Biomaterials 26 (2005) 6077–6086 A combinatorial approach to the selective capture of circulating malignant epithelial cells by peptide ligands Saurabh Aggarwal a,b , Samuel Janssen a , Randy M. Wadkins c , James L. Harden b , Samuel R. Denmeade a,b, a The Johns Hopkins University School of Medicine, The Sidney Kimmel Comprehensive Cancer Center, Baltimore, MD, USA b Department of Chemical and Biomolecular Engineering, Whiting School of Engineering, The Johns Hopkins University, Baltimore, MD, USA c Department of Chemistry and Biochemistry, University of Mississippi, Oxford, MS, USA Received 5 January 2005; accepted 10 March 2005 Available online 23 May 2005 Abstract Early detection is critical in the administration of definitive and curative therapy of cancer. However, current detection methods are ineffective at identifying the presence of circulating metastatic cancer cells in the blood because they typically sample only a relatively small volume of blood. One strategy for sampling larger blood volumes would be to capture circulating cells in vivo over an extended period of time. The development of such a method would be substantially facilitated by the identification of peptide ligands that bind selectively to metastatic cancer cells in the blood with high affinity. To identify such ligands a combinatorial peptide library was synthesized on polyethylene acrylamide (PEGA) resin and screened for binding to malignant epithelial cells. Using Biacore, cell binding assays were performed to demonstrate that peptides selected from PEGA bead screen can bind selectively to malignant epithelial cancer cells and not to circulating leukocytes under physiologic shear stress conditions. One peptide, with the sequence QMARIPKRLARH, was used to demonstrate selective labeling of malignant epithelial cells spiked in whole blood. When immobilized on appropriate surfaces, these peptides could be used in both in vivo and ex vivo cell separation devices to efficiently and selectively capture metastatic epithelial cancer cells from flowing blood. r 2005 Elsevier Ltd. All rights reserved. Keywords: Peptide; Biosensor; Cell adhesion; Epithelial cell; Catheter 1. Introduction All cancers of epithelial cell origin are incurable with currently available therapies once they have metasta- sized to distant sites. Chemotherapy, in some limited cases, may extend survival but remains for the most part palliative therapy. Currently, surgical removal or local radiation therapy of early stage cancers is the only curative therapy for epithelial cell cancers. Therefore, with present day treatment options, early detection is critical for the administration of definitive, curative therapy of cancer. In addition, limited clinical and extensive preclinical data support the idea that to have any chance of beneficial therapeutic effect, early administration of systemic therapy is of paramount importance. This may be particularly important for newer classes of agents such as anti-angiogenesis agents and signal transduction inhibitors. Therefore, besides the urgent need for the development of novel therapies for metastatic cancer, there is an equally critical need for the development of methods for early detection and for rapidly verifying a therapeutic response to new agents administered at earlier stages of disease. Currently, the diagnosis of cancer is made following detection of a mass that is either palpable to the patient or clinician, or is detected by imaging of the patient. ARTICLE IN PRESS www.elsevier.com/locate/biomaterials 0142-9612/$ - see front matter r 2005 Elsevier Ltd. All rights reserved. doi:10.1016/j.biomaterials.2005.03.040 Corresponding author. Department of Oncology, The Johns Hopkins School of Medicine, Cancer Research Building, 1650 Orleans Street, Baltimore, MD 21231, USA. Tel.: +1 410 502 3941; fax: +1 410 614 8397. E-mail address: [email protected] (S.R. Denmeade).

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ARTICLE IN PRESS
0142-9612/$ - se
doi:10.1016/j.bi
�CorrespondHopkins Schoo
Street, Baltimo
fax: +1410 614
E-mail addr
Biomaterials 26 (2005) 6077–6086
www.elsevier.com/locate/biomaterials
A combinatorial approach to the selective capture of circulatingmalignant epithelial cells by peptide ligands
Saurabh Aggarwala,b, Samuel Janssena, Randy M. Wadkinsc, James L. Hardenb,Samuel R. Denmeadea,b,�
aThe Johns Hopkins University School of Medicine, The Sidney Kimmel Comprehensive Cancer Center, Baltimore, MD, USAbDepartment of Chemical and Biomolecular Engineering, Whiting School of Engineering, The Johns Hopkins University, Baltimore, MD, USA
cDepartment of Chemistry and Biochemistry, University of Mississippi, Oxford, MS, USA
Received 5 January 2005; accepted 10 March 2005
Available online 23 May 2005
Abstract
Early detection is critical in the administration of definitive and curative therapy of cancer. However, current detection methods
are ineffective at identifying the presence of circulating metastatic cancer cells in the blood because they typically sample only a
relatively small volume of blood. One strategy for sampling larger blood volumes would be to capture circulating cells in vivo over
an extended period of time. The development of such a method would be substantially facilitated by the identification of peptide
ligands that bind selectively to metastatic cancer cells in the blood with high affinity. To identify such ligands a combinatorial
peptide library was synthesized on polyethylene acrylamide (PEGA) resin and screened for binding to malignant epithelial cells.
Using Biacore, cell binding assays were performed to demonstrate that peptides selected from PEGA bead screen can bind
selectively to malignant epithelial cancer cells and not to circulating leukocytes under physiologic shear stress conditions. One
peptide, with the sequence QMARIPKRLARH, was used to demonstrate selective labeling of malignant epithelial cells spiked in
whole blood. When immobilized on appropriate surfaces, these peptides could be used in both in vivo and ex vivo cell separation
devices to efficiently and selectively capture metastatic epithelial cancer cells from flowing blood.
r 2005 Elsevier Ltd. All rights reserved.
Keywords: Peptide; Biosensor; Cell adhesion; Epithelial cell; Catheter
1. Introduction
All cancers of epithelial cell origin are incurable withcurrently available therapies once they have metasta-sized to distant sites. Chemotherapy, in some limitedcases, may extend survival but remains for the most partpalliative therapy. Currently, surgical removal or localradiation therapy of early stage cancers is the onlycurative therapy for epithelial cell cancers. Therefore,with present day treatment options, early detection is
e front matter r 2005 Elsevier Ltd. All rights reserved.
omaterials.2005.03.040
ing author. Department of Oncology, The Johns
l of Medicine, Cancer Research Building, 1650 Orleans
re, MD 21231, USA. Tel.: +1410 502 3941;
8397.
ess: [email protected] (S.R. Denmeade).
critical for the administration of definitive, curativetherapy of cancer. In addition, limited clinical andextensive preclinical data support the idea that to haveany chance of beneficial therapeutic effect, earlyadministration of systemic therapy is of paramountimportance. This may be particularly important fornewer classes of agents such as anti-angiogenesis agentsand signal transduction inhibitors. Therefore, besidesthe urgent need for the development of novel therapiesfor metastatic cancer, there is an equally critical need forthe development of methods for early detection and forrapidly verifying a therapeutic response to new agentsadministered at earlier stages of disease.Currently, the diagnosis of cancer is made following
detection of a mass that is either palpable to the patientor clinician, or is detected by imaging of the patient.

