the Cl− hydration shell �solid�. The dotted curve shows the corre-sponding distribution for pure water �Ref. 28�. The inset shows the fre-quency distributions of bulk and hydration shell OD bonds each normalizedto the maximum value of 1. �b� The frequency distributions for differentvalues of the hydrogen bond angle �. The solid, dashed, dotted, and dashed-dotted curves are for OD groups with hydrogen bond angles of 5�5°,15�5°, 25�5°, and 35�5°, respectively. The top dashed curve representsaverages over all OD groups.
0
0.01
0.02
0.03(a)
P(R
Cl··
D)
1.8 2 2.2 2.4 2.6 2.8 32100
2200
2300
2400
2500
2600
RCl··D(A)
ω(c
m-1
) (b)
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
FIG. 3. �Color� �a� The distribution of the Cl¯D distance for fixed valuesof the OD frequency. The black solid, red dashed-dotted, and blue dashedcurves are for OD frequency �=0�5, −100�5, and 100�5 cm−1, re-spectively, where � represents the deviation from the average frequency.�b� Joint probability distribution of OD frequency and Cl¯D distance. Thecontour levels of different fractions of the maximum value are shown indifferent color codes. The results are for water molecules in the Cl− hydra-tion shell.
194512-5 Vibrational spectral diffusion in aqueous solutions J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
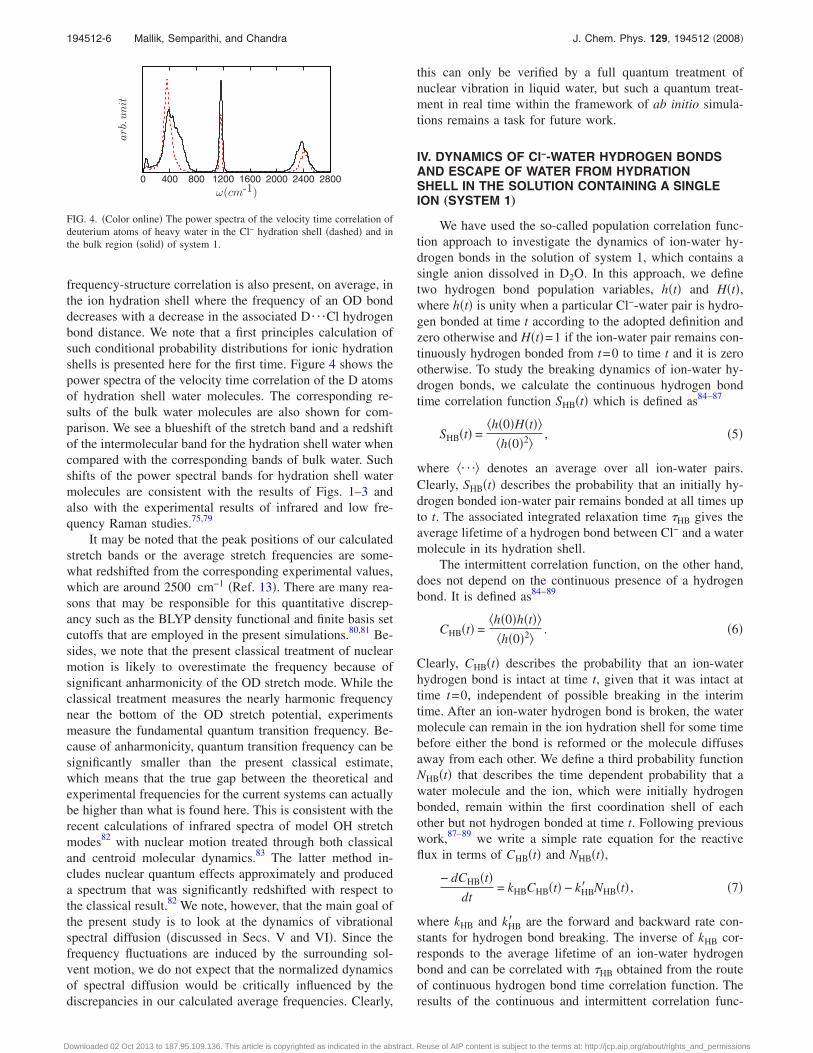
frequency-structure correlation is also present, on average, inthe ion hydration shell where the frequency of an OD bonddecreases with a decrease in the associated D¯Cl hydrogenbond distance. We note that a first principles calculation ofsuch conditional probability distributions for ionic hydrationshells is presented here for the first time. Figure 4 shows thepower spectra of the velocity time correlation of the D atomsof hydration shell water molecules. The corresponding re-sults of the bulk water molecules are also shown for com-parison. We see a blueshift of the stretch band and a redshiftof the intermolecular band for the hydration shell water whencompared with the corresponding bands of bulk water. Suchshifts of the power spectral bands for hydration shell watermolecules are consistent with the results of Figs. 1–3 andalso with the experimental results of infrared and low fre-quency Raman studies.75,79
It may be noted that the peak positions of our calculatedstretch bands or the average stretch frequencies are some-what redshifted from the corresponding experimental values,which are around 2500 cm−1 �Ref. 13�. There are many rea-sons that may be responsible for this quantitative discrep-ancy such as the BLYP density functional and finite basis setcutoffs that are employed in the present simulations.80,81 Be-sides, we note that the present classical treatment of nuclearmotion is likely to overestimate the frequency because ofsignificant anharmonicity of the OD stretch mode. While theclassical treatment measures the nearly harmonic frequencynear the bottom of the OD stretch potential, experimentsmeasure the fundamental quantum transition frequency. Be-cause of anharmonicity, quantum transition frequency can besignificantly smaller than the present classical estimate,which means that the true gap between the theoretical andexperimental frequencies for the current systems can actuallybe higher than what is found here. This is consistent with therecent calculations of infrared spectra of model OH stretchmodes82 with nuclear motion treated through both classicaland centroid molecular dynamics.83 The latter method in-cludes nuclear quantum effects approximately and produceda spectrum that was significantly redshifted with respect tothe classical result.82 We note, however, that the main goal ofthe present study is to look at the dynamics of vibrationalspectral diffusion �discussed in Secs. V and VI�. Since thefrequency fluctuations are induced by the surrounding sol-vent motion, we do not expect that the normalized dynamicsof spectral diffusion would be critically influenced by thediscrepancies in our calculated average frequencies. Clearly,
this can only be verified by a full quantum treatment ofnuclear vibration in liquid water, but such a quantum treat-ment in real time within the framework of ab initio simula-tions remains a task for future work.
IV. DYNAMICS OF Cl−-WATER HYDROGEN BONDSAND ESCAPE OF WATER FROM HYDRATIONSHELL IN THE SOLUTION CONTAINING A SINGLEION „SYSTEM 1…
We have used the so-called population correlation func-tion approach to investigate the dynamics of ion-water hy-drogen bonds in the solution of system 1, which contains asingle anion dissolved in D2O. In this approach, we definetwo hydrogen bond population variables, h�t� and H�t�,where h�t� is unity when a particular Cl−-water pair is hydro-gen bonded at time t according to the adopted definition andzero otherwise and H�t�=1 if the ion-water pair remains con-tinuously hydrogen bonded from t=0 to time t and it is zerootherwise. To study the breaking dynamics of ion-water hy-drogen bonds, we calculate the continuous hydrogen bondtime correlation function SHB�t� which is defined as84–87
SHB�t� =h�0�H�t��
h�0�2�, �5�
where ¯� denotes an average over all ion-water pairs.Clearly, SHB�t� describes the probability that an initially hy-drogen bonded ion-water pair remains bonded at all times upto t. The associated integrated relaxation time �HB gives theaverage lifetime of a hydrogen bond between Cl− and a watermolecule in its hydration shell.
The intermittent correlation function, on the other hand,does not depend on the continuous presence of a hydrogenbond. It is defined as84–89
CHB�t� =h�0�h�t��
h�0�2�. �6�
Clearly, CHB�t� describes the probability that an ion-waterhydrogen bond is intact at time t, given that it was intact attime t=0, independent of possible breaking in the interimtime. After an ion-water hydrogen bond is broken, the watermolecule can remain in the ion hydration shell for some timebefore either the bond is reformed or the molecule diffusesaway from each other. We define a third probability functionNHB�t� that describes the time dependent probability that awater molecule and the ion, which were initially hydrogenbonded, remain within the first coordination shell of eachother but not hydrogen bonded at time t. Following previouswork,87–89 we write a simple rate equation for the reactiveflux in terms of CHB�t� and NHB�t�,
− dCHB�t�dt
= kHBCHB�t� − kHB� NHB�t� , �7�
where kHB and kHB� are the forward and backward rate con-stants for hydrogen bond breaking. The inverse of kHB cor-responds to the average lifetime of an ion-water hydrogenbond and can be correlated with �HB obtained from the routeof continuous hydrogen bond time correlation function. Theresults of the continuous and intermittent correlation func-
0 400 800 1200 1600 2000 2400 2800ω(cm-1)
arb
.unit
FIG. 4. �Color online� The power spectra of the velocity time correlation ofdeuterium atoms of heavy water in the Cl− hydration shell �dashed� and inthe bulk region �solid� of system 1.
