3-Fluorotyrosine as a Complementary Probe of Hemoglobin Structure and Dynamics: A 19 F-NMR Study of Synechococcus sp. PCC 7002 GlbN by Matthew P. Pond, Belinda B. Wenke, Matthew R. Preimesberger, Selena L. Rice, and Juliette T. J. Lecomte* T.C. Jenkins Department of Biophysics, Johns Hopkins University, 3400 N. Charles St., Baltimore, MD 21218, USA (phone: þ 1-410-516-7019; fax: þ 1-410-516-4118; email: [email protected]) The hemoglobin from the cyanobacterium Synechococcus sp. PCC 7002 (GlbN) contains three tyrosines (Tyr5, Tyr22, and Tyr53), each of which undergoes a structural rearrangement when the protein binds an exogenous ligand such as cyanide. We explored the use of 3-fluorotyrosine and 19 F-NMR spectroscopy for the characterization of GlbN. Assignment of 19 F resonances in fluorinated GlbN (GlbN*) was achieved with individual Tyr5Phe and Tyr53Phe replacements. We observed marked variations in chemical shift and linewidth reflecting the dependence of structural and dynamic properties on oxidation state, ligation state, and covalent attachment of the heme group. The isoelectronic complexes of ferric GlbN* with cyanide and ferrous GlbN* with carbon monoxide gave contrasting spectra, the latter exhibiting heterogeneity and enhanced internal motions on a microsecond-to- millisecond time scale. The strength of the H-bond network involving Tyr22 (B10) and bound cyanide was tested at high pH. 3-Fluorotyrosine at position 22 had a pK a value at least 3 units higher than its intrinsic value, 8.5. In addition, evidence was found for long-range communication among the tyrosine sites. These observations demonstrated the utility of the 3-fluorotyrosine approach to gain insight in hemoglobin properties. Introduction. – Heme proteins offer special challenges in the elucidation of structure activity relationships. Globins, though extensively studied, are no exception in that respect. They possess the remarkable ability to bind O 2 reversibly, but they also can use O 2 in reactions such as NO dioxygenation [1]. Hemoglobins, like other heme proteins, achieve stereoelectronic control and balance among competing processes with residues near the heme group and at distant locations. Direct and remote interactions adjust ligand on and off rates, electron transfer rates, and other key features conditioning the reactivity of the heme group. The proteins from the 2/2 (truncated) lineage of the hemoglobin superfamily [2] are among the many non-vertebrate hemoglobins for which function and chemical properties remain largely unknown. We focus here on the 2/2 hemoglobin from the oxygenic cyanobacterium Synechococcus sp. PCC 7002 (GlbN), a protein thought to detoxify reactive oxygen/nitrogen species [ 3] . GlbN exhibits bis-histidine coordination of the heme iron in both the ferric and ferrous states [4] . His46 (distal ligand) is readily displaced by small exogenous ligands, whereas His70 (proximal ligand) remains bound. Under certain in vivo conditions, GlbN forms an irreversible covalent heme protein cross-link using His117 [ 3] . In vitro, GlbN can be prepared with and without the cross- link [5]. Bis-histidine coordination and post-translational modification are two CHEMISTRY & BIODIVERSITY – Vol. 9 (2012) 1703 # 2012 Verlag Helvetica Chimica Acta AG, Zɒrich

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

3-Fluorotyrosine as a Complementary Probe of Hemoglobin Structure andDynamics: A 19F-NMR Study of Synechococcus sp. PCC 7002 GlbN
by Matthew P. Pond, Belinda B. Wenke, Matthew R. Preimesberger, Selena L. Rice, andJuliette T. J. Lecomte*
T. C. Jenkins Department of Biophysics, Johns Hopkins University, 3400 N. Charles St., Baltimore,MD 21218, USA (phone: þ1-410-516-7019; fax: þ1-410-516-4118; email: [email protected])
The hemoglobin from the cyanobacterium Synechococcus sp. PCC 7002 (GlbN) contains threetyrosines (Tyr5, Tyr22, and Tyr53), each of which undergoes a structural rearrangement when the proteinbinds an exogenous ligand such as cyanide. We explored the use of 3-fluorotyrosine and 19F-NMRspectroscopy for the characterization of GlbN. Assignment of 19F resonances in fluorinated GlbN(GlbN*) was achieved with individual Tyr5Phe and Tyr53Phe replacements. We observed markedvariations in chemical shift and linewidth reflecting the dependence of structural and dynamic propertieson oxidation state, ligation state, and covalent attachment of the heme group. The isoelectroniccomplexes of ferric GlbN* with cyanide and ferrous GlbN* with carbon monoxide gave contrastingspectra, the latter exhibiting heterogeneity and enhanced internal motions on a microsecond-to-millisecond time scale. The strength of the H-bond network involving Tyr22 (B10) and bound cyanidewas tested at high pH. 3-Fluorotyrosine at position 22 had a pKa value at least 3 units higher than itsintrinsic value, 8.5. In addition, evidence was found for long-range communication among the tyrosinesites. These observations demonstrated the utility of the 3-fluorotyrosine approach to gain insight inhemoglobin properties.
Introduction. – Heme proteins offer special challenges in the elucidation ofstructure�activity relationships. Globins, though extensively studied, are no exceptionin that respect. They possess the remarkable ability to bind O2 reversibly, but they alsocan use O2 in reactions such as NO dioxygenation [1]. Hemoglobins, like other hemeproteins, achieve stereoelectronic control and balance among competing processes withresidues near the heme group and at distant locations. Direct and remote interactionsadjust ligand on and off rates, electron transfer rates, and other key featuresconditioning the reactivity of the heme group.
The proteins from the 2/2 (truncated) lineage of the hemoglobin superfamily [2] areamong the many non-vertebrate hemoglobins for which function and chemicalproperties remain largely unknown. We focus here on the 2/2 hemoglobin from theoxygenic cyanobacterium Synechococcus sp. PCC 7002 (GlbN), a protein thought todetoxify reactive oxygen/nitrogen species [3]. GlbN exhibits bis-histidine coordinationof the heme iron in both the ferric and ferrous states [4]. His46 (distal ligand) is readilydisplaced by small exogenous ligands, whereas His70 (proximal ligand) remains bound.Under certain in vivo conditions, GlbN forms an irreversible covalent heme�proteincross-link using His117 [3]. In vitro, GlbN can be prepared with and without the cross-link [5]. Bis-histidine coordination and post-translational modification are two
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012) 1703
� 2012 Verlag Helvetica Chimica Acta AG, Z�rich

characteristics shared by Synechocystis sp. PCC 6803 (S6803 GlbN hereafter) [5] [6]and illustrated in Fig. 1, a. The structure of the heme modification is shown in Fig. 1,b.
