A Common Haplotype within the PON1 Promoter Region is Associated with Sporadic ALS John E. Landers, PhD 1,* , Lijia Shi, MD 1 , Ting-Jan Cho, BS 1 , Jonathan D. Glass, MD 3 , Christopher E. Shaw, MD, FRACP 2 , P. Nigel Leigh, FRCP, PhD 2 , Frank Diekstra, BS 2 , Meraida Polak, RN 3 , Ildefonso Rodriguez-Leyza, MD 4 , Stephan Niemann, MD 1 , Bryan J. Traynor, MD 1 , Diane McKenna-Yasek, RN, BSN 1 , Peter C. Sapp, BS 1 , Ammar Al-Chalabi, PhD, FRCP 2 , Anne-Marie A. Wills, MD 1 , and Robert H. Brown Jr., MD, DPhil 1 1 Cecil B Day Laboratory for Neuromuscular Research, Massachusetts General Hospital East, Charlestown, MA, USA 2 MRC Centre for Neurodegeneration Research, King’s College London, Institute of Psychiatry, Department of Neurology, London SE5 8AF, UK 3 Center for Neurodegenerative Disease, School of Medicine, Emory University, Atlanta, GA, USA 4 Department of Neurology, Hospital Central, Colonia Universitaria, San Luis Potosi, Mexico Abstract Amyotrophic lateral sclerosis (ALS) is a progressive, neurodegenerative disorder of upper and lower motor neurons. Genetic variants in the paraoxonase gene cluster have been associated with susceptibility to sporadic ALS. Because these studies have yielded conflicting results, we have further investigated this association in a larger data set. Twenty SNPs spanning the paraoxonase gene cluster were genotyped on a panel of 835 case and 924 control samples and tested for association with risk of sporadic ALS and with ALS sub-phenotypes. Our study revealed 2 SNPs, rs2074351 and rs705382, within the paraoxonase gene cluster that are associated with susceptibility to sporadic ALS (uncorrected p=0.0016 and 0.0022, respectfully). None of the 20 SNPs displayed significant associations with age of onset, site of onset or disease survival. Using a sliding window approach, we have also identified a 5-SNP haplotype that is significantly associated with risk of sporadic ALS (p=2.42E-04). We conclude that a common haplotype within the PON1 promoter region is associated with susceptibility to sporadic ALS. Keywords ALS; PON1; SNP; haplotype; association study 1. Introduction ALS is an incurable, late-onset disease in which motor neurons deteriorate leading to muscular atrophy, weakness and death due to respiratory failure[1]. ALS typically develops in the fifth decade of life and is fatal within 3–5 years[2]. Familial ALS (FALS) accounts for 10% of all ALS cases[3]. Approximately 20% of FALS are due to mutations in the SOD1 gene and other genes[4–9]. Additional loci contributing to FALS have also been identified on chromosomes 9, 16, and 20 [10,11]. In contrast, 90% of ALS cases are sporadic in nature (SALS). Little is known about the factors contributing to the development of SALS. Twin studies have shown *To whom correspondence should be addressed. [email protected]. NIH Public Access Author Manuscript Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7. Published in final edited form as: Amyotroph Lateral Scler. 2008 October ; 9(5): 306–314. doi:10.1080/17482960802233177. NIH-PA Author Manuscript NIH-PA Author Manuscript NIH-PA Author Manuscript

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

A Common Haplotype within the PON1 Promoter Region isAssociated with Sporadic ALS
John E. Landers, PhD1,*, Lijia Shi, MD1, Ting-Jan Cho, BS1, Jonathan D. Glass, MD3,Christopher E. Shaw, MD, FRACP2, P. Nigel Leigh, FRCP, PhD2, Frank Diekstra, BS2, MeraidaPolak, RN3, Ildefonso Rodriguez-Leyza, MD4, Stephan Niemann, MD1, Bryan J. Traynor,MD1, Diane McKenna-Yasek, RN, BSN1, Peter C. Sapp, BS1, Ammar Al-Chalabi, PhD,FRCP2, Anne-Marie A. Wills, MD1, and Robert H. Brown Jr., MD, DPhil11 Cecil B Day Laboratory for Neuromuscular Research, Massachusetts General Hospital East,Charlestown, MA, USA2 MRC Centre for Neurodegeneration Research, King’s College London, Institute of Psychiatry,Department of Neurology, London SE5 8AF, UK3 Center for Neurodegenerative Disease, School of Medicine, Emory University, Atlanta, GA, USA4 Department of Neurology, Hospital Central, Colonia Universitaria, San Luis Potosi, Mexico
AbstractAmyotrophic lateral sclerosis (ALS) is a progressive, neurodegenerative disorder of upper and lowermotor neurons. Genetic variants in the paraoxonase gene cluster have been associated withsusceptibility to sporadic ALS. Because these studies have yielded conflicting results, we have furtherinvestigated this association in a larger data set. Twenty SNPs spanning the paraoxonase gene clusterwere genotyped on a panel of 835 case and 924 control samples and tested for association with riskof sporadic ALS and with ALS sub-phenotypes. Our study revealed 2 SNPs, rs2074351 and rs705382,within the paraoxonase gene cluster that are associated with susceptibility to sporadic ALS(uncorrected p=0.0016 and 0.0022, respectfully). None of the 20 SNPs displayed significantassociations with age of onset, site of onset or disease survival. Using a sliding window approach,we have also identified a 5-SNP haplotype that is significantly associated with risk of sporadic ALS(p=2.42E-04). We conclude that a common haplotype within the PON1 promoter region is associatedwith susceptibility to sporadic ALS.