194512-6 Mallik, Semparithi, and Chandra J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
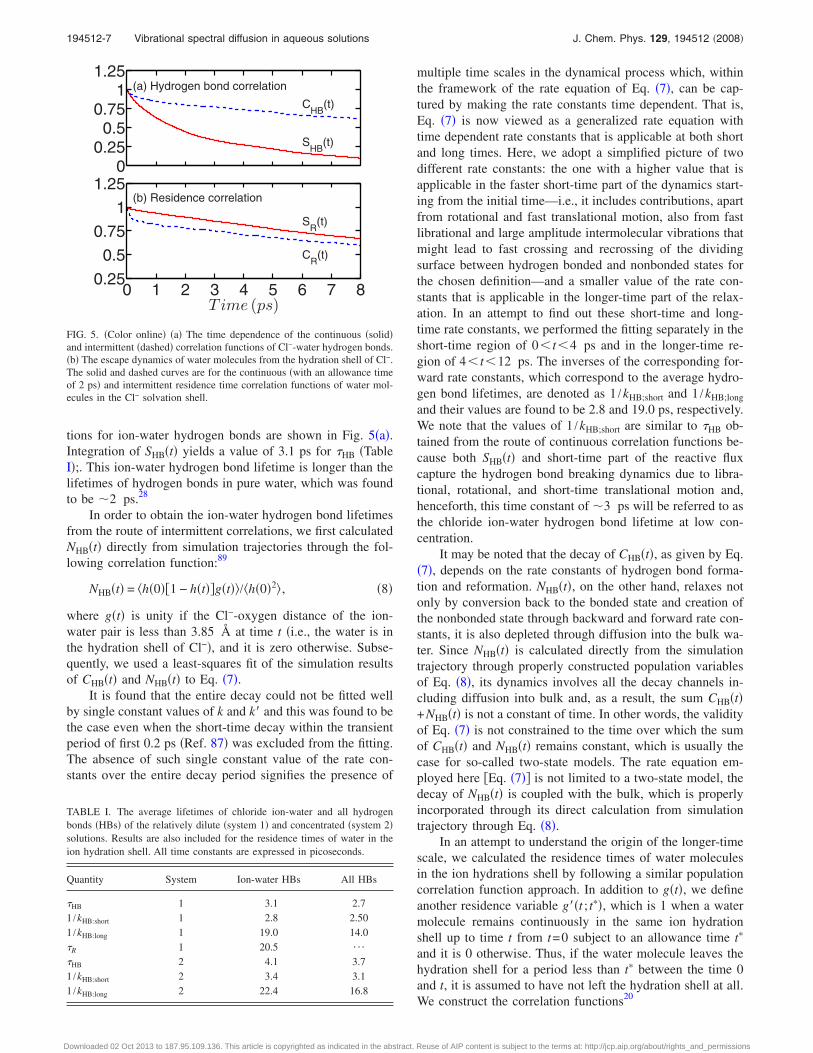
tions for ion-water hydrogen bonds are shown in Fig. 5�a�.Integration of SHB�t� yields a value of 3.1 ps for �HB �TableI�;. This ion-water hydrogen bond lifetime is longer than thelifetimes of hydrogen bonds in pure water, which was foundto be �2 ps.28
In order to obtain the ion-water hydrogen bond lifetimesfrom the route of intermittent correlations, we first calculatedNHB�t� directly from simulation trajectories through the fol-lowing correlation function:89
NHB�t� = h�0��1 − h�t��g�t��/h�0�2� , �8�
where g�t� is unity if the Cl−-oxygen distance of the ion-water pair is less than 3.85 Å at time t �i.e., the water is inthe hydration shell of Cl−�, and it is zero otherwise. Subse-quently, we used a least-squares fit of the simulation resultsof CHB�t� and NHB�t� to Eq. �7�.
It is found that the entire decay could not be fitted wellby single constant values of k and k� and this was found to bethe case even when the short-time decay within the transientperiod of first 0.2 ps �Ref. 87� was excluded from the fitting.The absence of such single constant value of the rate con-stants over the entire decay period signifies the presence of
multiple time scales in the dynamical process which, withinthe framework of the rate equation of Eq. �7�, can be cap-tured by making the rate constants time dependent. That is,Eq. �7� is now viewed as a generalized rate equation withtime dependent rate constants that is applicable at both shortand long times. Here, we adopt a simplified picture of twodifferent rate constants: the one with a higher value that isapplicable in the faster short-time part of the dynamics start-ing from the initial time—i.e., it includes contributions, apartfrom rotational and fast translational motion, also from fastlibrational and large amplitude intermolecular vibrations thatmight lead to fast crossing and recrossing of the dividingsurface between hydrogen bonded and nonbonded states forthe chosen definition—and a smaller value of the rate con-stants that is applicable in the longer-time part of the relax-ation. In an attempt to find out these short-time and long-time rate constants, we performed the fitting separately in theshort-time region of 0 t 4 ps and in the longer-time re-gion of 4 t 12 ps. The inverses of the corresponding for-ward rate constants, which correspond to the average hydro-gen bond lifetimes, are denoted as 1 /kHB;short and 1 /kHB;long
and their values are found to be 2.8 and 19.0 ps, respectively.We note that the values of 1 /kHB;short are similar to �HB ob-tained from the route of continuous correlation functions be-cause both SHB�t� and short-time part of the reactive fluxcapture the hydrogen bond breaking dynamics due to libra-tional, rotational, and short-time translational motion and,henceforth, this time constant of �3 ps will be referred to asthe chloride ion-water hydrogen bond lifetime at low con-centration.
It may be noted that the decay of CHB�t�, as given by Eq.�7�, depends on the rate constants of hydrogen bond forma-tion and reformation. NHB�t�, on the other hand, relaxes notonly by conversion back to the bonded state and creation ofthe nonbonded state through backward and forward rate con-stants, it is also depleted through diffusion into the bulk wa-ter. Since NHB�t� is calculated directly from the simulationtrajectory through properly constructed population variablesof Eq. �8�, its dynamics involves all the decay channels in-cluding diffusion into bulk and, as a result, the sum CHB�t�+NHB�t� is not a constant of time. In other words, the validityof Eq. �7� is not constrained to the time over which the sumof CHB�t� and NHB�t� remains constant, which is usually thecase for so-called two-state models. The rate equation em-ployed here �Eq. �7�� is not limited to a two-state model, thedecay of NHB�t� is coupled with the bulk, which is properlyincorporated through its direct calculation from simulationtrajectory through Eq. �8�.
In an attempt to understand the origin of the longer-timescale, we calculated the residence times of water moleculesin the ion hydrations shell by following a similar populationcorrelation function approach. In addition to g�t�, we defineanother residence variable g��t ; t��, which is 1 when a watermolecule remains continuously in the same ion hydrationshell up to time t from t=0 subject to an allowance time t�
and it is 0 otherwise. Thus, if the water molecule leaves thehydration shell for a period less than t� between the time 0and t, it is assumed to have not left the hydration shell at all.We construct the correlation functions20
TABLE I. The average lifetimes of chloride ion-water and all hydrogenbonds �HBs� of the relatively dilute �system 1� and concentrated �system 2�solutions. Results are also included for the residence times of water in theion hydration shell. All time constants are expressed in picoseconds.
Quantity System Ion-water HBs All HBs
�HB 1 3.1 2.71 /kHB:short 1 2.8 2.501 /kHB:long 1 19.0 14.0�R 1 20.5 ¯
�HB 2 4.1 3.71 /kHB:short 2 3.4 3.11 /kHB:long 2 22.4 16.8
00.25
0.50.75
11.25
(a) Hydrogen bond correlation
CHB
(t)
SHB
(t)
0 1 2 3 4 5 6 7 80.25
0.5
0.75
1
1.25
T ime (ps)
(b) Residence correlation
SR
(t)
CR
(t)
FIG. 5. �Color online� �a� The time dependence of the continuous �solid�and intermittent �dashed� correlation functions of Cl−-water hydrogen bonds.�b� The escape dynamics of water molecules from the hydration shell of Cl−.The solid and dashed curves are for the continuous �with an allowance timeof 2 ps� and intermittent residence time correlation functions of water mol-ecules in the Cl− solvation shell.
194512-7 Vibrational spectral diffusion in aqueous solutions J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

SR�t� = g�0�g��t;t���/g�0�2� , �9�
CR�t� = g�0�g�t��/g�0�2� , �10�
where SR�t� describes the probability that a water molecule,which was in the hydration shell of the ion at time t=0,remains continuously in the same hydration shell up to timet subject to the allowance time t�. The associated integratedrelaxation time �R gives the average residence time of a wa-ter molecule in the ion hydration shell. The intermittent resi-dence correlation function CR�t� gives the probability that awater molecule, which was in the hydration shell of an ion atthe initial time, is also found to be in the same hydrationshell at time t irrespective of what has happened in the in-terim period. In Fig. 5�b�, we have shown the decay of SR�t�and CR�t�. We calculated the residence time �R by explicitintegration of SR�t� from simulations until 10 ps and by cal-culating the integral for the tail part from fitted exponentialfunctions. Following previous work,20,90 we took the allow-ance time to be 2 ps for the continuous residence functionand found a value of 20.5 ps for the residence time of watermolecules in the Cl− solvation shell �Table I�. The error barof the residence time is about 25% of the average valuebecause of the rather short trajectory and a single ion that areinvolved in extracting this time scale. We note, however, thatthe present statistical averaging is better than many of theexisting ab initio simulation studies of residence time esti-mates where even shorter trajectories of aqueous ionic solu-tions were used.23,24 In the present study, we also extractedthe residence time from the intermittent correlation by thefollowing the method in Ref. 20 and a very similar value isfound. Thus, from the similarity of the time scales, we canconclude that the longer-time scale of 1 /kHB;long actually cor-responds to the residence time of a water molecule in thehydration shell of a tagged water to which it was hydrogenbonded at time t=0. Hence, this longer-time scale actuallycorresponds to the escape dynamics or slow diffusion of awater molecule from the hydration shell of the chloride ion.