Fig. 1. Stereoviews of ferric GlbN-A from Synechocystis sp. PCC 6803 in the a) bis-histidine state (1RTX[7]) and c) cyanide-bound state (1S69 [8]). The three tyrosines of Synechococcus sp. PCC 7002 GlbN(Y5, Y22, and Y53) are shown. b) The modified heme of GlbN-A with atom numbering used in the text.
GlbN-R contains a b heme (vinyl group at position 2).
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012)1704

Fig. 1, c, shows the conformational change caused by the displacement of the distalhistidine (His46) by an exogenous ligand (cyanide in this case). Fig. 1,a and c,highlights the three tyrosines of GlbN. In the bis-histidine complex, Tyr5 does not makestable contact with the rest of the protein; however, on binding cyanide, it becomesassociated with the EF corner. Tyr53 is also affected by cyanide binding, sliding awayfrom the heme periphery, as the E helix rotates to liberate His46 from the heme distalaxial position. More importantly, Tyr22 (at topological position referred to as B10 byanalogy to the vertebrate globins) reorients to form a H-bond to the bound cyanide. Atyrosine at position B10 is of special interest because it is highly conserved in the closerelatives of GlbN [9]. TyrB10, along with a glutamine in the E helix, establishes anetwork of H-bonds [10]. In various hemoglobins, the residue occupying position B10has attracted attention for its influence on ligand access and (de)stabilization [10a] [11].
GlbN offers an excellent opportunity to characterize the impact of ligand bindingand other alterations by using 3-fluorotyrosine (fY) and 19F-NMR spectroscopy. The19F nucleus (spin 1�2) occurs at 100% natural abundance; it has a high g ratio and asensitivity of 0.83 relative to 1H; it is not naturally found in amino acids and is asensitive probe of local environment. In addition, substitution at Ce1 of tyrosineincreases the acidity of the OH group, from pKa of 10.1 to 8.5 [12]. This thermodynamicperturbation can serve as a probe of the interactions of TyrB10.
The F-atom is neither a good H-bond acceptor [13] nor a good halogen bond donor[14]. The replacement of a 1H with 19F on a tyrosine ring is generally thought tointroduce a minimal perturbation [15]. However, there is evidence that multiple fYreplacements in the same polypeptide chain can affect stability [15], structure [16], anddynamics [17], cautioning that even single fY replacements may alter a protein.Because of the clear advantages of the 19F nucleus, 19F-NMR spectroscopy has beenapplied to proteins containing intrinsic 19F labels for many years [18]. Recentapplications and efforts have included in-cell experiments [19], the measurement ofsolvent and oxygen accessibility [20], and the development of reliable assignmentmethods [21].
In this report, we describe the incorporation of fY into GlbN and the analysis ofone-dimensional 19F-NMR spectra. The study allowed the continued investigation ofthe differences between distinct forms of GlbN. Long-range interactions and dynamicperturbations otherwise not readily detectable are discussed and demonstrate theutility of 19F as a spectroscopic probe of hemoglobins.
Results and Discussion.– 1. Relevant Forms of GlbN. In all, data were collected onmore than 20 versions of GlbN. For clarity, we adopted a nomenclature by which thevarious forms are described with the oxidation state of the iron (i.e., FeII or FeIII) , theiron axial ligands (e.g., His�His or His�X, where the left-hand His refers to theproximal His70, the right-hand His to the distal His46, and �X� to a generic non-histidine ligand), the amino acid sequence as coded by the glbN gene (i.e., WT, Y5F, orY53F), the post-translational status of the holoprotein (i.e., -R for the b heme or -A forthe covalently attached heme), and labeling with 3-fluorotyrosine (*). For example, fY-labeled Y5F GlbN in the reduced state with post-translational modification andhistidines as axial ligands is denoted as His�FeII�His Y5F GlbN*-A. As will beexplained below, GlbN* samples contained both labeled and unlabeled proteins. The
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012) 1705

FeIII complexes were paramagnetic with one unpaired electron (S¼1/2), whereas theFeII complexes were diamagnetic (S¼0). The Table lists the six forms of GlbNdiscussed in most detail below.
2. NMR Properties of GlbN Tyrosines. Analysis of 1H and heteronuclear (13C, 15N)NMR data provides information about the three tyrosines of GlbN in FeIII species andthe His�FeII�CO complex [3] [22], and constructs a framework in which GlbN* 19Fdata can be interpreted. The aromatic rings flip freely about their Cg�Cz axis on the 1Hchemical shift time scale, as tends to be the norm for tyrosines [23]. Tyr5, located at thebeginning of the A helix, has no long-range NOE in the His�FeIII�His states (GlbN-Rand GlbN-A), but, in the His�X states, it is in dipolar contact with the end of the Ehelix and the AB turn, specifically with Gly57 and Gly10. Tyr22 undergoes a rotamericstate change from c1 trans to gauche, when an exogenous ligand binds to the heme. Theproximity of OhH to the heme iron is demonstrated by the large downfield hyperfineshift detected in His�FeIII�CN complexes [22b]. Formation of an H-bond is supportedby solvent isotope effects on the heme electronic structure, slow exchange of the OH H-atom with solvent [22b], and a structural reorganization similar to that of S6803 GlbNupon ligand binding [22c] [8]. Finally, Tyr53 in His�FeIII�His GlbN-A has NOEs to theheme group (1-Me) and His117. These NOEs are not detected in His�FeIII�His GlbN-R nor the His�FeIII�CN complexes.
3. Incorporation of fY into GlbN. Fig. 2 shows the downfield-shifted region of1H-NMR spectra of WT GlbN-R (Fig. 2, a), WT GlbN*-R (Fig. 2, b), and Y53FGlbN*-R (Fig. 2,c) in the His�FeIII�His state. Comparison of Fig. 2, a and b, revealedadditional peaks in the WT GlbN* spectrum that disappeared in the Y53F GlbN*spectrum (Fig. 2, c). The additional peaks were also observed in the spectrum of Y5FGlbN* (not shown) and, therefore, attributed to the influence of fY53 on the hemegroup, in agreement with the location of the residue (Fig. 1, a).