KeywordsALS; PON1; SNP; haplotype; association study
1. IntroductionALS is an incurable, late-onset disease in which motor neurons deteriorate leading to muscularatrophy, weakness and death due to respiratory failure[1]. ALS typically develops in the fifthdecade of life and is fatal within 3–5 years[2]. Familial ALS (FALS) accounts for 10% of allALS cases[3]. Approximately 20% of FALS are due to mutations in the SOD1 gene and othergenes[4–9]. Additional loci contributing to FALS have also been identified on chromosomes9, 16, and 20 [10,11]. In contrast, 90% of ALS cases are sporadic in nature (SALS). Little isknown about the factors contributing to the development of SALS. Twin studies have shown
*To whom correspondence should be addressed. [email protected].
NIH Public AccessAuthor ManuscriptAmyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
Published in final edited form as:Amyotroph Lateral Scler. 2008 October ; 9(5): 306–314. doi:10.1080/17482960802233177.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

that the heritability of SALS is between 0.38 and 0.85[12], indicating that there is likely to bea significant contribution of non-genetic factors, such as exposure to adverse environmentalagents. Based on this evidence, we postulated that environmental response genes mightinfluence the risk of developing SALS. The human paraoxonases represent such anenvironmental response activity.
The paraoxonase gene cluster consists of three genes (cen-PON1-PON3-PON2-tel) locatedadjacent to each other in a region spanning ~140 kb on chromosome 7q21.3-q22.1[13]. Severallines of evidence indicate that this cluster may influence the risk of developing SALS. First,all three PON enzymes contain lipid antioxidant properties[14,15]. Heightened oxidativepathology has been implicated in both SALS and FALS [16], although its origin andsignificance are debated. Second, PON1 also functions to detoxify several organophosphatecompounds, many of which are neurotoxins found in insecticides, nerve gases, foods, and otherhousehold items[14,15]. Third, genetic variants in the PON genes have also been linked toenhanced risk for other neurological disorders such as Alzheimer’s disease [17,18], Parkinson’sdisease[19,20] and dementia [21], as well as atherosclerosis [22]. Last, four recent studies havedescribed an association between the PON gene cluster and SALS [23–26], although theconclusions of these studies are not entirely consistent. [27]. Each study observed a differentassociation peak; two were within differing PON1 amino acid variants [23,26], one was withinthe PON1 promoter region [24] and one was within a PON3/PON2 haplotype[25].
To further explore the hypothesis that paraoxonase is related to the risk of SALS, we havetested the potential association of multiple SNPs spanning the PON gene cluster with both ALSsusceptibility and phenotypes. Our results confirm the observation that several SNPs withinthis cluster are indeed over-represented in ALS cases as compared to controls.
2. Methods and Materials2.1 Study Subjects
All sporadic ALS patients fulfilled El Escorial criteria [28] for probable or definite ALS. Allpatients were self-reported Caucasians from either the United States or the United Kingdom.Informed consent was obtained from all individuals in accordance with the requirements ofthe participating institutional review boards. Age of onset and site of onset information wererequired for all samples. Duration information was known for 618 deceased cases. A portionof the control DNA samples was purchased from Coriell Cell Repositories. Whole blood fromanonymous individuals used for arylesterase assays was purchased from Innovative Research(Southfield, MI). The blood was isolated from apparently healthy individuals ranging in ages40–64 (average=47.2).
2.2 Tag SNP SelectionTag SNPs from the PON cluster were selected using an algorithm based on the r2 linkagedisequilibrium (LD) statistic [29]. Selection of SNPs was facilitated through the use of thesoftware SNPbrowser v. 2.0 using a pair wise r2 value of >=0.99 within the Caucasianpopulation. The PON cluster was defined as the region 10 kb downstream of the last exon ofPON1 to 10 kb downstream of the last exon of PON2.
2.3 SNP GenotypingAll genotyping was performed using a 5′ Nuclease Assay (TaqMan). Reactions volumes were5 ul and performed in 384-well format. Each reaction consisted of either genomic or wholegenome amplified DNA (5 ng/reaction), 1X TaqMan Universal PCR Master Mix, NoAmpErase UNG and 1X Validated TaqMan SNP Assay probes (Applied Biosystems).Reactions were thermocycled with an initial denaturation step of 95 degrees for 10 minutes,
Landers et al. Page 2
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

followed by 50 cycles of 95 degrees for 15 seconds 60 degrees for 1 minute. The assay resultswere collected using an ABI 7900HT Real-time PCR instrument and genotyping calls wereperformed using SDS 2.0 software (Applied Biosystems).