V. VIBRATIONAL SPECTRAL DIFFUSION OFHYDRATION SHELL WATER IN THE SOLUTIONCONTAINING A SINGLE ION „SYSTEM 1…
In this section, we calculate quantities related to the ex-perimental time dependent infrared signal associated with thehole dynamics in the ground OD vibrational state in aqueousionic solutions. In particular, we calculate the time evolutionof the average frequency for the nonequilibrium distributionof OD frequencies which is initially created from an equilib-rium distribution by the removal of a band of OD frequen-cies, called the hole modes, corresponding to their laser pro-motion. We also look at the time evolution of the averagefrequency of these hole modes. The present hole dynamicscalculations follow closely the approach suggested in Refs.18 and 78 and subsequently used in Ref. 28. As noted inthese earlier work,18,78 the time evolution of these initiallycreated nonequilibrium distributions is closely related to thetime evolution of the pump-probe signals of recent time de-
pendent infrared spectroscopic experiments designed to in-vestigate vibrational spectral diffusion in aqueous ionic solu-tions.
At t=0, the laser pump pulse, which is assumed to havea Gaussian frequency profile, burns a hole in the ground statefrequency distribution of the form
Ph�,0� = Peq��e−� − p�2/2�2, �11�
where p is the pulse center frequency and Peq�� denotesthe equilibrium frequency distribution of the OD modes ofinterest. Clearly, the initial distribution of the remaining ODfrequencies Pr� ,0�, i.e., the ones remaining in the groundstate in experimental situations, is equal to Peq��− Ph� ,0�. We use a Gaussian pulse of full width �2��140 cm−1 and calculate the time evolution of the nonequi-librium distributions Pr� , t� and Ph� , t� from a large set ofsystem trajectories reflecting the initial distributions Pr� ,0�and Ph� ,0�, respectively. The average frequency of the holemodes at time t is then calculated from the following rela-tion:
h�t� =1
Nh� dPh�,t� , �12�
where Nh=�dPh� ,0�. The average time dependent fre-quency of the remaining modes is calculated in a similar wayby using the remaining distribution in Eq. �12�.
We first considered the subset of hydration shell ODmodes only and carried out the above hole dynamics calcu-lations. We followed the dynamics of the average frequencyshifts of the hole and remaining modes of hydration shellwater molecules by creating holes in two different frequencyregions: one centered in the red side at p= hyd
−100 cm−1 and the other centered in the blue side at p
= hyd+100 cm−1 where, as defined earlier, hyd is the aver-age frequency of all the OD groups which are in the hydra-tion shell of the Cl− ion. We employed a Metropolis MonteCarlo-type algorithm91 to effect the creation of a chosenhole, red or blue, so as to satisfy the distribution of Eq. �11�.At a given time step along the trajectory, we first choose anOD group of frequency i in the hydration shell and, in orderto find out if this mode is included as a hole mode, we findthe value of the corresponding Gaussian probability Pi
=e−�i − p�2/2�2. We next generate a random number yi from a
uniform distribution between 0 and 1 and if yi comes out tobe less than the Gaussian probability Pi, then the chosenmode i is included as a hole mode. The above exercise isrepeated for all the OD groups in the Cl− hydration shell andover many different initial times to ensure that, on average,the hole modes satisfy the distribution of Eq. �11�.
We first show the time evolution of the average frequen-cies of the hole and the remaining modes in the hydrationshell after the hole is created at t=0. We note that in ourtheoretical calculations, the hole is created instantly unlike intrue experimental situations and, hence, its dynamics can befollowed immediately after t=0 in the present study. In trueexperiments, however, one requires laser pulses of higherwidth to meaningfully measure the dynamics within first
194512-8 Mallik, Semparithi, and Chandra J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
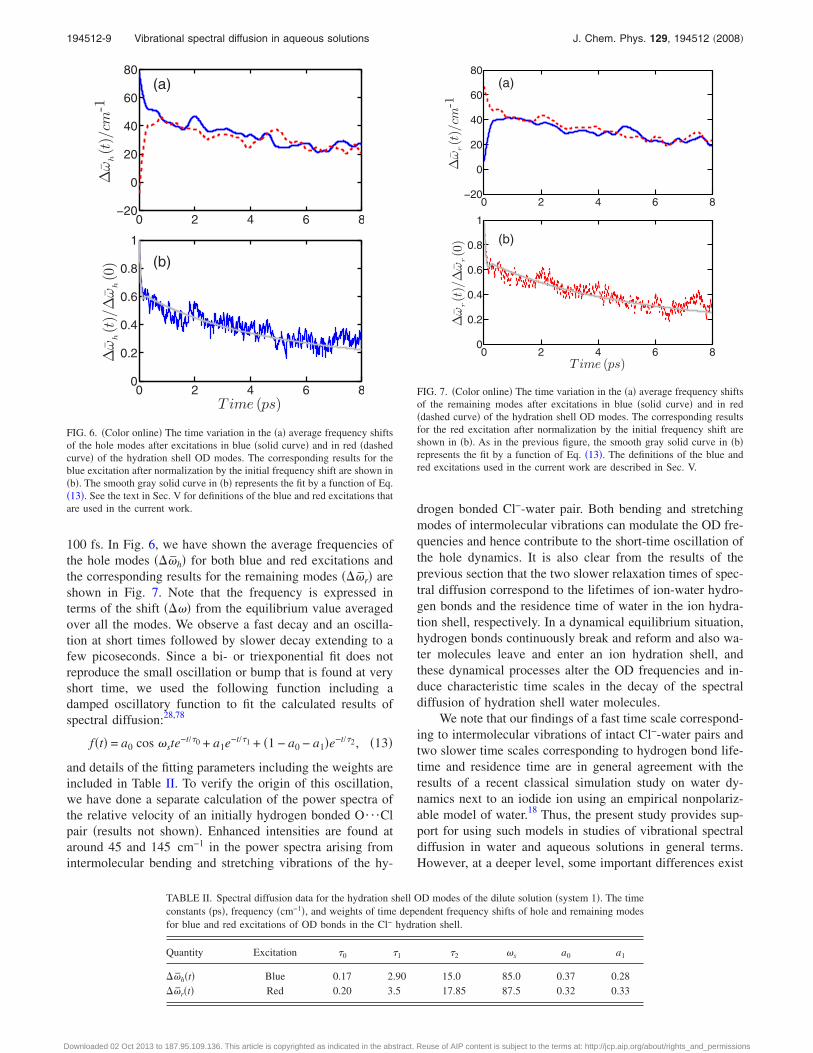
100 fs. In Fig. 6, we have shown the average frequencies ofthe hole modes ��h� for both blue and red excitations andthe corresponding results for the remaining modes ��r� areshown in Fig. 7. Note that the frequency is expressed interms of the shift ��� from the equilibrium value averagedover all the modes. We observe a fast decay and an oscilla-tion at short times followed by slower decay extending to afew picoseconds. Since a bi- or triexponential fit does notreproduce the small oscillation or bump that is found at veryshort time, we used the following function including adamped oscillatory function to fit the calculated results ofspectral diffusion:28,78
f�t� = a0 cos ste−t/�0 + a1e−t/�1 + �1 − a0 − a1�e−t/�2, �13�
and details of the fitting parameters including the weights areincluded in Table II. To verify the origin of this oscillation,we have done a separate calculation of the power spectra ofthe relative velocity of an initially hydrogen bonded O¯Clpair �results not shown�. Enhanced intensities are found ataround 45 and 145 cm−1 in the power spectra arising fromintermolecular bending and stretching vibrations of the hy-
drogen bonded Cl−-water pair. Both bending and stretchingmodes of intermolecular vibrations can modulate the OD fre-quencies and hence contribute to the short-time oscillation ofthe hole dynamics. It is also clear from the results of theprevious section that the two slower relaxation times of spec-tral diffusion correspond to the lifetimes of ion-water hydro-gen bonds and the residence time of water in the ion hydra-tion shell, respectively. In a dynamical equilibrium situation,hydrogen bonds continuously break and reform and also wa-ter molecules leave and enter an ion hydration shell, andthese dynamical processes alter the OD frequencies and in-duce characteristic time scales in the decay of the spectraldiffusion of hydration shell water molecules.
We note that our findings of a fast time scale correspond-ing to intermolecular vibrations of intact Cl−-water pairs andtwo slower time scales corresponding to hydrogen bond life-time and residence time are in general agreement with theresults of a recent classical simulation study on water dy-namics next to an iodide ion using an empirical nonpolariz-able model of water.18 Thus, the present study provides sup-port for using such models in studies of vibrational spectraldiffusion in water and aqueous solutions in general terms.However, at a deeper level, some important differences exist
TABLE II. Spectral diffusion data for the hydration shell OD modes of the dilute solution �system 1�. The timeconstants �ps�, frequency �cm−1�, and weights of time dependent frequency shifts of hole and remaining modesfor blue and red excitations of OD bonds in the Cl− hydration shell.
Quantity Excitation �0 �1 �2 s a0 a1
�h�t� Blue 0.17 2.90 15.0 85.0 0.37 0.28�r�t� Red 0.20 3.5 17.85 87.5 0.32 0.33
0 2 4 6 8−20
0
20
40
60
80
∆ω
h(t
)/cm
-1(a)
0 2 4 6 80
0.2
0.4
0.6
0.8
1
(b)
T ime (ps)
∆ω
h(t
)/∆
ωh(0
)
FIG. 6. �Color online� The time variation in the �a� average frequency shiftsof the hole modes after excitations in blue �solid curve� and in red �dashedcurve� of the hydration shell OD modes. The corresponding results for theblue excitation after normalization by the initial frequency shift are shown in�b�. The smooth gray solid curve in �b� represents the fit by a function of Eq.�13�. See the text in Sec. V for definitions of the blue and red excitations thatare used in the current work.