Assuming that there was no alteration of the signals due to interactions among the5, 22, and 53 sites in His�FeIII�His GlbN*-R, integration of peaks a and c in Fig. 2,b,returned an estimate of 20% incorporation of fY53 in GlbN*. This representedcontributions from one singly labeled form (fY53), two doubly labeled forms (fY5/fY53 and fY22/fY53), and one triply labeled form (fY5/fY22/fY53). Mass spectro-metric analysis revealed that ca. 63% of the protein was unlabeled, 20% carried one 19F,8% carried two, and 9% carried three. If uniform labeling at each site was assumed,these numbers corresponded to ca. 20% labeling at position 53, in agreement with the1H-NMR result. GlbN* from a different preparation was analyzed in the same fashionand yielded ca. 26% incorporation at position 53 (NMR) and 46% unlabeled(monoisotopic mass of 13718.0 Da), 27% with one 19F (þ18 Da), 14% with two(þ 36 Da), 12% with three (þ 54 Da), amounting to ca. 30% at position 53 (MS), and
Table. Main Forms of GlbN/GlbN* Discussed in the Text
His�His GlbN/GlbN* His�X GlbN/GlbN*
His�FeIII�His GlbN/*-R His�FeIII�CN GlbN/*-RHis�FeIII�His GlbN/*-A His�FeIII�CN GlbN/*-AHis�FeII�His GlbN/*-A His�FeII�CO GlbN/*-A
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012)1706

within error of the NMR estimate. The results confirmed that different growthsproduced varying amounts of labeled material, with the singly fluorinated species beingthe most abundant.
Although low incorporation precluded characterization with experimental methodsnecessitating homogeneous samples (e.g., optical spectroscopy for ligand binding), theinvolvement of tyrosines in GlbN conformational changes suggested that it was prudentto inspect the properties of mixtures of proteins carrying zero, one, two, or three labels.An advantage of mixtures is that their 19F spectra could reveal if individual labels orcombinations of them perturbed the structure of WT GlbN. The high sensitivity of the19F-NMR experiment allowed us to use 19F preparations below optimal level [17], albeitwith long acquisition times (h).
4. 19F-NMR Spectra of His�His GlbN*. Fig. 3 illustrates the effect of heme cross-linking and iron oxidation state on the 19F spectra of His�His WT, Y53F, and Y5FGlbN*: a –c are for His�FeIII�His GlbN*-R; d – f for His�FeIII�His GlbN*-A; and g – ifor His�FeII�His GlbN*-A. In all cases, it was straightforward to assign the WT 19Fsignals with the variant data. The Y53F replacement had a negligible effect on chemicalshift; in contrast, the Y5F replacement shifted the fY53 line in GlbN*-A (Fig. 3, f and i)in a hint of long-range 5 –53 interactions in the presence of the cross-link. Because Tyr5has few stable contacts with the rest of the protein in the His�His state, the effectsuggested a subtle rearrangement and the transmission of the perturbation.
A noteworthy feature of the GlbN*-R spectra 3,a and c, is the doubling of the fY53signal (labels 53M and 53m for major and minor species, resp.). This was attributed totwo holoprotein forms differing by a 1808 rotation of the heme about its a– g meso axis(heme orientational disorder [24]). The minor species amounts to ca. 8% of the WTprotein [4], a proportion also obtained from integration of 1H peaks a and b in Fig. 2,a
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012) 1707
Fig. 2. Downfield-shifted portion of the 1H-NMR spectrum of His�FeIII�His of a) WT GlbN-R, b) WTGlbN*-R, and c) Y53F GlbN*-R. Peaks labels: a, heme 5-Me, major orientational isomer, GlbN; b, heme8-Me, minor orientational isomer, GlbN; c, heme 5-Me, major orientational isomer, GlbN*. Data
recorded at 258 and pH 7.3.

and b. Likewise, integration of the 19F signals in Fig. 3, a, provided a ratio of ca. 10%,within error of the measurement. This indicated that fY53 was sensitive to hemedisorder in accordance with its location near the heme 1-Me (Fig.1,a), but did notaffect the relative stability of the two isomers.
In vitro, the heme�protein cross-link is formed by reduction of the heme iron inHis�FeIII�His GlbN-R [25]. Reaction of the minor isomer results in His117 addition tothe heme 4-vinyl; however, this modification occurs more slowly than hemereorientation, followed by reaction at the 2-vinyl [26]. Thus, nearly homogeneousGlbN-A samples are generally obtained. Large amounts of the alternative post-translational modification were not detected in Fig. 3, d– i.
Comparison of Fig. 3,a and d, revealed that fY53 was the most affected by the post-translational modification. This was not surprising, as the ring is located near His117and susceptible to structural as well as electronic (pseudocontact) perturbations.Interestingly, the signals of fY22 and fY53 were split in WT GlbN*-A (Fig. 3, d). The
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012)1708
Fig. 3. 19F-NMR Spectra of fY in His�His GlbN*. a) WT FeIII GlbN*-R; b) Y53F FeIII GlbN*-R; c) Y5FFeIII GlbN*-R; d) WT FeIII GlbN*-A; e) Y53F FeIII GlbN*-A; f) Y5F FeIII GlbN*-A; g) WT FeII GlbN*-
A; h) Y53F FeII GlbN*-A; i) Y5F FeII GlbN*-A. Data recorded at 258 and pH 7.3.

origin of this doubling was attributed to two different ring rotamers (c2 angle) and areduced ring flipping rate caused by specific interactions of the asymmetric fY ring withthe rest of the protein (see below). The finding that fY22 splitting vanished in the Y53Fvariant provided another indication of long-range communication among the sites.
Because of the rapid conversion of GlbN-R to GlbN-A upon iron reduction [26], itwas not possible to obtain His�FeII�His GlbN*-R data, and only the 19F spectra ofHis�FeII�His GlbN*-A are shown in Fig. 3, g – i. In this state as well, the Y5Freplacement induced a larger chemical-shift perturbation than the Y53F replacement.The striking feature of the reduced state, however, was broad linewidth. In WT GlbN*,the values changed from ca. 35 Hz (Fig. 3, a) to ca. 90 Hz (Fig. 3,g) on reduction (andelimination of the unpaired spin contribution to relaxation, as the heme iron remainshexacoordinated). Rigorous analysis of 19F T2 relaxation [27] is outside the scope of thisreport; nevertheless, the excess linewidth suggested that internal motions weredifferentially affected by the oxidation state of the iron. In a parallel dynamics study ofthe His�FeII�His state, 15N relaxation data indicated that backbone motions wereenhanced on the ms– ms time scale at the sites of the three tyrosines [22c]. The sidechain 19F data provided additional evidence for this consequence of iron reduction.