ARTICLE IN PRESSS. Aggarwal et al. / Biomaterials 26 (2005) 6077–60866078
With present techniques, the detection of cancer occurslong after the earliest stages of cancer development.Early detection should translate into increased cure ratesand application of systemic therapies at the earliest timepoint results in improved response rates and potentialfor cure. New methods are therefore needed to detecttumor cells at an early stage as an aid to detection andsubsequent curative intervention, and for followingresponse to the clinical intervention.The simplest method for obtaining tissue has been to
perform serial biopsies on patients with accessible softtissue metastases. Direct biopsy is not applicable forearly detection strategies because it is invasive anddifficult to accomplish without clearly defined targets tobiopsy. Serial biopsies are also not readily applicable tomost patients with advanced cancer because themajority of patients do not have readily accessible sitesthat can be easily biopsied. A potentially more easilyaccessible source of cancer cells is in blood samplesobtained from cancer patients. A number of groupshave demonstrated the presence of circulating cancercells in the blood either prior to or following definitivelocal therapy for cancer [1–8]. In most of these studies,peripheral blood (�20ml) is collected from patients atsome point following resection of their primary tumorand PCR analysis is performed in an attempt to identifypatients with minimal residual disease who might benefitfrom adjuvant therapies [7,8]. These PCR-based assays,however, only detect the presence or absence ofcirculating cells; these methods do not isolate cells forfurther characterization by various molecular profilingtechniques currently under development. These PCR-approaches have demonstrated however, that circulatingcancer cells are present and detectable in patients with avariety of cancer types [1–4]. These techniques have beenused to discriminate patients without cancer from thosewith localized disease [3,4].Thus far, the predominant strategy for isolating
cancer cells from blood has been to use a method thatutilizes antibodies toward epithelial cell specific cytoker-atins linked to magnetic beads [5–7]. The majorlimitation of the immunomagnetic bead method is thatsampling can only be done at one or, at most, a fewdiscrete time points and only on a relatively smallvolume of blood (i.e. typically 5–20ml). In early stagedisease, very few cells can be isolated by this method.The only testing that is performed on these few cells istypically PCR assays to detect cancer markers in orderto confirm presence of cancer cells within the sample.Few studies have attempted direct quantification ofcirculating cell numbers in early stage disease [7].Cancer cells are shed continuously into the circulation
to varying degrees according to stage. Instead ofsampling 5–20ml of blood at a given time point, theyield of circulating cancer cells could potentially beincreased if one could continually sample the blood over
a longer period of time, ‘‘catching’’ cells as they arereleased into the circulation. One such approach wouldbe to use a cell separations device, such as in in vivocatheter probe or an ex vivo blood filtration system,with an affinity for cancer cells to continuously extractcirculating cancer cells from blood over an appropriatediagnostic period. Such devices would have intrinsicallybioinert surfaces functionalized with ligands or anti-bodies that bind selectively to cell surface receptors oncancer cells. As an alternative approach to antibody-based cell binding we are proposing instead to use smallpeptides that bind selectively to epithelial cells. Smallpeptides of 12 amino acids are easily synthesized and arenot highly immunogenic. These peptides are relativelyinexpensive, stable, and easily coupled to a probesurface. In the present study, we have synthesized largecombinatorial peptide libraries and used them toidentify putative epithelial cell binding ligands. Fromthese studies, peptides were identified that boundselectively to epithelial cancer cells within a bloodsample but not to leukocytes.
2. Materials and methods
2.1. Materials
Fmoc-blocked amino acids and 200-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate(HBTU) were purchased from Advanced ChemTech,(Louisville, KY) Polyethylene Glycol Amide (PEGA)resin was from Polymer Labs (Amherst, MA). Vials forautomated peptide synthesis on PS-3 were from Anaspec(San Jose, CA). Polystyrene and polyethylene oxide(PEO–PS) comb polymer was purchased from PolymerSource (Quebec, Canada). Unless otherwise specified, allother reagents were from Sigma-Aldrich (St. Louis,MO).
2.2. Screening cell binding on peptide coated surfaces
Polystyrene dishes (35mm) were coated with PEO–PSdiblock comb polymer by incubating 2mg/ml solutionof PEO–PS in 80% Methanol and then drying at 37 1C.PEO–PS surface was thoroughly washed with ethanol toremove excess of comb polymer. Surface adsorption ofPEO–PS was confirmed by change in the contact angleof a drop of water. On a small marked spot on the dish,carboxyl groups at end of PEO arms were activated byplacing 1–2 ml of 10mg/ml of Sulfo-NHS with 5mg/mlof 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hy-drochloride (EDC) in 4-morpholineethanesulfonic acidmonohydrate (MES) buffer at 4 1C for 1 h. Dishes werewashed twice quickly with phosphate buffered saline(PBS) and then 1–2 ml of 1mg/ml peptide or antibodysolution in PBS was placed directly on the marked, dried

ARTICLE IN PRESSS. Aggarwal et al. / Biomaterials 26 (2005) 6077–6086 6079
spot and allowed to react for 2–4 h at 4 1C. Dishes wereagain washed with PBS� 2 and stored at 4 1C in PBS forlater use. Cell tracker rhodamine labeled PC-3 cells(20,000 cells/ml) suspended in serum free RPMI mediawere added to dish and incubated for 1 h at 37 1C.Dishes were washed with PBS� 3 to remove unboundcells and then cells fixed with 80% methanol/water forevaluation of binding using fluorescent microscopy.
2.3. Synthesis of the combinatorial peptide library
The peptide library was synthesized using the splitand mix synthesis method and assembled on a PEGAsupport (0.2mmol/g) using standard solid-phase Fmocchemistry [8]. The entire resin was first loaded byovernight coupling with 2mmoles of Fmoc–Glyci-ne–OH followed by Fmoc–Lysine(Fmoc)–OH. Fmo-c–Lysine(Fmoc)–OH was used to produce dimerizedpeptides. One synthetic cycle consisted of dividing theresin into 19 equal portions where each portion wastreated with 1 amino acid as follows: 2.5mmol ofderivatized amino acid and 2.5mmol of 1-hydroxyben-zotriazoile (HOBt) were dissolved in 2.5ml of 1-methyl-2-pyrrolidone (NMP), 2.5ml of a 1M solution of N,N0-diisopropylcarbodiimide (DIC) in NMP was added tothe above mentioned amino acid/HOBt solution. Theamino acid/HOBt/DIC solution was stirred for 2min,transferred to the resin, and incubated for 2 h at roomtemperature while mixing. The resin was filtered andrinsed three times with NMP and three times withmethanol. Each cycle was repeated to ensure completecouplings. The beads were repooled, mixed, rinsed threetimes with dichloromethane (DCM) and three timeswith dimethylformamide (DMF). Deprotection wascarried out by a 20-min treatment with 20% piper-idine/NMP, followed by filtration and rinsing threetimes with NMP, three times with Methanol. Couplingafter each amino acid was checked by a Kaiser test.This cycle was repeated 12 times to obtain a 12 amino
acid random peptide library. After synthesis wascompleted, side chain protecting groups attached tothe amino acids were cleaved by stirring the peptide-bearing resin in a solution of 90% trifluoroacetic acid,2.5% thioanisole, 5% H2O, and 2.5% ethanedithiol for3 h at room temperature.