0 2 4 6 8−20
0
20
40
60
80
∆ω
r(t
)/cm
-1
(a)
0 2 4 6 80
0.2
0.4
0.6
0.8
1
(b)
T ime (ps)
∆ω
r(t
)/∆
ωr(0
)
FIG. 7. �Color online� The time variation in the �a� average frequency shiftsof the remaining modes after excitations in blue �solid curve� and in red�dashed curve� of the hydration shell OD modes. The corresponding resultsfor the red excitation after normalization by the initial frequency shift areshown in �b�. As in the previous figure, the smooth gray solid curve in �b�represents the fit by a function of Eq. �13�. The definitions of the blue andred excitations used in the current work are described in Sec. V.
194512-9 Vibrational spectral diffusion in aqueous solutions J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
between the present first principles study and the existingclassical simulation studies. For example, empirical modelsalone, whether polarizable or nonpolarizable, cannot producethe time dependent fluctuating frequencies along a simula-tion trajectory. Another level of approximation, such as aperturbative approach18,78,92 or a different approximate po-tential of the solute HOD,76 or a relation between the fre-quency and electric field or electrostatic potential93 had to beemployed to extract the frequency fluctuations along thesimulation trajectory. On the contrary, the present methodol-ogy allows calculations of fluctuating frequencies directlyfrom real-time ab initio simulation trajectories by consider-ing all nuclear dynamical modes on equal footing. Hence, thepresent method of frequency calculations is more direct inthe sense that no additional approximation of the type dis-cussed above needs to be employed to extract the time de-pendent frequencies. However, the main drawback of thepresent methodology is that it treats OD vibration at classicallevel unlike in Refs. 18, 76, 78, 92, and 93, where a quantumtreatment of OH/OD vibration was employed at different lev-els of approximation. Although the absolute values of thevibrational frequencies obtained from this classical treatmentdiffer from the transition frequencies measured in experi-ments due to anharmonicity, our calculated blueshift in thestretch frequency of hydration shell D2O as compared to thatof bulk water agrees well with the experimental results ofsimilar ionic solutions,13 as has already been discussed inSec. III.
The residence times in the present calculations were cal-culated from the continuous residence correlation functionsby using a tolerance time of 2 ps, a prescription originallyproposed by Impey, Madden, and McDonald �IMM�.90 Veryrecently, it was argued that the tolerance time of 2 ps leads toan overestimation of the residence time.94 An alternate pre-scription based on stable state pictures95 was suggested,which led to residence times significantly shorter than thatobtained from IMM route. The difference was attributed tothe fact that there are some true escape events within thetolerance time of 2 ps which was not accounted for in theIMM prescription. In the present context of spectral diffu-sion, we note that any such fast escape event within 2 pswould show up in the shorter-time scale of spectral diffusion�characterized by �1�. Thus, the long-time dynamics of thespectral diffusion of hydration shell molecules correspondsmore to the longer-time escape events and, hence, goodagreement of �2 with the present residence times ��R� is ob-served even though, in the view of the results in Ref. 94, thecurrent values of �R might have actually overestimated thetrue average residence times.
VI. SPECTRAL DIFFUSION AND HYDROGEN BONDDYNAMICS OF ALL WATER MOLECULES:EFFECTS OF ION CONCENTRATION
Until now, we have looked at the spectral diffusion ofonly hydration shell water molecules. The dynamical re-sponse in an experimental situation would include contribu-tions from both hydration shell and bulk water molecules.Hence, it would be instructive to investigate the dynamics ofspectral diffusion of all the OD modes of the solutions. The
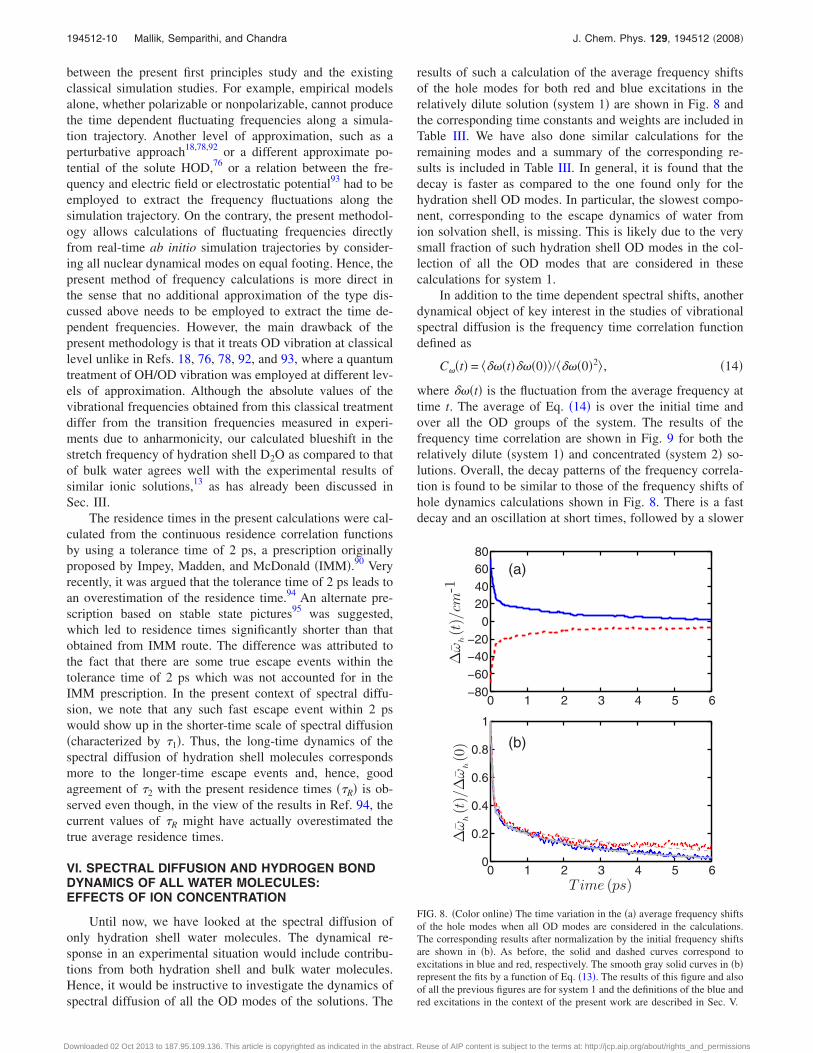
results of such a calculation of the average frequency shiftsof the hole modes for both red and blue excitations in therelatively dilute solution �system 1� are shown in Fig. 8 andthe corresponding time constants and weights are included inTable III. We have also done similar calculations for theremaining modes and a summary of the corresponding re-sults is included in Table III. In general, it is found that thedecay is faster as compared to the one found only for thehydration shell OD modes. In particular, the slowest compo-nent, corresponding to the escape dynamics of water fromion solvation shell, is missing. This is likely due to the verysmall fraction of such hydration shell OD modes in the col-lection of all the OD modes that are considered in thesecalculations for system 1.
In addition to the time dependent spectral shifts, anotherdynamical object of key interest in the studies of vibrationalspectral diffusion is the frequency time correlation functiondefined as
C�t� = ��t���0��/��0�2� , �14�
where ��t� is the fluctuation from the average frequency attime t. The average of Eq. �14� is over the initial time andover all the OD groups of the system. The results of thefrequency time correlation are shown in Fig. 9 for both therelatively dilute �system 1� and concentrated �system 2� so-lutions. Overall, the decay patterns of the frequency correla-tion is found to be similar to those of the frequency shifts ofhole dynamics calculations shown in Fig. 8. There is a fastdecay and an oscillation at short times, followed by a slower
0 1 2 3 4 5 6−80−60−40−20
020406080
∆ω
h(t
)/cm
- 1
(a)
0 1 2 3 4 5 60
0.2
0.4
0.6
0.8
1
(b)
T ime (ps)
∆ω
h(t
)/∆
ωh(0
)
FIG. 8. �Color online� The time variation in the �a� average frequency shiftsof the hole modes when all OD modes are considered in the calculations.The corresponding results after normalization by the initial frequency shiftsare shown in �b�. As before, the solid and dashed curves correspond toexcitations in blue and red, respectively. The smooth gray solid curves in �b�represent the fits by a function of Eq. �13�. The results of this figure and alsoof all the previous figures are for system 1 and the definitions of the blue andred excitations in the context of the present work are described in Sec. V.