2.4 Allelic Association and Linkage Disequilibrium AnalysesAllelic association testing and inferring individual haplotypes was performed using thesoftware application PLINK v0.99p [30] (http://pngu.mgh.harvard.edu/~purcell/plink/).Haploview v4.0 (http://www.broad.mit.edu/mpg/haploview/) was used to determine thelinkage disequilibrium in the PON region. Bonferroni multiple test correction was applied bymultiplying PLINK-derived p values by the number of SNPs or haplotypes assayed.
2.5 Serum Paraoxonase Activity LevelsThe levels of paraoxanse activity in human serum were determined using an arylesterase/paraoxonase assay kit (Zeptometerix). Briefly, arylesterase substrate buffer (20 mM Tris HCl,pH 8.0, 1 mM CaCl2, 4 mM phenyl acetate) was added to 2 ul serum within triplicate wells ofa 96 well UV transparent microtiter plate. The rate of hydrolysis of phenyl-acetate to phenolformation was monitored by measuring the absorbance at 260nm at 25°C. Data points werecollected every 45 seconds for a total of 6 time points. The change in absorbance per minutewas calculated and arylesterase activity was determined by comparing to known standards andadjusting for serum protein concentration as determined by a Bradford assay. Association ofserum levels to genotypes and haplotypes was performed using a 1-way ANOVA and unpairedt-test, respectively.
3. Results3.1 Sample Selection
To investigate the role of PON genes in sporadic ALS, 835 case and 924 control samples wereselected for genotyping. All samples chosen were Caucasian to prevent false-positives due toethnic-specific polymorphisms. Table 1 provides a detailed breakdown of the samples used inthis study.
3.2 Allelic Association of the PON genes to Sporadic ALSTag SNPs were selected from the PON gene cluster based on the r2 linkage disequilibrium(LD) statistic. To increase the power of our study, selection was performed at a conservativelevel of r2=0.99. Eighteen SNPs were selected over the PON cluster region. In addition, threeadditional SNPs were selected because of their previously established functional significancein the PON1 gene. A glutamine (Q) to arginine (R) polymorphism (rs662) located at codon192 (Q192R) has been shown to alter PON1 activity; the R isoform is more efficient atdetoxifying paraoxon whereas the Q isoform is more efficient at detoxifying sarin and soman[31]. Additionally, a L55M polymorphism (rs854560) influences PON1 serum levels with theM variant displaying lower plasma levels[31]. PON1 serum levels are also influenced by apromoter polymorphism located at position A–161G (rs705381) [32].
Each SNP was genotyped in the panel cases and controls. One SNP was subsequently removedfrom further analysis because of a low call rate. Allelic association analysis was performed foreach SNP and a Bonferroni multiple test correction was applied. The results of the analysis areshown in Table 2. Two SNPs within the PON cluster displayed significant association aftercorrection. SNP #6 (rs2074351), located ~90 bp upstream of the start of the exon 2 of PON1,was most significantly associated (p=0.0016). SNP #10 (rs705382), located 1.7 kb upstreamof the start of the exon 1 of PON1, displayed the second most significant association(p=0.0022). Interestingly, the R192Q, L55M and A–161G polymorphisms, represented by
Landers et al. Page 3
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

SNPs #4, #5 and #9, respectively, did not show significant association after multiple testcorrection (Table 2).
3.3 Phenotypic Association AnalysisWe also investigated the possibility that polymorphisms within the paraoxonase gene clusterare associated with ALS sub-phenotypes. Each of the 20 SNPs was tested for association withage of onset, site of onset (bulbar vs. spinal and upper limb vs. lower limb) and disease survival.The results from this analysis are shown in Table 3. Although four p-values were observedbelow 0.05 (0.0152, 0.0336, 0.0144, 0.0079), none was significant after Bonferroni multipletest correction. We therefore conclude that the paraoxonase gene cluster is not associated withage of onset, site of onset, or survival within sporadic ALS.
3.4 Linkage Disequilibrium within the PON ClusterThe foregoing association studies suggest that the PON1 gene is associated with SALS. It ispossible that this association is not due to the effects of the PON1 gene per se but rather reflectslinkage disequilibrium of the associated SNPs with another gene. To test this possibility, weinvestigated the extent of linkage disequilibrium within the PON region. Genotyping resultswere used to determine the linkage disequilibrium between each pair of SNPs within the PONcluster as shown in Figure 1. These results demonstrate that the PON cluster appears to bebroken into several smaller blocks of linkage disequilibrium. In particular, the SNPsdemonstrating significant association, rs2074351 and rs705382 are present within linkagedisequilibrium blocks consisting of SNPs #5-7 and SNPs #9-10, respectively.