5. 19F-NMR Spectra of His�CN GlbN*. Fig. 4 presents the 19F spectra ofHis�FeIII�CN GlbN*-R (Fig. 4, a –c) and GlbN*-A (Fig. 4,d – f). Again, the strongestsignals in the WT spectra were readily assigned with the aid of the two variants. In
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012) 1709
Fig. 4. 19F-NMR Spectra of fY in His�FeIII�CN GlbN*. a) WT GlbN*-R; b) Y53F GlbN*-R; c) Y5FGlbN*-R; d) WT GlbN*-A; e) Y53F GlbN*-A; f) Y5F GlbN*-A. Data recorded at 258 and pH 7.3. Line
broadening was applied to spectra d (5 Hz), and e and f (10 Hz).
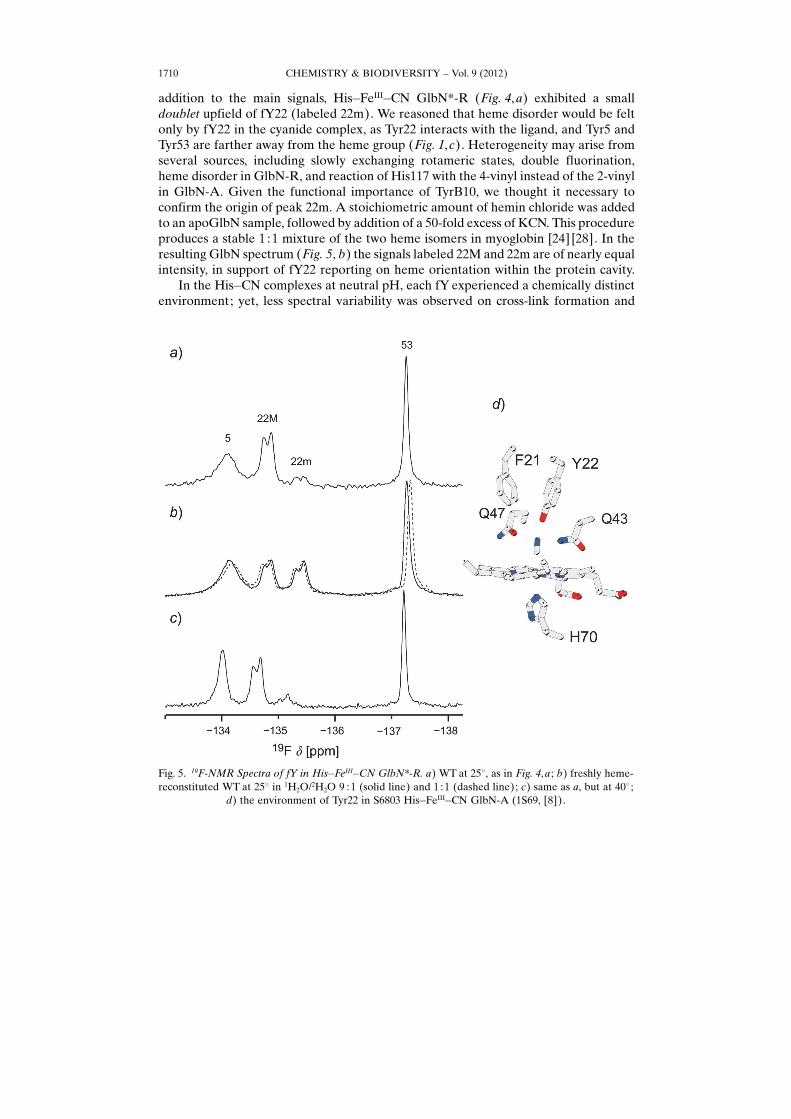
addition to the main signals, His�FeIII�CN GlbN*-R (Fig. 4, a) exhibited a smalldoublet upfield of fY22 (labeled 22m). We reasoned that heme disorder would be feltonly by fY22 in the cyanide complex, as Tyr22 interacts with the ligand, and Tyr5 andTyr53 are farther away from the heme group (Fig. 1,c). Heterogeneity may arise fromseveral sources, including slowly exchanging rotameric states, double fluorination,heme disorder in GlbN-R, and reaction of His117 with the 4-vinyl instead of the 2-vinylin GlbN-A. Given the functional importance of TyrB10, we thought it necessary toconfirm the origin of peak 22m. A stoichiometric amount of hemin chloride was addedto an apoGlbN sample, followed by addition of a 50-fold excess of KCN. This procedureproduces a stable 1 :1 mixture of the two heme isomers in myoglobin [24] [28]. In theresulting GlbN spectrum (Fig. 5, b) the signals labeled 22M and 22m are of nearly equalintensity, in support of fY22 reporting on heme orientation within the protein cavity.
In the His�CN complexes at neutral pH, each fY experienced a chemically distinctenvironment; yet, less spectral variability was observed on cross-link formation and
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012)1710
Fig. 5. 19F-NMR Spectra of fY in His�FeIII�CN GlbN*-R. a) WT at 258, as in Fig. 4, a ; b) freshly heme-reconstituted WT at 258 in 1H2O/2H2O 9 : 1 (solid line) and 1 : 1 (dashed line); c) same as a, but at 408 ;
d) the environment of Tyr22 in S6803 His�FeIII�CN GlbN-A (1S69, [8]).

Phe replacement than in the His�His complex. The chemical shift of fY53 (close to freefY, �136.6 ppm [29]) in the His�CN complexes suggested an exposed residue. 19F-NMR Spectra are sensitive to solvent composition and display upfield shifts of up to0.22 ppm in 2H2O compared to 1H2O [30]. Addition of 2H2O to 50% v/v gave thedashed-line spectrum in Fig. 5, b. The signal from fY53 shifted by ca. 0.07 ppm, inagreement with substantial solvent exposure. Interestingly, both minor and major formsof fY22 exhibited small shifts in the opposite direction. We will return to these shifts inthe next section.