194512-10 Mallik, Semparithi, and Chandra J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
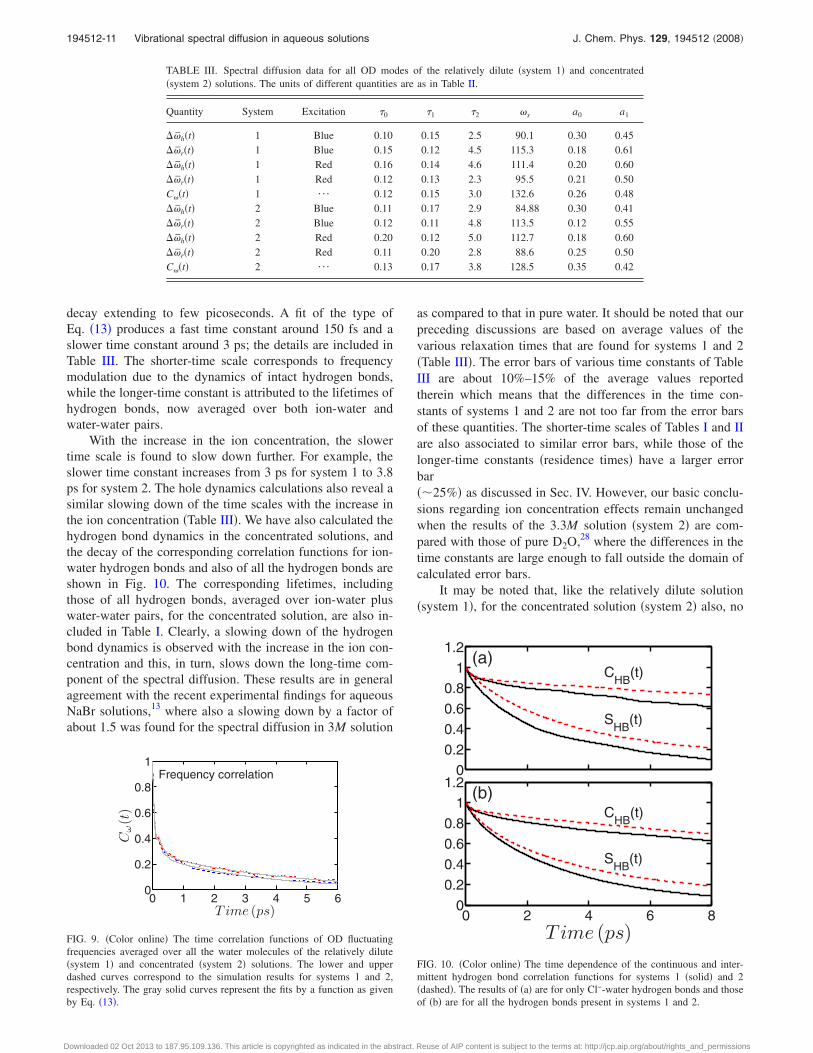
decay extending to few picoseconds. A fit of the type ofEq. �13� produces a fast time constant around 150 fs and aslower time constant around 3 ps; the details are included inTable III. The shorter-time scale corresponds to frequencymodulation due to the dynamics of intact hydrogen bonds,while the longer-time constant is attributed to the lifetimes ofhydrogen bonds, now averaged over both ion-water andwater-water pairs.
With the increase in the ion concentration, the slowertime scale is found to slow down further. For example, theslower time constant increases from 3 ps for system 1 to 3.8ps for system 2. The hole dynamics calculations also reveal asimilar slowing down of the time scales with the increase inthe ion concentration �Table III�. We have also calculated thehydrogen bond dynamics in the concentrated solutions, andthe decay of the corresponding correlation functions for ion-water hydrogen bonds and also of all the hydrogen bonds areshown in Fig. 10. The corresponding lifetimes, includingthose of all hydrogen bonds, averaged over ion-water pluswater-water pairs, for the concentrated solution, are also in-cluded in Table I. Clearly, a slowing down of the hydrogenbond dynamics is observed with the increase in the ion con-centration and this, in turn, slows down the long-time com-ponent of the spectral diffusion. These results are in generalagreement with the recent experimental findings for aqueousNaBr solutions,13 where also a slowing down by a factor ofabout 1.5 was found for the spectral diffusion in 3M solution
as compared to that in pure water. It should be noted that ourpreceding discussions are based on average values of thevarious relaxation times that are found for systems 1 and 2�Table III�. The error bars of various time constants of TableIII are about 10%–15% of the average values reportedtherein which means that the differences in the time con-stants of systems 1 and 2 are not too far from the error barsof these quantities. The shorter-time scales of Tables I and IIare also associated to similar error bars, while those of thelonger-time constants �residence times� have a larger errorbar��25%� as discussed in Sec. IV. However, our basic conclu-sions regarding ion concentration effects remain unchangedwhen the results of the 3.3M solution �system 2� are com-pared with those of pure D2O,28 where the differences in thetime constants are large enough to fall outside the domain ofcalculated error bars.
It may be noted that, like the relatively dilute solution�system 1�, for the concentrated solution �system 2� also, no
TABLE III. Spectral diffusion data for all OD modes of the relatively dilute �system 1� and concentrated�system 2� solutions. The units of different quantities are as in Table II.
Quantity System Excitation �0 �1 �2 s a0 a1
�h�t� 1 Blue 0.10 0.15 2.5 90.1 0.30 0.45�r�t� 1 Blue 0.15 0.12 4.5 115.3 0.18 0.61�h�t� 1 Red 0.16 0.14 4.6 111.4 0.20 0.60�r�t� 1 Red 0.12 0.13 2.3 95.5 0.21 0.50C�t� 1 ¯ 0.12 0.15 3.0 132.6 0.26 0.48�h�t� 2 Blue 0.11 0.17 2.9 84.88 0.30 0.41�r�t� 2 Blue 0.12 0.11 4.8 113.5 0.12 0.55�h�t� 2 Red 0.20 0.12 5.0 112.7 0.18 0.60�r�t� 2 Red 0.11 0.20 2.8 88.6 0.25 0.50C�t� 2 ¯ 0.13 0.17 3.8 128.5 0.35 0.42
0 1 2 3 4 5 60
0.2
0.4
0.6
0.8
1Frequency correlation
T ime (ps)
Cω(t
)
FIG. 9. �Color online� The time correlation functions of OD fluctuatingfrequencies averaged over all the water molecules of the relatively dilute�system 1� and concentrated �system 2� solutions. The lower and upperdashed curves correspond to the simulation results for systems 1 and 2,respectively. The gray solid curves represent the fits by a function as givenby Eq. �13�.
00.20.40.60.8
11.2
(a)C
HB(t)
SHB
(t)
0 2 4 6 80
0.20.40.60.8
11.2
(b)C
HB(t)
SHB
(t)
T ime (ps)FIG. 10. �Color online� The time dependence of the continuous and inter-mittent hydrogen bond correlation functions for systems 1 �solid� and 2�dashed�. The results of �a� are for only Cl−-water hydrogen bonds and thoseof �b� are for all the hydrogen bonds present in systems 1 and 2.
194512-11 Vibrational spectral diffusion in aqueous solutions J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
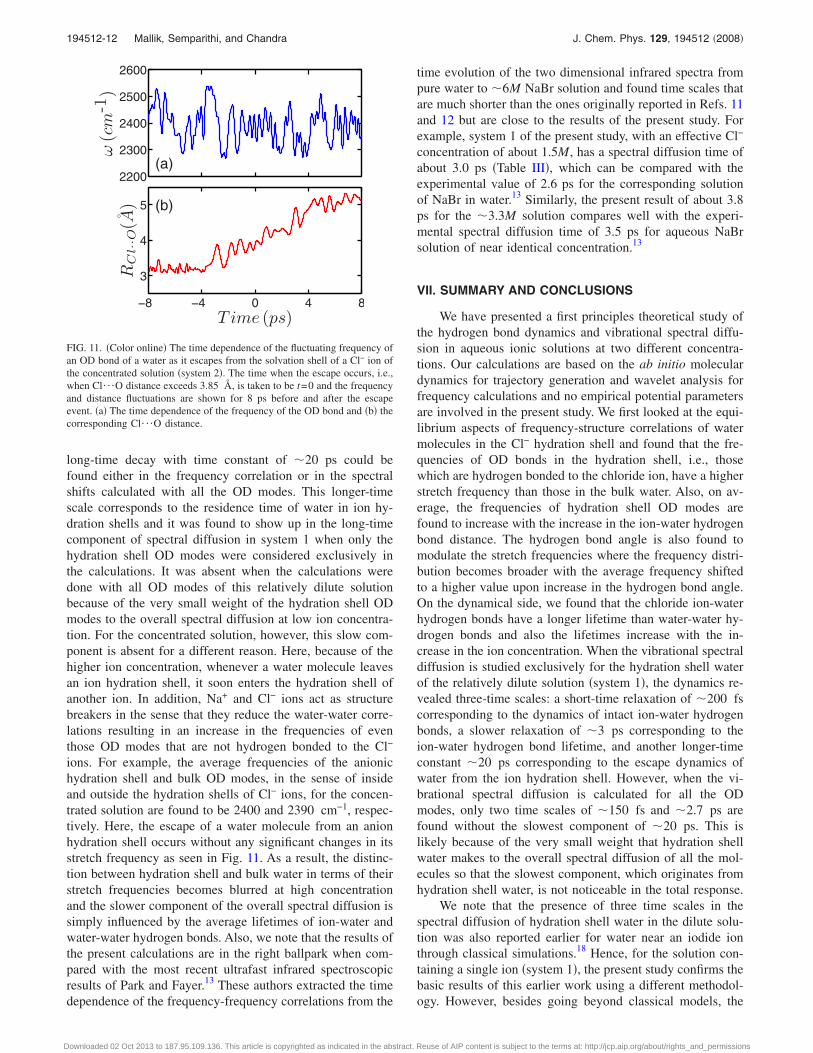
long-time decay with time constant of �20 ps could befound either in the frequency correlation or in the spectralshifts calculated with all the OD modes. This longer-timescale corresponds to the residence time of water in ion hy-dration shells and it was found to show up in the long-timecomponent of spectral diffusion in system 1 when only thehydration shell OD modes were considered exclusively inthe calculations. It was absent when the calculations weredone with all OD modes of this relatively dilute solutionbecause of the very small weight of the hydration shell ODmodes to the overall spectral diffusion at low ion concentra-tion. For the concentrated solution, however, this slow com-ponent is absent for a different reason. Here, because of thehigher ion concentration, whenever a water molecule leavesan ion hydration shell, it soon enters the hydration shell ofanother ion. In addition, Na+ and Cl− ions act as structurebreakers in the sense that they reduce the water-water corre-lations resulting in an increase in the frequencies of eventhose OD modes that are not hydrogen bonded to the Cl−
ions. For example, the average frequencies of the anionichydration shell and bulk OD modes, in the sense of insideand outside the hydration shells of Cl− ions, for the concen-trated solution are found to be 2400 and 2390 cm−1, respec-tively. Here, the escape of a water molecule from an anionhydration shell occurs without any significant changes in itsstretch frequency as seen in Fig. 11. As a result, the distinc-tion between hydration shell and bulk water in terms of theirstretch frequencies becomes blurred at high concentrationand the slower component of the overall spectral diffusion issimply influenced by the average lifetimes of ion-water andwater-water hydrogen bonds. Also, we note that the results ofthe present calculations are in the right ballpark when com-pared with the most recent ultrafast infrared spectroscopicresults of Park and Fayer.13 These authors extracted the timedependence of the frequency-frequency correlations from the
time evolution of the two dimensional infrared spectra frompure water to �6M NaBr solution and found time scales thatare much shorter than the ones originally reported in Refs. 11and 12 but are close to the results of the present study. Forexample, system 1 of the present study, with an effective Cl−
concentration of about 1.5M, has a spectral diffusion time ofabout 3.0 ps �Table III�, which can be compared with theexperimental value of 2.6 ps for the corresponding solutionof NaBr in water.13 Similarly, the present result of about 3.8ps for the �3.3M solution compares well with the experi-mental spectral diffusion time of 3.5 ps for aqueous NaBrsolution of near identical concentration.13
VII. SUMMARY AND CONCLUSIONS