2.4. Cell lines
The previously characterized LNCaP, PC-3 humanprostate cancer, TSU human bladder cancer and MCF-7 human breast cancer cell lines were obtained fromATCC (Rockville, MD). These lines were maintained byserial passage in RPMI 1640 media (Gibco, GrandIsland, NY) containing 10% fetal bovine serum (FBS)(Bio-Whittaker, Walkersville, MD). All standard mediaincluded 100U/ml penicillin G, and 100U/ml strepto-
mycin sulfate and all cells were grown in 5% CO2/95%air at 37 1C.
2.5. Screening peptide library to identify cell binding
ligands
For initial screening, the human prostate cancer cellline LNCaP was utilized. For this assay, cells weresuspended at concentration of 106 cells/ml and werefluorescently labeled using Cell Tracker Rhodamine(Molecular Probes, Eugene, OR) according to manu-facturer’s instructions. Cells suspended in 10% FBScontaining RPMI media were admixed with 100,000peptide-containing resin ‘‘beads’’ already suspended inserum containing media. Beads and cells were incubatedtogether with rotation at 37 1C for 40min. Unboundcells were removed by aspiration and gentle washing.Bound cells were fixed to the beads by 15min incubationin 80% Methanol/water. Subsequently, the beads werewashed vigorously with 80%Methanol to remove excessnon-bound cells. Beads whose surfaces were at least75% coated with cells were manually selected under adissecting microscope. Cells were stripped from selectedbeads using 8M Urea overnight. The peptide sequencefrom each individual selected bead was then determinedby Edman degradation performed by the University ofArizona Peptide Sequencing Core Facility.A similar screening procedure was utilized to analyze
ability of Ficoll separated white blood cells to bind toselected epithelial cell binding beads. Selected beadswere incubated with Cell Tracker green loaded whiteblood cell suspension at 106 cells/ml for 40min. Cellswere fixed and visualized as described above.
2.6. Binding analysis using surface plasmon resonance
Three of the selected peptides were resynthesized witha biotin at the C-terminus. Biotin was attached bysynthesizing the peptides on Fmoc–Lys–�–Biotin–WangResin (Anaspec). Peptides were purified on reversephase HPLC using a Luna C18 column (Phenomenex,Torrancem CA) and then suspended in PBS at 1mg/ml.Individual biotinylated peptides were incubated on astreptavidin coated surface plasmon resonance (SPR)chip (Biacore, Piscataway, NJ). Unbound peptide waswashed away with 1% Triton. Shear stress in the SPRflow chamber was calculated as 16 dynes/cm2 using theformula: tw ¼ 6mQ=h2w in which m ¼ 0:01 dynes s=cm2
is the fluid viscosity, Q ¼ 20ml=min is the flow rate, w ¼
0:5mm is the width of chamber, and h ¼ 0:05mm is theheight of chamber.
2.7. Surface binding visualization by confocal microscopy
Biotinylated peptide (30 nM) precomplexed with10 mg/ml of Neutravidin–Rhodamine (Molecular

ARTICLE IN PRESSS. Aggarwal et al. / Biomaterials 26 (2005) 6077–60866080
Probes) was incubated with LNCaP cells (106/ml) for 1 hat 37 1C in 10% FBS containing media. LNCaP cellswere pre-labeled with green cell tracker (MolecularProbes, OR). To determine the extent of non-specificbinding, green cell tracker labeled LNCaP cells wereincubated with Neutravidin–Rhodamine alone in theabsence of binding peptide.
2.8. Binding of peptide to LNCaP cells in whole blood
Green Cell Tracker labeled LNCaP cells, suspendedat 100,000 cells in 1ml of FBS containing media, weremixed with 1ml of whole blood. Biotinylated peptide(30 nM) was preincubated with Neutravidin–Rhodamine(10 mg/ml) and then added to the labeled cells/bloodmixture and incubated at 37 1C for 1 h. Cells were spundown and the cell pellet was stained with DAPI usingmounting media to visualize all cell nuclei prior toevaluation by fluorescent microscopy.
3. Results
3.1. Selection of epithelial cell binding peptides
Previous studies have identified peptides that can bindto cell surface proteins such as laminin (e.g. AG-73) andintegrins (e.g. RGD) [9,10]. In initial studies wesynthesized a series of these peptides to directly comparetheir binding characteristics when attached to a solidphase surface. To demonstrate cell binding, polystyrenedishes were coated with a functionalized PEO/PS combcopolymer. This comb copolymer consists of a PSbackbone copolymerized with carboxyl functionalizedPEO chains at the surface. Presentation of the PEO/PScomb polymer under appropriate solvent conditionsresults in spontaneous adsorption of the comb’s PSblock onto the PS dish producing a dish coated with athin coating of functionalized PEO at the surface. Dueto its hydrophilic nature, PEO inhibits protein adsorp-tion on surfaces and also prevents non-specific cellbinding that can occur with uncoated PS surfaces[11–13]. For these studies peptides were coupled tosmall preactivated spots on the PEO–PS coated surface.Peptides utilized in this study included: AG73, a 12amino acid peptide from the G domain of the laminin a1chain [9,14]; Peptide J, a 19 amino acid peptide derivedfrom a Trypanosoma cruzi surface glycoprotein thatbinds to cytokeratin 18 [15]; RGD that binds tointegrins [10,16]; luteinizing hormone releasing hormone(LHRH), a 10 amino acid peptide that binds to LHRHreceptors that are present on human prostate cancersand prostate cancer cell lines such as PC-3 line used inthis study [17]; and GRAGG, a 5 amino acid peptideselected from in vivo phage display based on it ability tobind to prostate tissue [18]. The binding of these
peptides was compared to binding of anti-humanepithelial antigen (HEA) antibody used extensively inmagnetic bead based applications to pull epithelial cellsout of blood samples [19]. To visualize binding, thehuman prostate cancer cell line PC-3 was pre-incubatedwith the fluorescent dye Cell-Tracker Orange. Fluores-cently labeled cells in 10% fetal calf serum containingtissue culture media were added to the dish for 10minfollowed by gentle washing with Hank’s balanced saltsolution. Cell-binding was visualized using a fluorescentmicroscope equipped with rhodamine filter set (Fig. 1a).These results demonstrate that some of these peptides,
when attached to a solid surface, can readily bind to theepithelial cancer cells while others show no bindingunder these conditions, as summarized in Table 1. Thebinding was qualitatively similar between the Peptide Jand AG-73 peptides and an anti-HEA antibody to apan-epithelial cell antigen (Table 1). In addition, the factthat cells bind only to peptide conjugated spotsemphasizes the non-absorbent characteristics of unmo-dified PEO/PS surface that are important to preventnon-specific cell binding (Fig. 1a).While these results demonstrate the feasibility of the
approach, the peptides utilized were initially selectedsolely for their ability to bind to a target antigen.Practical devices for capture of cancer cells from wholeblood, however, will require peptides that can bindselectively to circulating epithelial cancer cells while notbinding to highly abundant circulating leukocytes.Therefore, we opted to use a combinatorial basedpeptide library approach to identify peptides meetingthese two characteristics. This approach had been usedpreviously by Pennington et al. to identify lamininpeptides with sequence similar to AG-73 [20]. Thus, apeptide library was synthesized consisting of 12 randomamino acids coupled to a solid phase PEGA support.PEGA was selected for these studies because the outercoating of this resin consists of PEO and, therefore,selected cell-binding PEGA bound peptides shouldexhibit similar cell binding characteristics when incor-porated into the surface of a PEO-coated cell separa-tions device, such as a functionalized catheter.Prior to synthesizing a large random peptide library,
we synthesized the AG-73 peptide on the PEGA beadsto serve as a positive control. When these beads wereincubated with cell tracker labeled cells, no binding ofcells to beads was observed (data not shown). Afterconfirming that beads had the correct peptide sequence,we hypothesized that this negative result was due to thelower concentration of peptide present on the PEGAbead compared to concentrations achievable undersurface spotting conditions described above. Previously,Wrighton et al. had demonstrated that affinity forpeptides binding to the erythropoietin receptor could beincreased 100-fold when peptides were synthesized in adimeric form [21]. These dimeric peptides were gener-

ARTICLE IN PRESS
Table 1
Cell binding characteristics of selected peptides
Peptide/antibody Sequence Target Binding Ref.