In the cyanide complexes, the ring of Tyr5 inserts itself between the AB turn and theEF loop (Fig. 1, c). Unlike the 1H ring signals of the non-fluorinated Tyr, the 19F linewas broad and indicative of conformational exchange. Raising the temperature to 408(Fig. 5,c) sharpened the resonance, as the exchange rate increased toward the fastregime on the chemical-shift time scale. We also note that this increase in temperaturewas not sufficient to average the split signal of fY22, whereas the Y5F replacementcollapsed it (Fig. 4, c and f).
The origin of the three distinct line shapes (one broad, one split, one sharp) wasintriguing. The X-ray structures of tetradeca-(3-fluorotyrosyl)-glutathione S-trans-ferase [16] and (3-fluorotyrosyl)-manganese superoxide dismutase [31] contain amajority of fYs adopting a unique ring conformation (c2 angle). Rotameric hetero-geneity and rate of fY or (3-fluorophenyl)alanine (fF) ring flipping are discussed onlyrarely [21] [32], but it is possible that this particular type of internal motion contributedto GlbN*�s spectral appearance. In the case of fY53, the available data did not allow usto distinguish population of only one rotameric state from fast exchange (on 19Fchemical-shift time scale) between two such states. For fY5, broadening could arisefrom slow ring flipping or from some other source of slow motion (or both). Tyr22 isburied in the distal pocket (Fig. 5, d), and it is feasible that this restricted environmentboth equalized the standard free energy of two fY c2 rotamers and generated a highbarrier to ring flipping. The small difference in chemical shift between the two fY22peaks was consistent with calculated contributions from ring current shifts andpseudocontact shift at the He1 and He2 positions of Tyr22.
The related Chlamydomonas eugametos hemoglobin (1DLY [10b]) has a Phe at thelocation of GlbN�s Tyr5. This Phe side chain, along with those of TyrB10, PheB9, andmost other pocket residues, is also superimposable with the corresponding side chainsof S6803 GlbN and Mycobacterium tuberculosis trHbN (1RTE [33]), illustrating a well-defined and well-conserved region of the structure. Thus, a lowering of the rotationbarrier in Y5F GlbN* could not readily be rationalized. Alternatively, the effect of Y5Fon fY22 could be on rotamer selection or chemical shift rather than interconversionrate. The latter explanation seemed more likely because of the energies involved andthe location of fY22 near the heme. In His�FeIII�His GlbN*-A (Fig. 3,d), theappearance of fY22 and fY53 could also be linked to the ability to detect c2 fYrotameric states, although other slowly exchanging conformations could not be ruledout. In all cases, the data demonstrated the sensitivity of 19F signals to subtle dy-namic and thermodynamic perturbations caused by labeling and amino acid replace-ments.
6. pH Response of His�CN GlbN*-A. As a probe of H-bonding and conformationalproperties, a coarse pH titration of His�FeIII�CN WT GlbN*-A was performed
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012) 1711

(Fig. 6). As the pH was raised from 7.3 to 11.4, each of the three fYs exhibited a distinctresponse. The fY5 signal sharpened and moved upfield by more than 2 ppm with anapparent midpoint pH of ca. 9.3. The two components of fY22 shifted little, but mergedat high pH, possibly as a response to the titration of Y5. The fY53 signal moved upfieldby ca. 0.8 ppm with an apparent midpoint pH of ca. 8.9. This change in chemical shiftwas typical of the titration of a solvent-exposed fY [30] [34]. Thus, the response of fY53was consistent with a transition to phenolate at high pH; the behavior of fY5 likelyreflected deprotonation coupled with a conformational rearrangement.
fY22 remained protonated up to the highest pH despite the low intrinsic pKa of theunnatural residue. 1H-NMR Data did not indicate cyanide release nor reformation ofthe His�His complex (not shown). The high pKa value reflected the energetic cost ofplacing the phenolate in the distal pocket and the stability of the His�CN complex.Returning to Fig. 5,b, the downfield shift of the fY22 signals (dashed-line) matchedthat expected of an increase in pH by ca. 0.2 unit according to Fig. 6, b. This pHdifference was consistent with the effect of 2H2O on measured pH [35], leading to theconclusion that, despite the H-bond to cyanide, no net solvent isotope shift wasobserved in the 1H2O/2H2O mixture. The pH shift cannot account for the solventisotope effect on fY53.
7. 19F-NMR Spectra of His�CO GlbN*-A. Fig. 7 illustrates the 19F spectra ofHis�FeII�CO GlbN*. As for the His�FeII�His complex, it is practically difficult toobtain GlbN-R data because of cross-link formation in the reduced state, and onlyGlbN*-A data are shown. A comparison of the WT GlbN*-A spectrum (Fig. 7, a) withthat of the isoelectronic cyanide complex (Fig. 4, a) emphasized the surprisingcomplexity of the CO spectra. Attempts to assign the resonances using the variants
Fig. 6. pH Titration of His�FeIII�CN GlbN*-A. a) 19F-NMR Spectra; b) plot of chemical shift vs. pH. Thedotted lines are meant to guide the eye.