3.5 Haplotypic Association AnalysisTo further investigate the association of the PON genes with SALS, we considered thepossibility that a stronger association may be observed by establishing the association tohaplotypes within the PON region. Towards this end, we measured the association of 5 SNPhaplotypes in a moving window across the PON cluster to SALS. This analysis (Table 4)revealed a 5 SNP haplotype, consisting of SNPs #7-11 (rs854565, rs2299261, rs705381,rs705382, and rs4141217), that displayed a strong association with SALS (p=2.42E-04). Thenext lowest observed p-value is over 8 times higher and corresponds to the overlappingprevious 5 SNP window (SNP #6-10). The genomic region of the 5 SNP haplotype, termedHAP1 spans intron 1 of the PON1 gene to approximately 30 kb upstream of the start of thePON1 gene (Figure 1).
3.6 Serum Paraoxonase Activity Levels and PON Genotypes/HaplotypesBased on the location of the associated 5-SNP haplotype, it is reasonable to hypothesize thatthe expression of PON1 is altered in individuals harboring HAP1. To investigate thispossibility, we compared the PON1 expression levels of individuals to their respectivehaplotypes. Human blood was collected from 104 healthy control individuals. From eachsample, one aliquot was used to isolate DNA and while another provided a serum sample. TheDNA was genotyped for the 5 SNPs that compose the SALS-associated haplotype, as well asrs2074351 and rs705382, the SNPs displaying significant association. The arylesterase activitywas measured in each serum sample using phenylacetate as the substrate for hydrolysis.Previous studies have shown that the arylesterase activity directly reflects the protein levels ofPON1 in serum [32,33]. Haplotypes using the 5 SNPs that composed HAP1 were inferred usingthe software application PLINK and compared to the arylsterase activity (Figure 2). Noindividuals were observed to be homozygous for the HAP1 haplotypes. Using a one-sided t-test, no significant association was observed between the HAP1 haplotype and arylesteraseactivity (p=0.099). Similarly, we also compared the arylesterase activity observed forindividuals harboring each of the three genotypes for the two significantly associated SNPs
Landers et al. Page 4
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

(Figure 2). Although slight differences are observed between each genotype for each of thesemarkers, neither rs705382 (p=0.188) nor rs2074351 (p=0.458) were significantly different by1-way ANOVA testing. Based on these results, we cannot conclude that the rs2074351/rs705382 genotypes or HAP1 haplotypes modify PON1 arylesterase activity levels, asdetermined by phenyl acetate hydrolysis.
4. DiscussionThis study confirms previous reports that genetic variants within the PON1 gene are associatedwith susceptibility to SALS. This association prompts consideration of the biological functionof PON1 and mechanisms by which its variants may predispose to SALS. PON1 detoxifiesseveral neurotoxic organophosphate compounds often found in insecticides, nerve gas, foods,and other household items[14,15]. It therefore seemed reasonable at the outset of this study topostulate that decreased activity of PON1 increases ones risk with exposure to injuriousneurotoxins, leading over time to SALS. This concept is consistent with studies showing thatSALS is not fully explained by genetic factors (heritability=0.38–0.85)[12], and by theepidemiological observations that ALS cases have an increased exposure to insecticides andpesticides and that some occupations confer a higher ALS risk (e.g. farmers [34], members ofthe military in general and military personnel who were deployed in the first Gulf War [35,36]). If indeed PON1 influences SALS susceptibility through capacity to detoxify specifictoxins, then the identification of its environmental substrates as well as factors increasing theexpression/activity of PON1 may illuminate aspects of the pathogenesis of SALS andultimately be helpful in treating or reducing the risk of this disease.
Although the current study confirms the association of PON1 with sporadic ALS, it did notdocument a correlation between the disease-associated haplotype HAP1 and levels of PON1activity, as measured by phenylacetate hydrolysis. This parallels our observation reportedelsewhere that ALS sera also do not show reduced hydrolysis of paraoxon, diazoxon orphenylacetate compared to controls [37]. These findings raise the possibility that the ALS-related genetic variants in PON1 alter activity towards some substrate other than phenylacetateor paraxon. Indeed, PON1 is highly promiscuous, hydrolyzing hundreds of substrates.Moreover, it is highly polymorphic with more than 160 known polymorphisms, many of whichdifferentially affect hydrolysis of different substrates [38].
We also note that PON1 is a potent inhibitor of lipid oxidation; it is tightly associated withHDL particles and is substantially anti-atherogenic[39–41]. It may therefore also be thatvariations in the lipid anti-oxidant properties of ALS-associated PON1 polymorphismsunderlie the SALS susceptibility. There are several lines of inquiry incriminating oxidativecytotoxicity as a causative factor in ALS, although this remains controversial. By analogy,heightened oxidative toxicity is reported in other neurodegenerative disorders (e.g.Alzheimer’s and Parkinson’s disease [42]). Reports have also shown that the risk of SALS canbe reduced by the intake of antioxidants [43]. It is possible the detoxification and antioxidantproperties both contribute to influencing the susceptibility of SALS. The study of PON1variants that have lost their antioxidant properties, but not their detoxification properties, maybe useful in dissecting this question.