AG-73 RKRLQVQSIRTG Integrins +++ [21]
Peptide J KGVTVTNVFLYNRPL Cytokeratin-18 +++ [22]
RGD YGRGD Integrins � [23]
LHRH pEHWSYGLRPG LHRH receptor + [24,25]
AGG GRAGGS Prostate tissue � [26]
Anti-HEA Antibody HEA +++ [27]
(b)(a)
(c) (d)
Fig. 1. (a) Rhodamine cell tracker labeled human PC3 prostate cancer cells binding to AG-73 peptide coupled to derivatized spot on PEO–PS coated
35mm dish. Cells bind only to spotted area (lower right-hand corner) and not to underivatized PEO–PS surface (upper left-hand corner). (b) FITC
cell tracker labeled LNCaP cells binding to PEGA beads conjugated to dimeric AG-73 peptide after 1 h incubation. Majority of beads demonstrate
some cell binding while �20% of beads have no cells bound. (c) Representative cell bound bead from screen of dimeric, random peptide library.
Rhodamine labeled LNCaP cells coat bead with putative cell binding peptide where other peptide-conjugated beads in field have no cells bound. (d)
Representative resynthesized cell binding peptide in which all peptide beads become heavily coated with rhodamine labeled LNCaP cells after 1 h
incubation.
S. Aggarwal et al. / Biomaterials 26 (2005) 6077–6086 6081
ated using a C-terminal lysine residue as a branch point[21]. Therefore, we used a similar strategy to synthesizethe dimeric form of AG-73 on PEGA beads anddetermined that this dimeric peptide was now able tobind to labeled PC-3 cells (Fig. 1b).On the basis of these results we next synthesized a
library of approximately 100,000 random dimericpeptides using a lysine residue at the C-terminus as abranch point. Peptides were synthesized using the splitand mix synthetic approach [8]. This synthetic schemegenerates a library in which each individual resin ‘‘bead’’has multiple copies of a single amino acid sequence.These beads were suspended in serum containing media
and incubated with LNCaP cells labeled with rhodamineCell Tracker at a cell density of 106 cells/ml. LNCaPcells were selected as a representative human epithelialcancer cell line for this initial screen. LNCaP is anandrogen sensitive human prostate cancer cell line thatexpresses many of the same differentiation products ashuman prostate cancers (i.e. PSA, PSMA, hK2) [22].Incubation of fluorescently labeled LNCaP cells with thepeptide library yielded a small number of beads thatwere extensively coated with rhodamine labeled cells in abackground of beads with few to no cells bound, asshown in Fig. 1c. Approximately 100 cell coated beadswere obtained from this 100,000 bead library (i.e. 0.1%).

ARTICLE IN PRESSS. Aggarwal et al. / Biomaterials 26 (2005) 6077–60866082
Ten of the brightest beads containing the highestnumber of cells were selected manually from this pooland the amino acid sequence of peptides on eachindividual bead was determined (see Table 2).No consensus binding sequence was discernible
from these ten sequences. A BLAST search of eachsequence did not yield any close match with anypreviously sequenced protein. In addition, none of theselected peptides contained integrin-binding peptidemotifs such as the RGD tripeptide. Each selectedpeptide listed in Table 2 contained 4–6 of the basicamino acids Arg, His, and Lys. Five of these peptideshad 3–4 sequential basic amino acids near either the N-or C-terminus.Following this initial screen, these 10 LNCaP binding
peptides were resynthesized on PEGA resin andrescreened to confirm binding to LNCaP and todetermine ability of these peptides to bind to leukocytes.Aliquots of beads containing a unique peptide sequencewere incubated with labeled LNCaP cells and evaluatedby fluorescent microscopy (Fig. 1d). For each peptidetested, beads rapidly became coated with large numbersof LNCaP cells confirming that peptides selected frominitial screens were not false positives and were truly cellbinding peptides (Fig. 1d). Semi-quantitative compar-ison of the selected peptides to the AG-73 controlrevealed that a larger percentage of selected peptidebeads became coated with cells and number of cells/beadappeared higher than AG-73, suggesting that theselected peptides possess superior binding characteristicscompared to AG-73.The next experiments were designed to determine
whether the selected peptides could also bind leukocytes.For this assay the leukocyte fraction was separated fromwhole blood using a Ficoll gradient. Leukocytes wereloaded with Cell Tracker green and then incubated withbeads at 37 1C for 40min. The majority of the tenpeptides bound a small amount of leukocytes (i.e.o20% total bead surface coated). Three peptides withsequences QMARIPKRLARH, RHNGKPDHRKSVand QVKDYHVAPRRK, however, were identifiedwhich had no discernible leukocyte binding (data notshown).