We have presented a first principles theoretical study ofthe hydrogen bond dynamics and vibrational spectral diffu-sion in aqueous ionic solutions at two different concentra-tions. Our calculations are based on the ab initio moleculardynamics for trajectory generation and wavelet analysis forfrequency calculations and no empirical potential parametersare involved in the present study. We first looked at the equi-librium aspects of frequency-structure correlations of watermolecules in the Cl− hydration shell and found that the fre-quencies of OD bonds in the hydration shell, i.e., thosewhich are hydrogen bonded to the chloride ion, have a higherstretch frequency than those in the bulk water. Also, on av-erage, the frequencies of hydration shell OD modes arefound to increase with the increase in the ion-water hydrogenbond distance. The hydrogen bond angle is also found tomodulate the stretch frequencies where the frequency distri-bution becomes broader with the average frequency shiftedto a higher value upon increase in the hydrogen bond angle.On the dynamical side, we found that the chloride ion-waterhydrogen bonds have a longer lifetime than water-water hy-drogen bonds and also the lifetimes increase with the in-crease in the ion concentration. When the vibrational spectraldiffusion is studied exclusively for the hydration shell waterof the relatively dilute solution �system 1�, the dynamics re-vealed three-time scales: a short-time relaxation of �200 fscorresponding to the dynamics of intact ion-water hydrogenbonds, a slower relaxation of �3 ps corresponding to theion-water hydrogen bond lifetime, and another longer-timeconstant �20 ps corresponding to the escape dynamics ofwater from the ion hydration shell. However, when the vi-brational spectral diffusion is calculated for all the ODmodes, only two time scales of �150 fs and �2.7 ps arefound without the slowest component of �20 ps. This islikely because of the very small weight that hydration shellwater makes to the overall spectral diffusion of all the mol-ecules so that the slowest component, which originates fromhydration shell water, is not noticeable in the total response.
We note that the presence of three time scales in thespectral diffusion of hydration shell water in the dilute solu-tion was also reported earlier for water near an iodide ionthrough classical simulations.18 Hence, for the solution con-taining a single ion �system 1�, the present study confirms thebasic results of this earlier work using a different methodol-ogy. However, besides going beyond classical models, the
2200
2300
2400
2500
2600
(a)
ω(c
m- 1
)
−8 −4 0 4 8
3
4
5 (b)
T ime (ps)
RC
l··O(A
)
FIG. 11. �Color online� The time dependence of the fluctuating frequency ofan OD bond of a water as it escapes from the solvation shell of a Cl− ion ofthe concentrated solution �system 2�. The time when the escape occurs, i.e.,when Cl¯O distance exceeds 3.85 Å, is taken to be t=0 and the frequencyand distance fluctuations are shown for 8 ps before and after the escapeevent. �a� The time dependence of the frequency of the OD bond and �b� thecorresponding Cl¯O distance.
194512-12 Mallik, Semparithi, and Chandra J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

present study also considers spectral diffusion in a concen-trated ionic solution ��3M� including counterions whichwas not considered in the earlier study. Interestingly, for thespectral diffusion of all OD modes in the concentrated solu-tion, the slowest component of �20 ps is not found eventhough the weight of hydration shell water is clearly signifi-cant at this concentration. At this high concentration regime,whenever a water molecule leaves a hydration shell, it soonenters the hydration shell of another ion. Also, the presenceof Na+ and Cl− ions reduces the water-water correlationsresulting in an increase in the frequency of even those ODmodes that are not hydrogen bonded to the Cl−. Hence, adistinct frequency separation between the hydration shell andbulk water does not exist at high ion concentration and, as aresult, the very slow escape events are not manifested in thespectral diffusion of OD modes in the concentrated solutioneven though not all OD modes are hydrogen bonded to thenegative ions. The present results can also be connected wellto recent ultrafast infrared spectroscopic studies on a seriesof high concentration aqueous NaBr solutions.13 The timeconstants of the slowest component of spectral diffusionwere found to change from 1.7 ps for pure water to 2.6, 3.5,and 4.8 ps for 1.5, 3.5, and 6.0M NaBr solutions, respec-tively. These time scales are much shorter than the originallymeasured in Refs. 11 and 12 but are close to the ones pre-dicted by the present calculations for solutions of Cl− /NaClin D2O and in Ref. 28 for pure heavy water.
The present study is based on a time dependent fre-quency analysis of either solvation shell or all OD modes ofa D2O liquid containing one or more number of ions. Thecorresponding experiments on liquid water or aqueous ionicsolutions have, however, considered dilute mixtures of HODin H2O or D2O. Our choice of the system makes the presentcalculations computationally more efficient. For diluteHOD /H2O or HOD /D2O systems, one necessarily needs toconsider a very large system to have sufficient number ofHOD molecules in the ion hydration shells but simulations ofsuch large systems through ab initio molecular dynamics is avery difficult task at present. Hence, following our previouswork on neat water,28 we considered a liquid of only D2O�with ions immersed in it� and considered every OD bond asa local vibration for the purpose of frequency calculations.This greatly enhanced the number of OD vibrations of inter-est compared to that of a dilute HOD /H2O system of similarsize and thus allowed us to obtain statistically meaningfulresults for spectral diffusion from our simulations. Since theinstantaneous frequencies are calculated directly from thesimulated time dependence of OD oscillations and thus anyeffects of intermode coupling on the vibration of an ODgroup are implicitly taken into account in our calculations,the calculated frequencies can be treated as effective localfrequencies of OD oscillators. Since the experimental com-plications of vibrational energy relaxation from excitedquantum states through intermolecular resonant channels arenot there in the present simulation systems involving a clas-sical treatment of nuclear motion, we expect that the presentresults of the spectral diffusion are not significantly affectedby the above choice of treating every OD bond as an effec-tive local oscillator of interest. As has been stated earlier,28
although one can use normal modes to analyze the spectraldiffusion, such an analysis is complicated by the fact that thefrequency perturbations of such modes arise from two donorhydrogen bonds as compared to only one when the analysisis made in terms of local modes. Also, we note that thedegeneracy of the two OD modes of an isolated water isgenerally broken in the liquid phase due to asymmetric sol-vation and the resultant frequency nondegeneracy is ex-pected to provide some local character to the OD vibrations.This effect of asymmetric solvation is expected to be evenmore significant for anionic solvation shell water moleculeswhere one OD is generally hydrogen bonded to the negativeion, while the other OD of the same water is hydrogenbonded to a second water giving rise to quite different vibra-tional frequencies of the two OD modes of the solvationshell water.
We note that, for a given solute, the normalized dynam-ics of the spectral diffusion depends on the molecular dy-namics of the solvent, and thus our results would correspondto vibrational spectral diffusion in deuterated aqueous ionicsolutions. However, the experimental results of spectral dif-fusion in liquid H2O and D2O are not very different fromeach other,5,8 especially for the long-time part of the dynam-ics; hence our calculated results can be used to understandthe basic aspects of vibrational spectral diffusion in aqueoussolutions containing ions dissolved in either H2O or D2O.Also, since the present study is based on frequency calcula-tions directly from ab initio molecular dynamics trajectorieswithout involving any empirical potentials, it can be readilyextended to investigate vibrational spectral diffusion and as-sociated dynamical properties of other chemically interestingsystems such as acidic and basic solutions andclusters,47,68,96,97 aqueous surfaces and interfaces,61,98–101 andwater in confinement.102–104 The present approach can alsobe extended to aqueous solutions of molecular ions, peptides,and polypeptides105–108 and look at the spectral diffusion ofselective vibrational modes of these solutes rather than thestretch modes of the solvent. Although simulations of inter-faces and peptides require rather large systems, handlingsuch systems through first principles simulations is no longertotally intractable with modern computing facilities.61,108 Wehope to address some of these systems in future publications.