This is the fifth report implicating variants in paraoxonases as susceptibility factors for SALS[23–26]. It is potentially of importance that there are inconsistencies in these studies. Eachstudy observed a different association peak; Two studies observed association peaks atdiffering PON1 amino acid variants, L55M and Q192R [23,26], neither which were significantin our study. One study observed an association within the PON1 promoter region [24] andone was within a PON3/PON2 haplotype [25]. Thus, three of the previous reports implicatePON1 as an ALS risk factor while the fourth implicates PON2 and 3. Further, even among the
Landers et al. Page 5
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

reports associating PON1 with SALS there is no consistency in the linked variants. At leastthree factors may explain this. First, the SALS patients were derived from different populationsin the studies (Poland, Australia, Ireland, North American Caucasians, USA/UK Caucasians).Conceivably, linkage disequilibrium patterns within the PON cluster may differ in eachpopulation, leading to divergent results. Second, it is possible that more than one variant in thePON1 gene can increase SALS susceptibility; differing observations may reflect thefrequencies of the causal variants in that given population. And third, each population maydiffer in the patterns of exposure to environmental toxins and to compounds that influenceparaoxonase expression. For example, smoking [44], diet[45], lipid-controlling medicationexposure[46] and organophosphate exposure [47](both the amount and type) are all known tomodify levels of PON1 expression; each is likely to vary among different populations. Thus,if a particular variant within the PON1 gene demonstrates a decreased activity in detoxifyingcigarette smoke, the association with SALS may be more prominent in populations with highsmoking rates. Obviously, the combination of several of these factors further complicates anydissection of the association.
AcknowledgmentsThe Day Neuromuscular Research Laboratory (RHB) receives support from the National Institute of NeurologicalDisease and Stroke, the National Institute for Aging, the Angel Fund, the ALS Association, Project ALS, the PierreL. de Bourgknect ALS Research Foundation and the Al-Athel ALS Foundation. RHB and JEL received support forthis project from the ALS Therapy Alliance. JDG receives support from the Packard Center for ALS Research. AA-C is supported by the Medical Research Council, Motor Neurone Disease Association, ALS Association and ProjectALS. PS is supported through the auspices of Dr. H. R. Horvitz, an Investigator of the Howard Hughes MedicalInstitute, Department of Biology, Massachusetts Institute of Technology. A.-M.W. is supported by the AmericanAcademy of Neurology Foundation and ALS Association.
References1. Tandan R, Bradley WG. Amyotrophic lateral sclerosis: Part 1. Clinical features, pathology, and ethical
issues in management. Ann Neurol 1985;18(3):271–80. [PubMed: 4051456]2. Mulder DW, et al. Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology
1986;36(4):511–7. [PubMed: 3960325]3. Camu W, et al. Genetics of familial ALS and consequences for diagnosis. French ALS Research Group.
J Neurol Sci 1999;165(Suppl 1):S21–6. [PubMed: 10448977]4. Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial
amyotrophic lateral sclerosis. Nature 1993;362(6415):59–62. [PubMed: 8446170]5. Chen YZ, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis
(ALS4). Am J Hum Genet 2004;74(6):1128–35. [PubMed: 15106121]6. Hadano S, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral
sclerosis 2. Nat Genet 2001;29(2):166–73. [PubMed: 11586298]7. Nishimura AL, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal
muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 2004;75(5):822–31. [PubMed:15372378]
8. Yang Y, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factordomains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 2001;29(2):160–5. [PubMed: 11586297]
9. Hafezparast M, et al. Mutations in dynein link motor neuron degeneration to defects in retrogradetransport. Science 2003;300(5620):808–12. [PubMed: 12730604]
10. Morita M, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporaldementia. Neurology 2006;66(6):839–44. [PubMed: 16421333]
11. Sapp PC, et al. Identification of two novel loci for dominantly inherited familial amyotrophic lateralsclerosis. Am J Hum Genet 2003;73(2):397–403. [PubMed: 12858291]
12. Graham AJ, Macdonald AM, Hawkes CH. British motor neuron disease twin study. J NeurolNeurosurg Psychiatry 1997;62(6):562–9. [PubMed: 9219739]
Landers et al. Page 6
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

13. Kent WJ, et al. The human genome browser at UCSC. Genome Res 2002;12(6):996–1006. [PubMed:12045153]
14. Aldridge WN. Serum esterases. II. An enzyme hydrolysing diethyl p-nitrophenyl phosphate (E600)and its identity with the A-esterase of mammalian sera. Biochem J 1953;53(1):117–24. [PubMed:13032042]
15. Furlong CE, et al. Spectrophotometric assays for the enzymatic hydrolysis of the active metabolitesof chlorpyrifos and parathion by plasma paraoxonase/arylesterase. Anal Biochem 1989;180(2):242–7. [PubMed: 2479288]
16. Beal MF, et al. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis.Ann Neurol 1997;42(4):644–54. [PubMed: 9382477]
17. Cellini E, et al. Association analysis of the paraoxonase-1 gene with Alzheimer’s disease. NeurosciLett 2006;408(3):199–202. [PubMed: 16996683]