Table 2
Peptide sequences of selected cell binding peptides
1. RHNGKPDHRKSV
2. QVKDYHVAPRRK
3. RRKKVRYGMIQP
4. QMARIPKRLARH
5. YKKKHVVRDTPR
6. GKHKFRISKGIR
7. KKFGHERGTRKG
8. KXQKAERWWPWK
9. GIKKLMRSYGRR
10. IGKTKVKNQKYY
3.2. Affinity and selectivity of peptide binding to
malignant epithelial cell line
One requirement for effective separation of cancercells from flowing blood (e.g. by an indwelling catheterdevice) is the ability to capture cells under physiologicshear conditions. Previously it has been demonstratedthat physiologic shear can be up to �25 dynes/cm2 [23].To model such shear conditions, peptides were resynthe-sized with the introduction of biotin at the aminoterminus and then attached to the surface of astreptavidin coated surface plasmon resonance (SPR)chamber. LNCaP cells were then flowed over thissurface at a rate of 20 ml/min, which, given thedimensions of the SPR chamber, resulted in a calculatedshear force of 16 dynes/cm2. The Biacore SPR ‘‘chips’’have capacity to analyze four different conditionssimultaneously [24]. Therefore, cell binding to the threepeptides and a control surface could be analyzed in thesame experiment. In this SPR system, binding ismeasured in Response Units. Using this approach, allthree peptides demonstrated comparable ability tocapture flowing cells (50,000 cells/ml injected at 20 ml/min) as measured by change in response units overbaseline, as shown in Fig. 2a. Of the three, theQMARIPKRLARH peptide exhibited the steepest slopeduring the cell binding phase, indicating the strongesttendency for peptide–cell association. Binding to allthree peptides was linear over cell concentrationsranging from 2500 to 50,000 cells/ml (data not shown).No significant non-specific binding to the streptavidincoated control channel was observed (Fig. 2a).On the basis of no demonstrated leukocyte binding
and slightly superior binding in the SPR system, thepeptide QMARIPKRLARH was further screened todetermine whether the peptide could bind to a panel ofepithelial cancer cells or was only capable of binding tothe LNCaP cell line used in the original screen.Therefore, aliquots of peptide containing beads wereincubated with a second human prostate cancer cell line,PC-3, and with human MCF-7 breast cancer cells andthe human TSU bladder cancer cells. In each case, cellbinding was of similar magnitude to that observed withLNCaP cells (data not shown) suggesting that selectedpeptides may bind to an antigen shared by epithelialcancer cells from a variety of tissues.An additional crucial requirement for effective cancer
cell separation from blood is the ability of the peptide toselectively bind to epithelial cells and not bind tocirculating leukocytes. Therefore, to confirm qualitativeresults from bead binding experiments described above,Ficoll separated leukocytes were flowed over the threeselected peptides. In these traces a ‘‘square wave’’ isobserved in which the initial sharp increase is due to cellsrolling across the plasmon resonance surface. The sharpdecrease back to baseline observed when cell containing
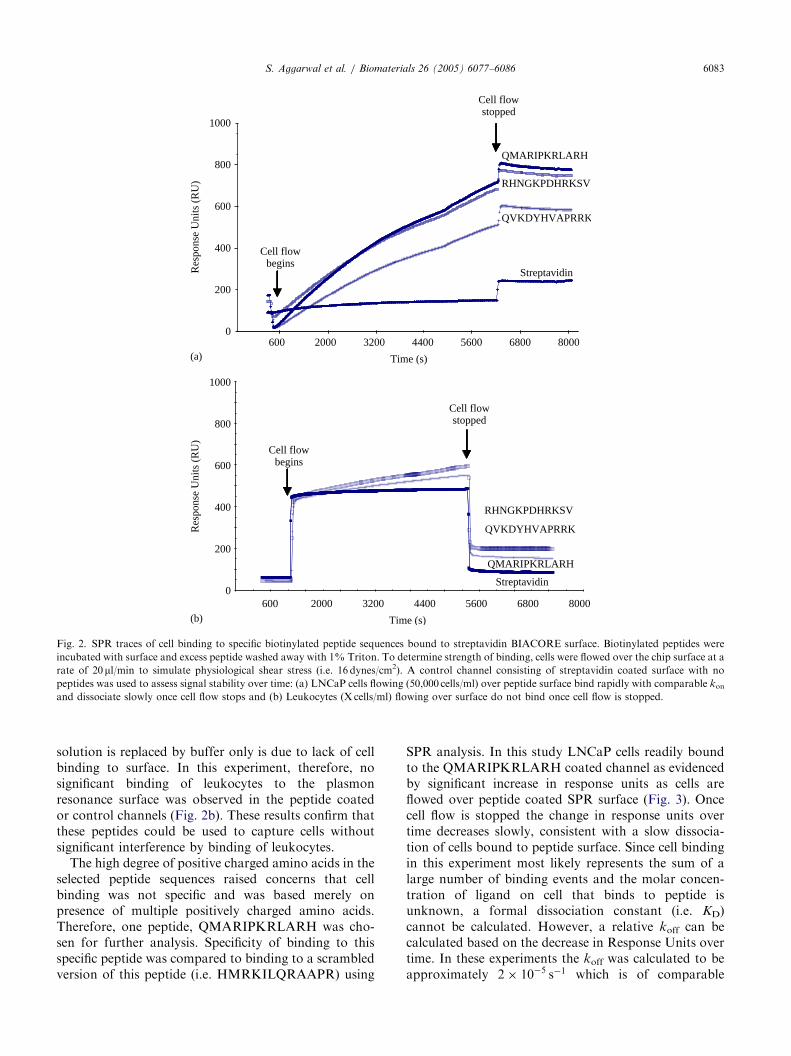
ARTICLE IN PRESS
1000
800
600
400
200
0
Res
pons
e U
nits
(R
U)
1000
800
600
400
200
0
Res
pons
e U
nits
(R
U)
Cell flowbegins
Cell flowbegins
Cell flowstopped
Cell flowstopped
QMARIPKRLARH
RHNGKPDHRKSV
QVKDYHVAPRRK
Streptavidin
800068005600440032002000600
Time (s)
800068005600440032002000600
Time (s)
RHNGKPDHRKSV
QVKDYHVAPRRK
QMARIPKRLARH
Streptavidin
(a)
(b)
Fig. 2. SPR traces of cell binding to specific biotinylated peptide sequences bound to streptavidin BIACORE surface. Biotinylated peptides were
incubated with surface and excess peptide washed away with 1% Triton. To determine strength of binding, cells were flowed over the chip surface at a
rate of 20ml/min to simulate physiological shear stress (i.e. 16 dynes/cm2). A control channel consisting of streptavidin coated surface with no
peptides was used to assess signal stability over time: (a) LNCaP cells flowing (50,000 cells/ml) over peptide surface bind rapidly with comparable konand dissociate slowly once cell flow stops and (b) Leukocytes (X cells/ml) flowing over surface do not bind once cell flow is stopped.