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012)1712
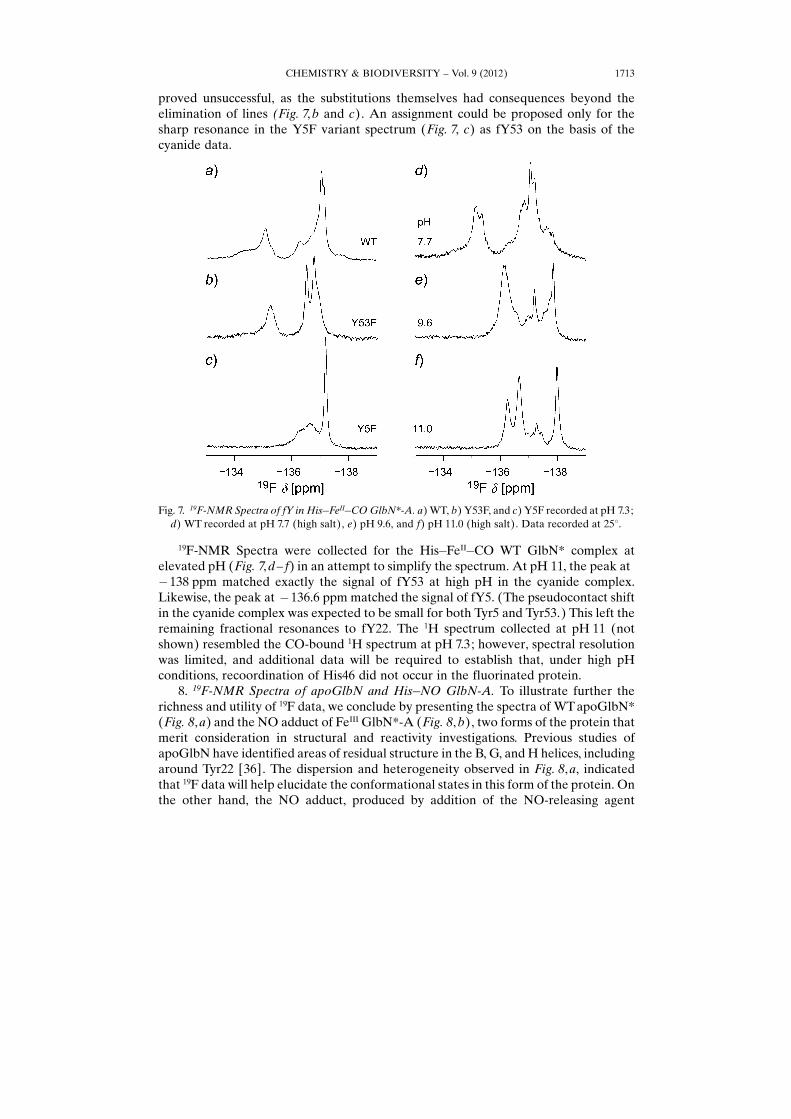
proved unsuccessful, as the substitutions themselves had consequences beyond theelimination of lines (Fig. 7, b and c). An assignment could be proposed only for thesharp resonance in the Y5F variant spectrum (Fig. 7, c) as fY53 on the basis of thecyanide data.
19F-NMR Spectra were collected for the His�FeII�CO WT GlbN* complex atelevated pH (Fig. 7, d– f) in an attempt to simplify the spectrum. At pH 11, the peak at�138 ppm matched exactly the signal of fY53 at high pH in the cyanide complex.Likewise, the peak at �136.6 ppm matched the signal of fY5. (The pseudocontact shiftin the cyanide complex was expected to be small for both Tyr5 and Tyr53.) This left theremaining fractional resonances to fY22. The 1H spectrum collected at pH 11 (notshown) resembled the CO-bound 1H spectrum at pH 7.3; however, spectral resolutionwas limited, and additional data will be required to establish that, under high pHconditions, recoordination of His46 did not occur in the fluorinated protein.
8. 19F-NMR Spectra of apoGlbN and His�NO GlbN-A. To illustrate further therichness and utility of 19F data, we conclude by presenting the spectra of WT apoGlbN*(Fig. 8,a) and the NO adduct of FeIII GlbN*-A (Fig. 8, b), two forms of the protein thatmerit consideration in structural and reactivity investigations. Previous studies ofapoGlbN have identified areas of residual structure in the B, G, and H helices, includingaround Tyr22 [36]. The dispersion and heterogeneity observed in Fig. 8,a, indicatedthat 19F data will help elucidate the conformational states in this form of the protein. Onthe other hand, the NO adduct, produced by addition of the NO-releasing agent
Fig. 7. 19F-NMR Spectra of fY in His�FeII�CO GlbN*-A. a) WT, b) Y53F, and c) Y5F recorded at pH 7.3;d) WT recorded at pH 7.7 (high salt), e) pH 9.6, and f) pH 11.0 (high salt). Data recorded at 258.
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012) 1713

DPTA-NONOate to His�FeIII�His GlbN*-A, resulted in 1H and 19F spectra reminis-cent of the diamagnetic His�FeII�CO GlbN*-A. Studies are in progress to characterizethe nature of the nitrosyl adduct.
Conclusions. – We showed that 19F-NMR spectroscopy of fY-substituted GlbN is aninexpensive and readily implemented avenue to gain information at key positions in thestructure. We found that the Y5F substitution had subtle effects on the spectralproperties of fY22 and fY53. Such long-range communications among the three sitesvalidated the use of samples with a high proportion of single-site fY incorporation. Oneof the aims of the study was to examine the strength of the H-bond network involvingTyr22. We found that the pKa value of fY22 was higher than 11.5 in the cyanidecomplex. Taking the pKa value of fY as 8.5, this implied a shift of at least 3 units and astabilization greater than 17 kJ/mol. In addition, the FeII forms of GlbN* yieldedbroader lines or heterogeneous spectra, which was in line with expectations based onthe 1H and 15N linewidth data on GlbN. The results revealed specific responses to thenature of the distal ligand and other perturbations, and provided a complementary viewof GlbN dynamics. However, they also cautioned that the introduction of a 19F labelmay perturb delicate equilibria or favor states not accessible in the unlabeled protein.Extension of some of the observations from GlbN* to GlbN will require additionalexperiments.
This work was supported by grant MCB-0349409 of the National Science Foundation, USA. Mass-spectrometric data were collected at the Pacific Northwest National Laboratory (thanks to Drs. Bigelowand Kelly) and at the University of Maryland Baltimore County (thanks to Dr. Wilhide). The authors alsothank Dr. Ananya Majumdar for assistance in NMR data collection, and Dr. Christopher Falzone foruseful discussions and reading of the manuscript.
Fig. 8. 19F-NMR Spectra of fY in a) WT apoGlbN* and b) WT His�FeIII�His GlbN*-A to which NO wasadded. Data recorded at 258.
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012)1714

Experimental Part
General. Cell lysis: French Pressure Cell (Slm-Aminco, Rochester, NY). UV/VIS Spectra: Cary 50Bio (Agilent Technologies, Santa Clara, CA). NMR Spectra: Varian Inova spectrometer operating at a 1Hresonance frequency of 499.91 MHz (19F resonating at 470.31 MHz) equipped with a tunable broadbandprobe (Agilent Technologies, Santa Clara, CA). Abbreviations: DPTA-NONOate, (Z)-1-[N-(3-amino-propyl)-N-(3-ammoniopropyl)amino]diazen-1-ium-1,2-diolate; fY, 3-fluoro-l-tyrosine; S6803, Synecho-cystis sp. PCC 6803.