18. Erlich PM, et al. Polymorphisms in the PON gene cluster are associated with Alzheimer disease. HumMol Genet 2006;15(1):77–85. [PubMed: 16319130]
19. Akhmedova SN, Yakimovsky AK, Schwartz EI. Paraoxonase 1 Met--Leu 54 polymorphism isassociated with Parkinson’s disease. J Neurol Sci 2001;184(2):179–82. [PubMed: 11239953]
20. Kondo I, Yamamoto M. Genetic polymorphism of paraoxonase 1 (PON1) and susceptibility toParkinson’s disease. Brain Res 1998;806(2):271–3. [PubMed: 9739148]
21. Dantoine TF, et al. Paraoxonase 1 activity: a new vascular marker of dementia? Ann N Y Acad Sci2002;977:96–101. [PubMed: 12480737]
22. Wheeler JG, et al. Four paraoxonase gene polymorphisms in 11212 cases of coronary heart diseaseand 12786 controls: meta-analysis of 43 studies. Lancet 2004;363(9410):689–95. [PubMed:15001326]
23. Cronin S, et al. Paraoxonase promoter and intronic variants modify risk of sporadic amyotrophiclateral sclerosis. J Neurol Neurosurg Psychiatry 2007;78(9):984–6. [PubMed: 17702780]
24. Morahan JM, et al. A gene-environment study of the paraoxonase 1 gene and pesticides in amyotrophiclateral sclerosis. Neurotoxicology 2007;28(3):532–40. [PubMed: 17204329]
25. Saeed M, et al. Paraoxonase cluster polymorphisms are associated with sporadic ALS. Neurology2006;67(5):771–6. [PubMed: 16822964]
26. Slowik A, et al. Paraoxonase gene polymorphisms and sporadic ALS. Neurology 2006;67(5):766–70. [PubMed: 16822965]
27. Shaw CE, Al-Chalabi A. Susceptibility genes in sporadic ALS: separating the wheat from the chaffby international collaboration. Neurology 2006;67(5):738–9. [PubMed: 16966531]
28. Brooks BR, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateralsclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1(5):293–9. [PubMed:11464847]
29. Carlson CS, et al. Selecting a maximally informative set of single-nucleotide polymorphisms forassociation analyses using linkage disequilibrium. Am J Hum Genet 2004;74(1):106–20. [PubMed:14681826]
30. Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkageanalyses. Am J Hum Genet 2007;81(3):559–75. [PubMed: 17701901]
31. Adkins S, et al. Molecular basis for the polymorphic forms of human serum paraoxonase/arylesterase:glutamine or arginine at position 191, for the respective A or B allozymes. Am J Hum Genet 1993;52(3):598–608. [PubMed: 7916578]
32. Brophy VH, et al. Effects of 5′ regulatory-region polymorphisms on paraoxonase-gene (PON1)expression. Am J Hum Genet 2001;68(6):1428–36. [PubMed: 11335891]
33. Garin MC, et al. Paraoxonase polymorphism Met-Leu54 is associated with modified serumconcentrations of the enzyme. A possible link between the paraoxonase gene and increased risk ofcardiovascular disease in diabetes. J Clin Invest 1997;99(1):62–6. [PubMed: 9011577]
34. McGuire V, et al. Occupational exposures and amyotrophic lateral sclerosis. A population-based case-control study. Am J Epidemiol 1997;145(12):1076–88. [PubMed: 9199537]
35. Horner RD, et al. Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology2003;61(6):742–9. [PubMed: 14504315]
Landers et al. Page 7
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

36. Weisskopf MG, et al. Prospective study of military service and mortality from ALS. Neurology2005;64(1):32–7. [PubMed: 15642900]
37. Wills A-M, et al. Paraoxonase 1 (PON1) organophosphate hydrolysis is not reduced in ALS.Neurology. 2007In Press
38. Costa LG, et al. Modulation of paraoxonase (PON1) activity. Biochem Pharmacol 2005;69(4):541–50. [PubMed: 15670573]
39. Mackness MI, Arrol S, Durrington PN. Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett 1991;286(1–2):152–4. [PubMed: 1650712]
40. Aviram M, et al. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions.A possible peroxidative role for paraoxonase. J Clin Invest 1998;101(8):1581–90. [PubMed:9541487]
41. Oda MN, et al. Paraoxonase 1 overexpression in mice and its effect on high-density lipoproteins.Biochem Biophys Res Commun 2002;290(3):921–7. [PubMed: 11798161]
42. Reynolds A, et al. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int RevNeurobiol 2007;82:297–325. [PubMed: 17678968]
43. Veldink JH, et al. Intake of polyunsaturated fatty acids and vitamin E reduces the risk of developingamyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2007;78(4):367–71. [PubMed:16648143]
44. Nishio E, Watanabe Y. Cigarette smoke extract inhibits plasma paraoxonase activity by modificationof the enzyme’s free thiols. Biochem Biophys Res Commun 1997;236(2):289–93. [PubMed:9240427]
45. Shih DM, et al. Genetic-dietary regulation of serum paraoxonase expression and its role inatherogenesis in a mouse model. J Clin Invest 1996;97(7):1630–9. [PubMed: 8601628]
46. Gouedard C, et al. Opposite regulation of the human paraoxonase-1 gene PON-1 by fenofibrate andstatins. Mol Pharmacol 2003;63(4):945–56. [PubMed: 12644596]
47. Sozmen EY, et al. Effect of organophosphate intoxication on human serum paraoxonase. Hum ExpToxicol 2002;21(5):247–52. [PubMed: 12141395]
Landers et al. Page 8
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 1. Linkage disequilibrium plot for SNPs within the PON clusterPairwise linkage disequilibrium values (D′) were calculated for SNPs spanning the PONcluster. The color key for D′ values is shown. The −log10(p) values for association with riskare shown above each SNP. The location of the three paraoxonase genes and their genomicposition on chromosome 7 is shown above the plot.