S. Aggarwal et al. / Biomaterials 26 (2005) 6077–6086 6083
solution is replaced by buffer only is due to lack of cellbinding to surface. In this experiment, therefore, nosignificant binding of leukocytes to the plasmonresonance surface was observed in the peptide coatedor control channels (Fig. 2b). These results confirm thatthese peptides could be used to capture cells withoutsignificant interference by binding of leukocytes.The high degree of positive charged amino acids in the
selected peptide sequences raised concerns that cellbinding was not specific and was based merely onpresence of multiple positively charged amino acids.Therefore, one peptide, QMARIPKRLARH was cho-sen for further analysis. Specificity of binding to thisspecific peptide was compared to binding to a scrambledversion of this peptide (i.e. HMRKILQRAAPR) using
SPR analysis. In this study LNCaP cells readily boundto the QMARIPKRLARH coated channel as evidencedby significant increase in response units as cells areflowed over peptide coated SPR surface (Fig. 3). Oncecell flow is stopped the change in response units overtime decreases slowly, consistent with a slow dissocia-tion of cells bound to peptide surface. Since cell bindingin this experiment most likely represents the sum of alarge number of binding events and the molar concen-tration of ligand on cell that binds to peptide isunknown, a formal dissociation constant (i.e. KD)cannot be calculated. However, a relative koff can becalculated based on the decrease in Response Units overtime. In these experiments the koff was calculated to beapproximately 2� 10�5 s�1 which is of comparable
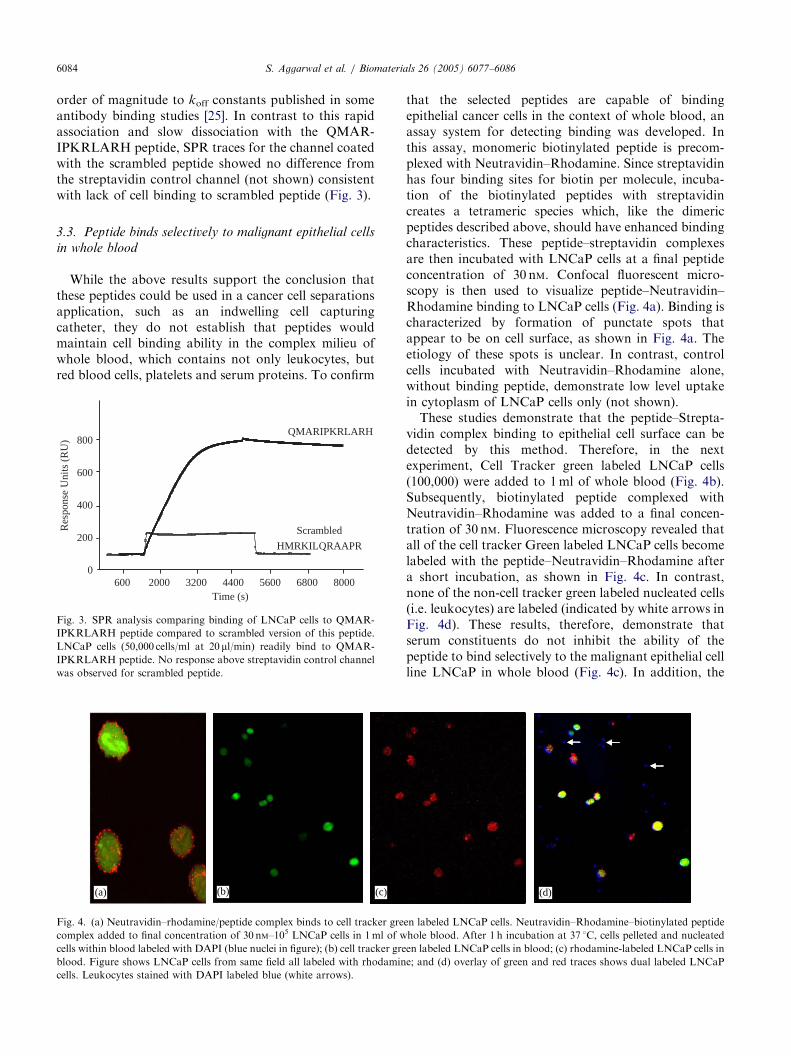
ARTICLE IN PRESSS. Aggarwal et al. / Biomaterials 26 (2005) 6077–60866084
order of magnitude to koff constants published in someantibody binding studies [25]. In contrast to this rapidassociation and slow dissociation with the QMAR-IPKRLARH peptide, SPR traces for the channel coatedwith the scrambled peptide showed no difference fromthe streptavidin control channel (not shown) consistentwith lack of cell binding to scrambled peptide (Fig. 3).
3.3. Peptide binds selectively to malignant epithelial cells
in whole blood
While the above results support the conclusion thatthese peptides could be used in a cancer cell separationsapplication, such as an indwelling cell capturingcatheter, they do not establish that peptides wouldmaintain cell binding ability in the complex milieu ofwhole blood, which contains not only leukocytes, butred blood cells, platelets and serum proteins. To confirm
600 2000 3200 4400 5600 6800 8000Time (s)
800
600
400
200
0
Res
pons
e U
nits
(R
U)
QMARIPKRLARH
Scrambled
HMRKILQRAAPR
Fig. 3. SPR analysis comparing binding of LNCaP cells to QMAR-
IPKRLARH peptide compared to scrambled version of this peptide.
LNCaP cells (50,000 cells/ml at 20 ml/min) readily bind to QMAR-
IPKRLARH peptide. No response above streptavidin control channel
was observed for scrambled peptide.
(c)(b)(a)
Fig. 4. (a) Neutravidin–rhodamine/peptide complex binds to cell tracker gre
complex added to final concentration of 30 nM–105 LNCaP cells in 1ml of w
cells within blood labeled with DAPI (blue nuclei in figure); (b) cell tracker gre
blood. Figure shows LNCaP cells from same field all labeled with rhodamin
cells. Leukocytes stained with DAPI labeled blue (white arrows).