Materials. Molecular biology: mutagenesis with QuikChange (Qiagen, Valencia, CA), primers fromIntegrated DNA Technologies (Coralville, IA), plasmid purification with Quiaprep Spin Miniprep Kit(Qiagen), sequencing by GENEWIZ, Inc. (South Plainfield, NJ), E. coli BL21(DE3) cells from NewEngland Biolabs (Ipswich, MA), 3-fluoro-l-tyrosine (Astatech, Inc., Bristol, PA) purity checked byNMR, ampicillin (Sigma). Chemistry: bovine hemin chloride (FeIII-protoporphyrin IX, Sigma), carbonmonoxide (CP grade, Airgas), DPTA-NONOate (Cayman Chemical, Ann Arbor, MI), glucose oxidase(Sigma), catalase (Sigma), KCN (J. T. Baker), Na2S2O4 (Sigma).
Protein Expression and Purification. Site-directed mutagenesis was performed on the parent plasmidto substitute Tyr5 and Tyr53 for Phe following the QuikChange protocol provided by the manufacturer.Plasmid preparation of the glbN gene was carried out as discussed in [4].
GlbN*-Containing ferric heme was produced as described for GlbN in [4] with only slightmodifications to achieve fY incorporation. Specifically, a 60-ml starter culture was incubated in M9minimal medium to an OD600 of ca. 2 prior to transfer into 4�500 ml volumes of identical medium. Theculture was further incubated until an OD600 of 0.3 was reached, at which time 3-fluoro-l-tyrosine wasadded to a final concentration of 18 mg/l. We note that the expression system does not require induction[4], and that the cells were sensitive to this level of 3-fluoro-l-tyrosine. The cultures were allowed toreach a final OD600 of ca. 1.5 before harvesting. To raise incorporation levels, the growth medium can besupplemented with tryptophan and phenylalanine to repress the shikimate pathway; glyphosate can alsobe added to inhibit it [15]. Future studies will determine whether these approaches are advantageous forGlbN* production.
Lysis was performed by two rounds of four cycles by French Pressure Cell, and four wash steps wereperformed, but purification was otherwise identical to previous descriptions. The concentration of GlbN*was estimated by UV/VIS spectrophotometry using the unmodified ferric GlbN-R extinction coefficient(e411 nm 96 mm
�1 cm�1 [4]). GlbN* was concentrated to ca. 15 ml and exchanged into a low-concentrationphosphate buffer (ca. 100 mm, pH 7.2) prior to lyophilization. Typical yields were ca. 15 mg/l of growthmedium.
NMR Sample Preparation. Lyophilized GlbN* samples were resuspended in 100 or 250 mm
phosphate buffer containing 10% 2H2O. Protein concentrations varied between 1.5 and 3.5 mm GlbN*(see text for 19F incorporation percentages). If required, iron reduction was carried out by adding a five-fold excess of Na2S2O4 to GlbN* samples under microoxic conditions. His�FeIII�CN GlbN* wasproduced by adding 50-fold excess of KCN to His�FeIII�His GlbN*, and His�FeII�CO GlbN* wasproduced by gently bubbling CO in a soln. of reduced GlbN*-A. The NO adduct was prepared byaddition of DPTA-NONOate to a soln. of His�FeIII�His GlbN*-A in the presence of the glucose oxidase/d-glucose/catalase enzymatic O2 elimination system [26] [37]. The addition was such that 6�NO wouldbe produced over the course of the experiment. The soln. was allowed to incubate for several h beforeNMR data collection.
Mass Spectrometry. MS analysis of desalted GlbN was performed at the Pacific Northwest NationalLaboratory with a Bruker 12T SolariX FTICR mass spectrometer and electrospray emitters custom madeusing 150 mm o.d. �20 mm i.d. chemically etched fused silica [38], and at the University of MarylandBaltimore County with a Bruker 12T FT-ICR Apex IV qQ mass spectrometer.
NMR Spectroscopy. 19F-NMR Spectra were collected at pH 7.3 and r.t. unless otherwise specified.Data were referenced to a fY standard in 2H2O by setting the resonance frequency of the peak to�136.6 ppm with respect to CFCl3 [29]. Spectra were acquired for 1 to 5 h with 1200 complex points, aspectral width of 6 kHz, and an inter-scan delay of 1 s. Data were processed with a cosine-squared bellwindow function and zero filled prior to Fourier transform.
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012) 1715

REFERENCES
[1] P. R. Gardner, J. Inorg. Biochem. 2005, 99, 247.[2] J. B. Wittenberg, M. Bolognesi, B. A. Wittenberg, M. Guertin, J. Biol. Chem. 2002, 277, 871; S. N.
Vinogradov, L. Moens, J. Biol. Chem. 2008, 283, 8773.[3] N. L. Scott, Y. Xu, G. Shen, D. A. Vuletich, C. J. Falzone, Z. Li, M. Ludwig, M. P. Pond, M. R.
Preimesberger, D. A. Bryant, J. T. J. Lecomte, Biochemistry 2010, 49, 7000.[4] N. L. Scott, C. J. Falzone, D. A. Vuletich, J. Zhao, D. A. Bryant, J. T. J. Lecomte, Biochemistry 2002,
41, 6902.[5] B. C. Vu, A. D. Jones, J. T. J. Lecomte, J. Am. Chem. Soc. 2002, 124, 8544.[6] N. L. Scott, J. T. J. Lecomte, Protein Sci. 2000, 9, 587.[7] J. A. Hoy, S. Kundu, J. T. Trent 3rd, S. Ramaswamy, M. S. Hargrove, J. Biol. Chem. 2004, 279, 16535.[8] J. T. Trent 3rd, S. Kundu, J. A. Hoy, M. S. Hargrove, J. Mol. Biol. 2004, 341, 1097.[9] D. A. Vuletich, J. T. J. Lecomte, J. Mol. Evol. 2006, 62, 196.
[10] a) M. Bolognesi, D. Bordo, M. Rizzi, C. Tarricone, P. Ascenzi, Prog. Biophys. Mol. Biol. 1997, 68, 29;b) A. Pesce, M. Couture, S. Dewilde, M. Guertin, K. Yamauchi, P. Ascenzi, L. Moens, M. Bolognesi,EMBO J. 2000, 19, 2424; c) R. E. Weber, S. N. Vinogradov, Physiol. Rev. 2001, 81, 569; d) M. Milani,A. Pesce, M. Nardini, H. Ouellet, Y. Ouellet, S. Dewilde, A. Bocedi, P. Ascenzi, M. Guertin, L.Moens, J. M. Friedman, J. B. Wittenberg, M. Bolognesi, J. Inorg. Biochem. 2005, 99, 97.