Landers et al. Page 9
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
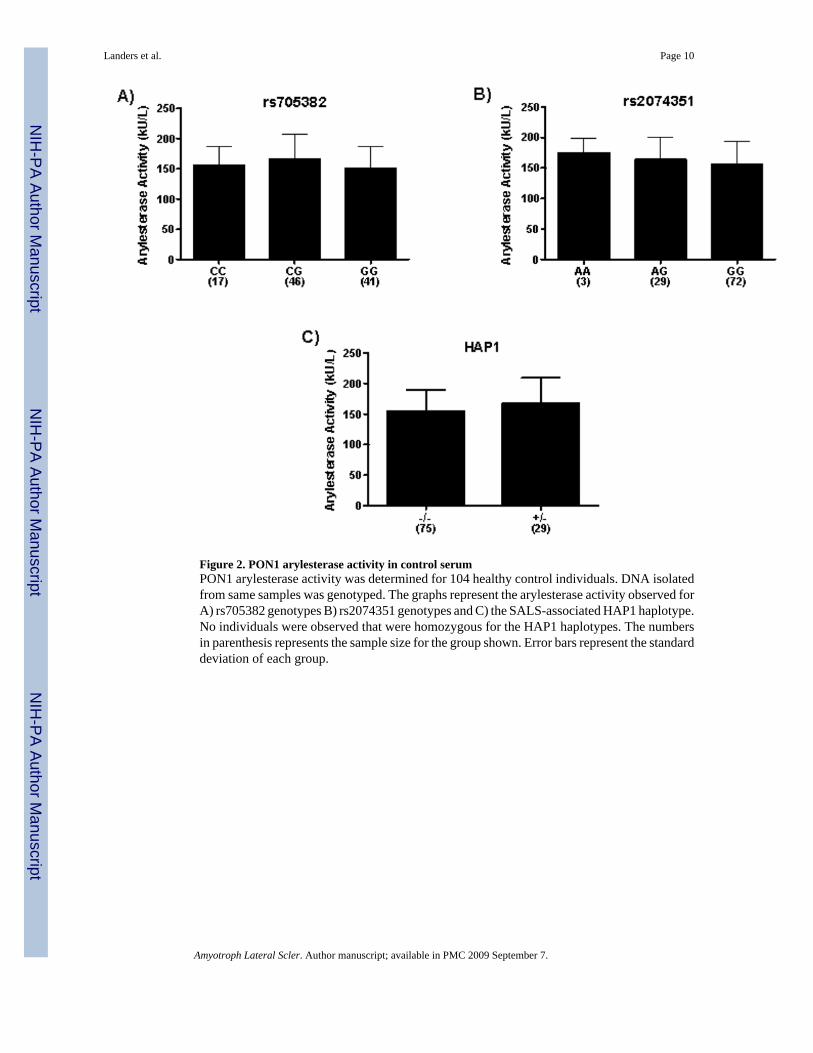
Figure 2. PON1 arylesterase activity in control serumPON1 arylesterase activity was determined for 104 healthy control individuals. DNA isolatedfrom same samples was genotyped. The graphs represent the arylesterase activity observed forA) rs705382 genotypes B) rs2074351 genotypes and C) the SALS-associated HAP1 haplotype.No individuals were observed that were homozygous for the HAP1 haplotypes. The numbersin parenthesis represents the sample size for the group shown. Error bars represent the standarddeviation of each group.
Landers et al. Page 10
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Landers et al. Page 11Ta
ble
1D
istri
butio
n of
Spo
radi
c A
LS C
ases
and
Con
trol.
Tot
alM
ales
Fem
ales
Age
+/−
SD
Con
trol
sTo
tal
924
569
355
57.8
7 +/−
13.5
9
Cas
esTo
tal
835
539
296
56.6
8 +/−
12.4
5
Bul
bar
214
122
9258
.25
+/−
12.5
7
Upp
er L
imb
315
248
6751
.58
+/−
13.1
3
Low
er L
imb
271
142
129
55.3
2 +/−
12.2
4
Mul
tiple
/Oth
er35
278
57.5
0 +/−
12.4
5
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.

NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Landers et al. Page 12Ta
ble
2A
ssoc
iatio
n te
stin
g of
PO
N c
lust
er v
aria
nts w
ith sp
orad
ic A
LS.
SNP
IDC
ase
Freq
.C
ont.
Freq
.A
llele
sp-
valu
eA
dj. p
-val
ueC
hi S
quar
eO
RH
WE
1rs
8545
430.
208
0.20
1A
>C0.
6219
10.
241.
040.
419
2rs
8545
490.
336
0.36
0C
>A0.
1284
12.
310.
900.
430
3rs
2237
582
0.31
30.
288
A>G
0.10
011
2.70
1.13
0.82
0
4rs
662
0.31
10.
287
T>C
0.11
611
2.47
1.13
1
5rs
8545
600.
355
0.37
1A
>T0.
3285
10.
950.
930.
499
6rs
2074
351
0.31
80.
269
G>A
0.00
160.
032
9.99
1.27
0.22
1
7rs
8545
650.
282
0.31
5G
>A0.
0352
0.70
44.
440.
850.
954
8rs
2299
261
0.34
60.
377
A>G
0.06
201
3.48
0.88
0.43
7
9rs
7053
810.
225
0.25
7C
>T0.
0286
0.57
14.
790.
840.
742
10rs
7053
820.
339
0.38
9G
>C0.
0022
0.04
49.
370.
810.
439
11rs
4141
217
0.48
20.
446
C>T
0.03
300.
661
4.54
1.16
0.56
2
12rs
9168
640.
207
0.17
9C
>T0.
0354
0.70
94.
421.
200.
757
13rs
3757
708
0.47
20.
441
T>G
0.06
591
3.38
1.13
0.59
6
14rs
1048
7132
0.41
40.
427
A>G
0.45
411
0.56
0.95
0.80
6
15rs
2072
200
0.21
40.
182
G>C
0.01
660.
333
5.73
1.23
0.94
0
16rs
9875
390.
478
0.42
9C
>T0.
0037
0.07
38.
441.
220.
469
17rs
2286
233
0.11
30.
123
A>T
0.35
861
0.84
0.91
0.25
1
18rs
1198
1433
0.41
10.
445
T>C
0.04
150.
830
4.16
0.87
0.55
8
19rs
4303
70.
396
0.41
8T>
C0.
1770
11.
820.
910.
197
20rs
1095
3147
0.47
10.
438
A>G
0.05
211
3.77
1.14
1
Bol
d te
xt re
pres
ents
SN
Ps w
ith c
orre
cted
p<0
.05.
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.

NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Landers et al. Page 13Ta
ble
3U
ncor
rect
ed P
-val
ues f
or a
ssoc
iatio
n w
ith A
LS su
b-ph
enot
ypes
SNP
IDO
nset
Age
Bul
bar
vs. S
pina
lU
pper
vs.
Low
er L
imb
Surv
ival
1rs
8545
430.
2537
0.36
230.
9547
0.07
86
2rs
8545
490.
8842
0.84
790.
1436
0.66
67
3rs
2237
582
0.86
260.
5160
0.82
640.
0079
4rs
662
0.61
970.
6295
0.76
950.
0144
5rs
8545
600.
8139
0.04
750.
1014
0.22
65
6rs
2074
351
0.65
960.
0152
0.82
850.
3902
7rs
8545
650.
3766
0.84
220.
2657
0.85
75
8rs
2299
261
0.48
850.
3230
0.94
200.
4919
9rs
7053
810.
4498
0.63
570.
0336
0.41
79
10rs
7053
820.
5544
0.79
850.
2130
0.64
89
11rs
4141
217
0.82
610.
9053
0.48
050.
7253
12rs
9168
640.
7620
0.25
170.
6566
0.12
53
13rs
3757
708
0.84
990.
8332
0.46
070.
4810
14rs
1048
7132
0.94
460.
8646
0.25
910.
4522
15rs
2072
200
0.70
400.
1691
0.53
430.
1032
16rs
9875
390.
6624
0.47
600.
5576
0.33
30
17rs
2286
233
0.22
280.
3310
0.29
430.
7104
18rs
1198
1433
0.82
610.
9539
0.33
150.
2762
19rs
4303
70.
3929
0.93
480.
3427
0.19
57
20rs
1095
3147
0.05
650.
8006
0.89
810.
4408
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.

NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Landers et al. Page 14Ta
ble
4To
p ha
plot
ype
with
in th
e PO
N c
lust
er a
ssoc
iate
d w
ith sp
orad
ic A
LS.
SNPs
Hap
loty
peC
ase
Freq
.C
ont.
Freq
.p-
valu
eA
djus
ted
p-va
lue
Chi
Squ
are
OR
rs85
4565
|rs22
9926
1|rs
7053
81|rs
7053
82|rs
4141
217
GA
CG
T0.
229
0.17
72.
42E-
040.
034
13.4
71.
29
Amyotroph Lateral Scler. Author manuscript; available in PMC 2009 September 7.
Related Documents