that the selected peptides are capable of bindingepithelial cancer cells in the context of whole blood, anassay system for detecting binding was developed. Inthis assay, monomeric biotinylated peptide is precom-plexed with Neutravidin–Rhodamine. Since streptavidinhas four binding sites for biotin per molecule, incuba-tion of the biotinylated peptides with streptavidincreates a tetrameric species which, like the dimericpeptides described above, should have enhanced bindingcharacteristics. These peptide–streptavidin complexesare then incubated with LNCaP cells at a final peptideconcentration of 30 nM. Confocal fluorescent micro-scopy is then used to visualize peptide–Neutravidin–Rhodamine binding to LNCaP cells (Fig. 4a). Binding ischaracterized by formation of punctate spots thatappear to be on cell surface, as shown in Fig. 4a. Theetiology of these spots is unclear. In contrast, controlcells incubated with Neutravidin–Rhodamine alone,without binding peptide, demonstrate low level uptakein cytoplasm of LNCaP cells only (not shown).These studies demonstrate that the peptide–Strepta-
vidin complex binding to epithelial cell surface can bedetected by this method. Therefore, in the nextexperiment, Cell Tracker green labeled LNCaP cells(100,000) were added to 1ml of whole blood (Fig. 4b).Subsequently, biotinylated peptide complexed withNeutravidin–Rhodamine was added to a final concen-tration of 30 nM. Fluorescence microscopy revealed thatall of the cell tracker Green labeled LNCaP cells becomelabeled with the peptide–Neutravidin–Rhodamine aftera short incubation, as shown in Fig. 4c. In contrast,none of the non-cell tracker green labeled nucleated cells(i.e. leukocytes) are labeled (indicated by white arrows inFig. 4d). These results, therefore, demonstrate thatserum constituents do not inhibit the ability of thepeptide to bind selectively to the malignant epithelial cellline LNCaP in whole blood (Fig. 4c). In addition, the
(d)
en labeled LNCaP cells. Neutravidin–Rhodamine–biotinylated peptide
hole blood. After 1 h incubation at 37 1C, cells pelleted and nucleated
en labeled LNCaP cells in blood; (c) rhodamine-labeled LNCaP cells in
e; and (d) overlay of green and red traces shows dual labeled LNCaP

ARTICLE IN PRESSS. Aggarwal et al. / Biomaterials 26 (2005) 6077–6086 6085
peptide only binds to LNCaP cells in the blood and doesnot bind to leukocytes or red blood cells (Fig. 4d).
4. Discussion
The goal of this study was to use a combinatorialapproach to identify small peptides that bind selectivelyto circulating epithelial cancer cells in the blood that canthen be incorporated into devices for separating cancercells from whole blood. Such a device must meet severalimportant requirements. First, as we have demonstratedin this study, the cell binding component must bindselectively to the target, in this case circulating epithelialcells, and not bind non-specifically to other cellularcomponents such as leukocytes. The binding ligandmust also be able to bind to cells when the ligand isattached to a solid surface. Thus, we initially selectedour peptides based on the ability of cells to bind topeptides covalently attached to PEGA beads. Particu-larly for in vivo devices, such as an indwelling catheter,the ligand must also be able to capture circulating cellsunder physiological shear conditions, as we havedemonstrated using the SPR flow chamber. The surfaceof the probe must be relatively inert and not adsorbproteins and/or cells non-specifically. PEO was selectedas the screening surface because it has been demon-strated to be one of the most effective biomaterials forcoating surfaces to prevent non-specific adsorption[11–13]. As our long-term goal is to produce a cathetercoated with ligand-functionalized PEO, we chose resinbeads that have a PEO outer surface in order to mimiccatheter surface conditions from the outset in theselection of cell-binding peptides.In this study, no cell binding was observed on PEGA
beads when a peptide was synthesized as a monomer. Ashas been previously shown, dimerization of peptidesthrough the use of a lysine residue at the C-terminus canmarkedly increase binding affinity due to an avidityeffect [21]. While monomeric, biotinylated peptides wereused in the SPR experiments and to demonstrate cellbinding in whole blood, in both cases peptides werebound to streptavidin to generate a complex with fourpeptides per streptavidin protein. This tetrameric com-plex should increase binding affinity and may beresponsible for the favorable binding characteristicsobserved under shear conditions in SPR chamber and inwhole blood. In future studies we will compare bindingaffinities of a dimeric peptide to a tetrameric peptidesynthesized through use of two branched lysine residuesat the C-terminus of the peptide.SPR has proven to be a versatile tool with a wide
variety of applications. SPR has been used extensively tostudy antibody binding to antigen but has also beenused to study protein–protein, peptide–protein andpeptide–membrane interactions [25–29]. However, SPR
is not commonly used to study cell–ligand interaction aswe have done in this study. In the only publicationknown to us, Quinn et al. compared several SPR-basedbiosensors including a fiber optic SPR-based (FOSPR)biosensor, employing wavelength-dependent SPR, amodified commercially available integrated angle-de-pendent SPR-based refractometer (ISPR) and BIA-CORE [30]. This group utilized affinity-captured bloodgroup specific antibodies to detect binding of whole redblood cells (RBCs) and determined that, of the threedetection methods, the BIACORE system performed thebest with lowest level of non-specific cell binding tocontrol surface. We used the BIACORE system for ourstudies and found negligible cell binding to thestreptavidin surface in the absence of binding peptides.This SPR system does adequately simulate cell binding
under physiologic shear conditions and can be used tocompare binding characteristics of a series of peptidessimultaneously. However, interpretation of binding andcalculation of binding constants is complicated using theSPR system because the system cannot discriminate highaffinity interactions from a summation of multiple lowaffinity interactions between peptide and cell surface.Although the dissociation of cells from the peptide wasvery slow in these experiments (i.e. estimated half-life withkoff of 2� 10�5 s�1 is 500min), this dissociation was mostlikely due to summation of large number of interactionsbetween the peptide ligand and the cognate receptor on thecell surface. This receptor appeared to be present on avariety of epithelial cell types, but not present onleukocytes or red blood cells. The identity of the receptorremains to be determined in future experiments, butcandidates would include cytokeratins, laminin or theglycocalyx. The lack of RGD motif in the peptides makesthe integrin family an unlikely candidate.
5. Conclusions
Using BIACORE, cell binding assays were performedto demonstrate that peptides selected from PEGA beadscreen can bind selectively to malignant epithelial cancercells and not to circulating leukocytes under physiologicshear stress conditions. One peptide, with the sequenceQMARIPKRLARH, was used to demonstrate selectivelabeling of malignant epithelial cells spiked in wholeblood. When immobilized on appropriate surfaces, thesepeptides could be used in both in vivo and ex vivo cellseparation devices to efficiently and selectively capturemetastatic epithelial cancer cells from flowing blood.
Acknowledgements
This work was supported by funding from Alexanderand Margaret Stewart Trust to SRD, an Aegon

ARTICLE IN PRESSS. Aggarwal et al. / Biomaterials 26 (2005) 6077–60866086
Research Fellowship to SJ and partial support from theNASA Human Exploration and Development of Spaceprogram NAG 9-1345 to JLH.
References
[1] Gilbey AM, Burnett D, Coleman RE, Holen I. The detection of
circulating breast cancer cells in blood. J Clin Pathol
2004;57:903–11.
[2] Schuster R, Max N, Mann B, Heufelder K, Thilo F, Grone J,
Rokos F, Buhr HJ, Thiel E, Keilholz U. Quantitative real-time
RT-PCR for detection of disseminated tumor cells in peripheral
blood of patients with colorectal cancer using different mRNA
markers. Int J Cancer 2004;108:219–27.
[3] Ennis RD, Katz AE, de Vries GM, Heitjan DF, O’Toole KM,
Rubin M, Buttyan R, Benson MC, Schiff PB. Detection of
circulating prostate carcinoma cells via an enhanced reverse
transcriptase–polymerase chain reaction assay in patients with
early stage prostate carcinoma. Independence from other
pretreatment characteristics. Cancer 1997;79:2402–8.