ACKNOWLEDGMENTS
Financial supports from the Department of Science andTechnology �DST�, Department of Atomic Energy �DAE�,and Council of Scientific and Industrial Research �CSIR�,Government of India, and Alexander von Humboldt Founda-tion are gratefully acknowledged. We are also thankful toProfessor S. Keshavamurthy for many useful discussions andhelp in the frequency calculations.
1 R. Laenen, C. Rauscher, and A. Laubereau, Phys. Rev. Lett. 80, 2622�1998�; R. Laenen, K. Simeonidis, and A. Laubereau, J. Phys. Chem. B106, 408 �2002�.
2 S. Woutersen, U. Emmerichs, and H. J. Bakker, Science 278, 658 �1997�;S. Woutersen and H. J. Bakker, Phys. Rev. Lett. 83, 2077 �1999�.
3 G. M. Gale, G. Gallot, F. Hache, N. Lascoux, and S. Bratos, Phys. Rev.Lett. 82, 1068 �1999�; S. Bratos, G. M. Gale, G. Gallot, F. Hache, N.Lascoux, and J.-C. Leickman, Phys. Rev. E 61, 5211 �2000�.
194512-13 Vibrational spectral diffusion in aqueous solutions J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

4 Z. Wang, A. Pakoulev, Y. Pang, and D. D. Dlott, Chem. Phys. Lett. 378,281 �2003�; A. Pakoulev, Z. Wang, Y. Pang, and D. D. Dlott, ibid. 371,594 �2003�.
5 J. B. Asbury, T. Steinel, C. Stromberg, S. A. Corcelli, C. P. Lawrence, J.L. Skinner, and M. D. Fayer, J. Phys. Chem. A 108, 1107 �2004�.
6 T. Steinel, J. B. Asbury, S. A. Corcelli, C. P. Lawrence, J. L. Skinner, andM. D. Fayer, Chem. Phys. Lett. 386, 295 �2004�; J. B. Asbury, T. Steinel,K. Kwak, S. A. Corcelli, C. P. Lawrence, J. L. Skinner, and M. D. Fayer,J. Chem. Phys. 121, 12431 �2004�.
7 C. J. Fecko, J. D. Eaves, J. J. Loparo, A. Tokmakoff, and P. L. Geissler,Science 301, 1698 �2003�; J. D. Eaves, J. J. Loparo, C. J. Fecko, S. T.Roberts, A. Tokmakoff, and P. L. Geissler, Proc. Natl. Acad. Sci. U.S.A.102, 13019 �2005�.
8 C. J. Fecko, J. J. Loparo, S. T. Roberts, and A. Tokmakoff, J. Chem.Phys. 122, 054506 �2005�; S. T. Roberts, J. J. Loparo, and A. Tokmakoff,ibid. 125, 084502 �2006�.
9 J. J. Loparo, S. T. Roberts, and A. Tokmakoff, J. Chem. Phys. 125,194521 �2006�.
10 J. Stenger, D. Madsen, P. Hamm, E. T. J. Nibbering, and T. Elsaesser, J.Phys. Chem. A 106, 2341 �2002�; M. L. Cowan, B. D. Bruner, N. Huse,J. R. Dwyer, B. Chugh, E. T. J. Nibbering, T. Elsaesser, and R. J. D.Miller, Nature �London� 434, 199 �2005�.
11 M. F. Kropman and H. J. Bakker, Science 291, 2118 �2001�.12 M. F. Kropman and H. J. Bakker, J. Chem. Phys. 115, 8942 �2001�.13 S. Park and M. D. Fayer, Proc. Natl. Acad. Sci. U.S.A. 104, 16731
�2007�.14 H. S. Tan, I. R. Piletic, R. E. Riter, N. E. Levinger, and M. D. Fayer,
Phys. Rev. Lett. 94, 057405 �2005�.15 S. Woutersen and H. J. Bakker, Phys. Rev. Lett. 96, 138305 �2006�.16 H.-K. Nienhuys, A. J. Lock, R. A. van Santen, and H. J. Bakker, J. Chem.
Phys. 117, 8021 �2002�.17 M. Rini, B.-Z. Magnes, E. Pines, and E. T. J. Nibbering, Science 301,
349 �2003�.18 B. Nigro, S. Re, D. Laage, R. Rey, and J. T. Hynes, J. Phys. Chem. A
110, 11237 �2006�.19 D. Laage and J. T. Hynes, Proc. Natl. Acad. Sci. U.S.A. 104, 11167
�2007�.20 S. Chowdhuri and A. Chandra, J. Phys. Chem. B 110, 9674 �2006�; A.
Chandra, ibid. 107, 3899 �2003�.21 R. Car and M. Parrinello, Phys. Rev. Lett. 55, 2471 �1985�.22 D. Marx and J. Hutter, in Modern Methods and Algorithms of Quantum
Chemistry, edited by J. Grotendorst �NIC, FZ Jülich, 2000�, seewww.theochem.ruhr-uni-bochum.de/go/cprev.html
23 S. Raugei and M. L. Klein, J. Am. Chem. Soc. 123, 9484 �2001�; J.Chem. Phys. 116, 196 �2002�.
24 J. M. Heuft and E. J. Meijer, J. Chem. Phys. 119, 11788 �2003�; 123,094506 �2005�.
25 M. Fuentes, P. Guttorp, and P. D. Sampson, in Statistical Methods forSpatio-Temporal Systems, edited by B. Finkenstädt, L. Held, and V.Isham �Chapman and Hall, London/CRC, Boca Raton, 2007�, Chap. 3.
26 L. V. Vela-Arevalo and S. Wiggins, Int. J. Bifurcation Chaos Appl. Sci.Eng. 11, 1359 �2001�.
27 A. Semparithi and S. Keshavamurthy, Phys. Chem. Chem. Phys. 5, 5051�2003�, see Sec. IV for a calculation of time dependent frequencies usingthe wavelet method.
28 B. S. Mallik, A. Semparithi, and A. Chandra, J. Phys. Chem. A 112, 5104�2008�.
29 J. Hutter, A. Alavi, T. Deutsch, M. Bernasconi, S. Goedecker, D. Marx,M. Tuckerman, and M. Parrinello, CPMD program, MPI für Festkörperfor-schung and IBM Zurich Research Laboratory.
30 Handbook of Chemistry and Physics, edited by D. R. Lide �CRC, BocaRaton, FL/Taylor & Francis, London, 2006�, Vol. 87.
31 M. Laliberte and W. E. Cooper, J. Chem. Eng. Data 49, 1141 �2004�.32 W. Kohn and L. J. Sham, Phys. Rev. 140, A1133 �1965�.33 N. Troullier and J. L. Martins, Phys. Rev. B 43, 1993 �1991�.34 See, for example, P. Hunt and M. Sprik, ChemPhysChem 6, 1805 �2005�;
H.-S. Lee and M. E. Tuckerman, J. Chem. Phys. 125, 154507 �2006�, andreferences therein.
35 J. VandeVondele, F. Mohamed, M. Krack, J. Hutter, M. Sprik, and M.Parrinello, J. Chem. Phys. 122, 014515 �2005�.
36 I.-F. W. Kuo, C. J. Mundy, M. J. McGrath, J. I. Siepmann, J. VandeVon-dele, M. Sprik, J. Hutter, B. Chen, M. L. Klein, F. Mohamed, M. Krack,and M. Parrinello, J. Phys. Chem. B 108, 12990 �2004�.
37 J. C. Grossman, E. Schwegler, E. W. Draeger, F. Gygi, and G. Galli, J.
Chem. Phys. 120, 300 �2004�; E. Schwegler, C. Grossman, F. Gygi, andG. Galli, ibid. 121, 5400 �2004�.
38 Y. A. Mantz, B. Chen, and G. J. Martyna, Chem. Phys. Lett. 405, 294�2005�.
39 A. D. Becke, Phys. Rev. A 38, 3098 �1988�; C. Lee, W. Yang, and R. G.Parr, Phys. Rev. B 37, 785 �1988�.
40 K. Laasonen, M. Sprik, M. Parrinello, and R. Car, J. Chem. Phys. 99,9080 �1993�.
41 M. Sprik, J. Hutter, and M. Parrinello, J. Chem. Phys. 105, 1142 �1996�.42 P. L. Silvestrelli and M. Parrinello, Phys. Rev. Lett. 82, 3308 �1999�; J.
Chem. Phys. 111, 3572 �1999�; P. L. Silvestrelli, M. Bernasconi, and M.Parrinello, Chem. Phys. Lett. 277, 478 �1997�.
43 M. Krack, A. Gambirasio, and M. Parrinello, J. Chem. Phys. 117, 9409�2002�; B. Chen, I. Ivanov, M. L. Klein, and M. Parrinello, Phys. Rev.Lett. 91, 215503 �2003�.
44 S. Izvekov and G. A. Voth, J. Chem. Phys. 116, 10372 �2002�.45 M. Boero, K. Terakura, T. Ikeshoji, C. C. Liew, and M. Parrinello, Phys.
Rev. Lett. 85, 3245 �2000�; J. Chem. Phys. 115, 2219 �2001�.46 M. Boero, J. Phys. Chem. A 111, 12248 �2007�.47 D. Marx, M. E. Tuckerman, J. Hutter, and M. Parrinello, Nature
�London� 397, 601 �1999�; M. E. Tuckerman, D. Marx, and M. Par-rinello, ibid. 417, 925 �2002�.