[11] A. Pesce, L. Tilleman, S. Dewilde, P. Ascenzi, M. Coletta, C. Ciaccio, S. Bruno, L. Moens, M.Bolognesi, M. Nardini, IUBMB Life 2011, 63, 287; S. Kundu, G. C. Blouin, S. A. Premer, G. Sarath,J. S. Olson, M. S. Hargrove, Biochemistry 2004, 43, 6241; F. Draghi, A. E. Miele, C. Travaglini-Allocatelli, B. Vallone, M. Brunori, Q. H. Gibson, J. S. Olson, J. Biol. Chem. 2002, 277, 7509.
[12] B. Brooks, R. S. Phillips, W. F. Benisek, Biochemistry 1998, 37, 9738.[13] P. Murray-Rust, W. C. Stallings, C. T. Monti, R. K. Preston, J. P. Glusker, J. Am. Chem. Soc. 1983,
105, 3206; J. A. K. Howard, V. J. Hoy, D. O� Hagan, G. T. Smith, Tetrahedron 1996, 52, 12613.[14] P. Auffinger, F. A. Hays, E. Westhof, P. S. Ho, Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 16789.[15] C. Frieden, S. D. Hoeltzli, J. G. Bann, Methods Enzymol. 2004, 380, 400.[16] G. Xiao, J. F. Parsons, K. Tesh, R. N. Armstrong, G. L. Gilliland, J. Mol. Biol. 1998, 281, 323.[17] J. L. Kitevski-LeBlanc, F. Evanics, R. S. Prosser, J. Biomol. NMR 2010, 48, 113.[18] M. P. Gamcsik, J. T. Gerig, R. B. Swenson, Biochim. Biophys. Acta 1986, 874, 372; J. T. Gerig, Prog.
Nucl. Magn. Reson. Spectrosc. 1994, 26, 293; M. A. Danielson, J. J. Falke, Annu. Rev. Biophys.Biomol. Struct. 1996, 25, 163; J. T. Gerig, �Fluorine NMR�, in �Nuclear Magnetic Resonance(NMR)�, Ed. D. Gorenstein, in �Biophysical Textbook On-Line�, Biophysical Society, 1997, http://biosci.cbs.umn.edu/biophys/OLTB/NMR/jtg-nmr3.pdf; N. C. Yoder, K. Kumar, Chem. Soc. Rev.2002, 31, 335; S. L. Cobb, C. D. Murphy, J. Fluorine Chem. 2009, 130, 132.
[19] C. Li, G.-F. Wang, Y. Wang, R. Creager-Allen, E. A. Lutz, H. Scronce, K. M. Slade, R. A. S. Ruf,R. A. Mehl, G. J. Pielak, J. Am. Chem. Soc. 2010, 132, 321.
[20] J. L. Kitevski-LeBlanc, F. Evanics, R. S. Prosser, J. Biomol. NMR 2009, 45, 255.[21] J. L. Kitevski-Leblanc, F. Evanics, R. S. Prosser, J. Biomol. NMR 2010, 47, 113.[22] a) M. P. Pond, D. A. Vuletich, C. J. Falzone, A. Majumdar, J. T. J. Lecomte, Biomol. NMR
Assignments 2009, 3, 211; b) B. C. Vu, H. J. Nothnagel, D. A. Vuletich, C. J. Falzone, J. T. J. Lecomte,Biochemistry 2004, 43, 12622; c) M. P. Pond, A. Majumdar, J. T. J. Lecomte, Biochemistry 2012, 51,5733; d) M. R. Preimesberger, M. P. Pond, A. Majumdar, J. T. J. Lecomte, J. Biol. Inorg. Chem.2012, 17, 599.
[23] S. J. Baturin, M. Okon, L. P. McIntosh, J. Biomol. NMR 2011, 51, 379; J. J. Skalicky, J. L. Mills, S.Sharma, T. Szyperski, J. Am. Chem. Soc. 2001, 123, 388.
[24] G. N. La Mar, N. L. Davis, D. W. Parish, K. M. Smith, J. Mol. Biol. 1983, 168, 887.[25] B. C. Vu, D. A. Vuletich, S. A. Kuriakose, C. J. Falzone, J. T. J. Lecomte, J. Biol. Inorg. Chem. 2004,
9, 183.[26] H. J. Nothnagel, M. R. Preimesberger, M. P. Pond, B. Y. Winer, E. M. Adney, J. T. J. Lecomte, J.
Biol. Inorg. Chem. 2011, 16, 539.[27] W. E. Hull, B. D. Sykes, J. Mol. Biol. 1975, 98, 121.
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012)1716

[28] G. N. La Mar, H. Toi, R. Krishnamoorthi, J. Am. Chem. Soc. 1984, 106, 6395.[29] R. Chirakal, K. L. Brown, G. Firnau, E. S. Garnett, D. W. Hughes, B. G. Sayer, R. W. Smith, J.
Fluorine Chem. 1987, 37, 267.[30] W. E. Hull, B. D. Sykes, Biochemistry 1976, 15, 1535.[31] I. Ayala, J. J. Perry, J. Szczepanski, J. A. Tainer, M. T. Vala, H. S. Nick, D. N. Silverman, Biophys. J.
2005, 89, 4171.[32] F. Khan, I. Kuprov, T. D. Craggs, P. J. Hore, S. E. Jackson, J. Am. Chem. Soc. 2006, 128, 10729.[33] M. Milani, Y. Ouellet, H. Ouellet, M. Guertin, A. Boffi, G. Antonini, A. Bocedi, M. Mattu, M.
Bolognesi, P. Ascenzi, Biochemistry 2004, 43, 5213.[34] P. Lu, M. Jarema, K. Mosser, W. E. Daniel, Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 3471; F. Sixl, R. W.
King, M. Bracken, J. Feeney, Biochem. J. 1990, 266, 545.[35] P. K. Glasoe, F. A. Long, J. Phys. Chem. 1960, 64, 188.[36] D. A. Vuletich, C. J. Falzone, J. T. J. Lecomte, Biochemistry 2006, 45, 14075.[37] S. W. Englander, D. B. Calhoun, J. J. Englander, Anal. Biochem. 1987, 161, 300.[38] R. T. Kelly, J. S. Page, Q. Z. Luo, R. J. Moore, D. J. Orton, K. Q. Tang, R. D. Smith, Anal. Chem.
2006, 78, 7796.
Received December 30, 2011
CHEMISTRY & BIODIVERSITY – Vol. 9 (2012) 1717
Related Documents