[4] Racila E, Euhus D, Weiss AJ, Rao C, McConnell J, Terstappen
LWMM, Uhr JW. Detection and characterization of carcinoma
cells in the blood. Proc Natl Acad Sci 1998;95:4589–94.
[5] Rye PD, Hoifodt HK, Overli GE, Fodstad O. Immunobead
filtration: a novel approach for the isolation and propagation of
tumor cells. Am J Pathol 1997;150:99–106.
[6] Hardingham JE, Kotasek D, Farmer B, Butler RN, Mi J, Sage
RE, Dobrovic A. Immunobead-PCR: a technique for the
detection of circulating tumor cells using immunomagnetic beads
and the polymerase chain reaction. Cancer Res 1993;53:3455–8.
[7] Wang ZP, Eisenberger MA, Carducci MA, Partin AW, Scher HI,
Ts’o PO. Identification and characterization of circulating
prostate carcinoma cells. Cancer 2000;88:2787–95.
[8] Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM,
Knapp RJ. A new type of synthetic peptide library for identifying
ligand-binding activity. Nature 1991;354:82–4.
[9] Nomizu M, Kim WH, Yamamura K, Utani A, Song SY, Otaka
A, Roller PP, Kleinman HK, Yamada Y. Identification of cell
binding sites in the laminin alpha 1 chain carboxyl-terminal
globular domain by systematic screening of synthetic peptides. J
Biol Chem 1995;270:20583–90.
[10] Pytela R, Pierschbacher MD, Ginsberg MH, Plow EF, Ruoslahti
E. Platelet membrane glycoprotein IIb/IIIa: member of a family
of Arg–Gly–Asp—specific adhesion receptors. Science
1986;231:1559–62.
[11] Zhang M, Desai T, Ferrari M. Proteins and cells on PEG
immobilized silicon surfaces. Biomaterials 1998;19:953–60.
[12] VandeVondele S, Voros J, Hubbell JA. RGD-grafted poly-L-
lysine-graft-(polyethylene glycol) copolymers block non-specific
protein adsorption while promoting cell adhesion. Biotechnol
Bioeng 2003;82:784–90.
[13] Frederix F, Bonroy K, Reekmans G, Laureyn W, Campitelli A,
Abramov MA, Dehaen W, Maes G. Reduced nonspecific
adsorption on covalently immobilized protein surfaces using
poly(ethylene oxide) containing blocking agents. Biochem Bio-
phys Methods 2004;58:67–74.
[14] DeRoock IB, Pennington ME, Sroka TC, Lam KS, Bowden GT,
Bair EL, Cress AE. Synthetic peptides inhibit adhesion of human
tumor cells to extracellular matrix proteins. Cancer Res
2001;61:3308–13.
[15] Magdesian MH, Giordano R, Ulrich H, Juliano MA, Juliano L,
Schumacher RI, Colli W, Alves MJ. Infection by Trypanosoma
cruzi. Identification of a parasite ligand and its host cell receptor.
J Biol Chem 2001;276:19382–9.
[16] Ruoslahti E. RGD and other recognition sequences for integrins.
Annu Rev Cell Dev Biol 1996;12:697–715.
[17] Bahk JY, Hyun JS, Lee H, Kim MO, Cho GJ, Lee BH, Choi WS.
Expression of gonadotropin-releasing hormone (GnRH) and
GnRH receptor mRNA in prostate cancer cells and effect of
GnRH on the proliferation of prostate cancer cells. Urol Res
1998;26:259–64.
[18] Arap W, Kolonin MG, Trepel M, Lahdenranta J, Cardo-Vila M,
Giordano RJ, Mintz PJ, Ardelt PU, Yao VJ, Vidal CI, Chen L,
Flamm A, Valtanen H, Weavind LM, Hicks ME, Pollock RE,
Botz GH, Bucana CD, Koivunen E, Cahill D, Troncoso P,
Baggerly KA, Pentz RD, Do KA, Logothetis CJ, Pasqualini R.
Steps toward mapping the human vasculature by phage display.
Nat Med 2002;8:121–7.
[19] Kemmner W, Moldenhauer G, Schlag P, Brossmer R. Sepa-
ration of tumor cells from a suspension of dissociated human
colorectal carcinoma tissue by means of monoclonal anti-
body-coated magnetic beads. J Immunol Methods 1992;147:
197–200.
[20] Pennington ME, Lam KS, Cress AE. The use of a combinatorial
library method to isolate human tumor cell adhesion peptides.
Mol Divers 1996;2:19–28.
[21] Wrighton NC, Balasubramanian P, Barbone FP, Kashyap AK,
Farrell FX, Jolliffe LK, Barrett RW, Dower WJ. Increased
potency of an erythropoietin peptide mimetic through covalent
dimerization. Nat Biotechnol 1997;15:1261–5.
[22] Denmeade SR, Sokoll LJ, Dalrymple S, Rosen DM, Gady AM,
Bruzek D, Ricklis RM, Isaacs JT. Dissociation between androgen
responsiveness for malignant growth vs. expression of prostate
specific differentiation markers PSA, hK2, and PSMA in human
prostate cancer models. Prostate 2003;54:249–57.
[23] Karlsson R. SPR for molecular interaction analysis: a
review of emerging application areas. J Mol Recognit 2004;17:
151–61.
[24] Fivash M, Towler EM, Fisher RJ. BIACORE for macromole-
cular interaction. Curr Opin Biotechnol 1998;9:97–101.
[25] Campagnolo C, Meyers KJ, Ryan T, Atkinson RC, Chen YT,
Scanlan MJ, Ritter G, Old LJ, Batt CA. Real-time, label-free
monitoring of tumor antigen and serum antibody interactions.
J Biochem Biophys Methods 2004;61:283–98.
[26] Malmqvist M. Surface plasmon resonance for detection and
measurement of antibody–antigen affinity and kinetics. Curr Opin
Immunol 1993;5:282–6.
[27] van der Merwe PA, Barclay AN. Analysis of cell-adhesion
molecule interactions using surface plasmon resonance. Curr
Opin Immunol 1996;8:257–61.
[28] O’Shannessy DJ. Determination of kinetic rate and equilibrium
binding constants for macromolecular interactions: a critique of
the surface plasmon resonance literature. Curr Opin Biotechnol
1994;5:65–71.
[29] Mozsolits H, Aguilar MI. Surface plasmon resonance spectro-
scopy: an emerging tool for the study of peptide–membrane
interactions. Biopolymers 2002;66:3–18.
[30] Quinn JG, O’Neill S, Doyle A, McAtamney C, Diamond D,
MacCraith BD, O’Kennedy R. Development and appli-
cation of surface plasmon resonance-based biosensors for the
detection of cell–ligand interactions. Anal Biochem 2000;281:
135–43.
Related Documents