48 B. Kirchner, J. Stubbs, and D. Marx, Phys. Rev. Lett. 89, 215901 �2002�.49 J. M. Heuft and E. J. Meijer, Phys. Chem. Chem. Phys. 8, 3116 �2006�.50 M. Cavallari, C. Cavazzoni, and M. Ferrario, Mol. Phys. 102, 959
�2004�.51 T. Ikeda, M. Hirata, and T. Kimura, J. Chem. Phys. 119, 12386 �2003�.52 K. Leung and S. B. Rempe, J. Am. Chem. Soc. 126, 344 �2004�.53 L. M. Ramaniah, M. Barnasconi, and M. Parrinello, J. Chem. Phys. 111,
1587 �1999�; A. P. Lyubartsev, K. Laasonen, and A. Laaksonen, ibid.114, 3120 �2001�.
54 M.-P. Gaigeot and M. Sprik, J. Phys. Chem. B 108, 7458 �2004�.55 E. Tsuchida, Y. Kanada, and M. Tsukada, Chem. Phys. Lett. 311, 236
�1999�.56 M. Pagliai, G. Cardini, R. Righini, and V. Schettino, J. Chem. Phys. 119,
6655 �2003�.57 J. A. Morrone and M. E. Tuckerman, J. Chem. Phys. 117, 4403 �2002�.58 M. Diraison, G. J. Martyna, and M. E. Tuckerman, J. Chem. Phys. 111,
1096 �1999�.59 A. D. Boese, A. Chandra, J. M. L. Martin, and D. Marx, J. Chem. Phys.
119, 5965 �2003�.60 J. A. Morrone, K. E. Haslinger, and M. E. Tuckerman, J. Phys. Chem. B
110, 3712 �2006�.61 I.-F. W. Kuo and C. J. Mundy, Science 303, 658 �2004�.62 J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865
�1996�.63 J. P. Perdew and Y. Wang, Phys. Rev. B 45, 13244 �1992�; J. P. Perdew,
J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh,and C. Fiolhais, ibid. 46, 6671 �1992�.
64 J. M. Tao, J. P. Perdew, V. N. Staroverov, and G. E. Scuseria, Phys. Rev.Lett. 91, 146401 �2003�.
65 N. C. Handy and A. J. Cohen, J. Chem. Phys. 116, 5411 �2002�.66 F. A. Hamprecht, A. J. Cohen, D. J. Tozer, and N. C. Handy, J. Chem.
Phys. 109, 6264 �1998�; A. D. Boese and N. C. Handy, ibid. 114, 5497�2001�.
67 See, for example, M. Boero, T. Ikeshoji, C. C. Liew, K. Terakura, and M.Parrinello, J. Am. Chem. Soc. 126, 6280 �2004�; C. J. Mundy, J. Hutter,and M. Parrinello, ibid. 122, 4837 �2000�.
68 M. E. Tuckerman, A. Chandra, and D. Marx, Acc. Chem. Res. 39, 151�2006�; A. Chandra, M. E. Tuckerman, and D. Marx, Phys. Rev. Lett. 99,145901 �2007�.
69 M. J. McGrath, J. I. Siepmann, I.-F. W. Kuo, C. J. Mundy, J. VandeVon-dele, J. Hutter, F. Mohamed, and M. Krack, ChemPhysChem 6, 1894�2005�; J. Phys. Chem. A 110, 640 �2006�.
70 H. J. C. Berendsen, J. R. Grigera, and T. P. Straatsma, J. Phys. Chem. 91,6269 �1987�.
71 L. X. Dang, Chem. Phys. Lett. 200, 21 �1992�; L. X. Dang and B. C.Garrett, J. Chem. Phys. 99, 2972 �1993�.
72 S. Koneshan, J. C. Rasaiah, R. M. Lynden-Bell, and S. H. Lee, J. Phys.Chem. B 102, 4193 �1998�.
73 R. Carmona, W. Hwang, and B. Torresani, Practical Time-FrequencyAnalysis: Gabor and Wavelet Transforms with an Implementation �Aca-demic, New York, 1998�.
194512-14 Mallik, Semparithi, and Chandra J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions

74MATLAB, Version-R2007a, The Math Works �http://www.mathworks.com�.
75 J.-J. Max and C. Chapados, J. Chem. Phys. 115, 2664 �2001�.76 C. P. Lawrence and J. L. Skinner, J. Chem. Phys. 118, 264 �2003�; Chem.
Phys. Lett. 369, 472 �2003�.77 S. A. Corcelli, C. P. Lawrence, and J. L. Skinner, J. Chem. Phys. 120,
8107 �2004�.78 R. Rey, K. B. Moller, and J. T. Hynes, J. Phys. Chem. A 106, 11993
�2002�; K. B. Moller, R. Rey, and J. T. Hynes, ibid. 108, 1275 �2004�.79 Y. Wang and Y. Tominaga, J. Chem. Phys. 101, 3453 �1994�; K. Mizogu-
chi, T. Ujike, and Y. Tominaga, ibid. 109, 1867 �1998�; Y. Amo and Y.Tominaga, Phys. Rev. E 58, 7553 �1998�.
80 I.-F. W. Kuo and D. J. Tobias, J. Phys. Chem. A 106, 10969 �2002�.81 H.-S. Lee and M. E. Tuckerman, J. Chem. Phys. 126, 164501 �2007�.82 R. Ramirez, T. Lopez-Ciudad, P. Kumar, and D. Marx, J. Chem. Phys.
121, 3973 �2004�.83 J. Cao and G. A. Voth, J. Chem. Phys. 99, 10070 �1993�; 100, 5106
�1994�; G. A. Voth, Adv. Chem. Phys. 93, 135 �1996�.84 D. Rapaport, Mol. Phys. 50, 1151 �1983�.85 A. Chandra, Phys. Rev. Lett. 85, 768 �2000�.86 S. Balasubramanian, S. Pal, and B. Bagchi, Phys. Rev. Lett. 89, 115505
�2002�.87 A. Luzar, J. Chem. Phys. 113, 10663 �2000�.88 A. Luzar and D. Chandler, Phys. Rev. Lett. 76, 928 �1996�; Nature
�London� 379, 55 �1996�.89 H. Xu and B. J. Berne, J. Phys. Chem. B 105, 11929 �2001�; H. Xu, H.
A. Stern, and B. J. Berne, ibid. 106, 2054 �2002�.90 R. W. Impey, P. A. Madden, and I. R. McDonald, J. Phys. Chem. 87,
5071 �1983�.91 M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids �Oxford,
New York, 1987�.92 S. Roychowdhury and B. Bagchi, Chem. Phys. 343, 76 �2008�.93 J. R. Schmidt, S. A. Corcelli, and J. L. Skinner, J. Chem. Phys. 121,
8887 �2004�.94 D. Laage and J. T. Hynes, J. Phys. Chem. B 112, 7697 �2008�.95 S. H. Northrup and J. T. Hynes, J. Chem. Phys. 73, 2700 �1980�.96 J. M. J. Swanson, C. M. Maupin, H. Chen, M. K. Petersen, J. Xu, Y. Wu,
and G. A. Voth, J. Phys. Chem. B 111, 4300 �2007�.97 D. Wei, J. Truchon, S. Sirois, and D. Salahub, J. Chem. Phys. 116, 6028
�2002�.98 L. F. Scatena, M. G. Brown, and G. L. Richmond, Science 292, 908
�2001�.99 M. J. Shultz, S. Aldelli, C. Schnitzer, and D. Simonelli, J. Phys. Chem. B
106, 5313 �2002�.100 S. Paul and A. Chandra, J. Chem. Phys. 123, 174712 �2005�; J. Chem.
Theory Comput. 1, 1221 �2005�.101 S. Senapati and M. L. Berkowitz, Phys. Rev. Lett. 87, 176101 �2001�; R.
S. Taylor, L. X. Dang, and B. C. Garrett, J. Phys. Chem. 100, 11720�1996�.
102 J. J. Gilijamse, A. J. Lock, and H. J. Bakker, Proc. Natl. Acad. Sci.U.S.A. 102, 3202 �2005�.
103 S. Vaitheeswaran, H. Yin, J. C. Rasaiah, and G. Hummer, Proc. Natl.Acad. Sci. U.S.A. 101, 17002 �2004�; C. Dellago and G. Hummer, Phys.Rev. Lett. 97, 245901 �2006�.
104 K. Sahu, S. K. Mondal, S. Ghosh, and K. Bhattacharyya, Bull. Chem.Soc. Jpn. 80, 1033 �2007�.
105 S. Li, J. R. Schmidt, A. Piryatinski, C. P. Lawrence, and J. L. Skinner, J.Phys. Chem. B 110, 18933 �2006�.
106 Y. Mu and G. Stock, J. Phys. Chem. B 106, 5294 �2002�; R. D. Gor-bunov, P. H. Nguyen, M. Kobus, and G. Stock, J. Chem. Phys. 126,054509 �2007�.
107 P. Hamm, M. Lim, W. F. DeGrado, and R. M. Hochstrasser, Proc. Natl.Acad. Sci. U.S.A. 96, 2036 �1999�.
108 M. D. Peraro, S. Raugei, P. Carloni, and M. L. Klein, ChemPhysChem 6,1715 �2005�.
194512-15 Vibrational spectral diffusion in aqueous solutions J. Chem. Phys. 129, 194512 �2008�
Downloaded 02 Oct 2013 to 187.95.109.136. This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://jcp.aip.org/about/rights_and_permissions
Related Documents











