Hematopoietic (bone marrow) failure with pancytopenia (bi- or tricytopenia) of the peripheral blood. Characteristics: hypocellular bone marrow with fatty substitution; no bone marrow stro- mal cell defects; no malignant cells. D61 Incidence worldwide: 2–6 cases per 1,000,000 population/year; much higher in China, the Far East, and South East Asia. Age distribution: two peaks, around 20 years and 65 years. Pathophysiological Model Destruction / suppression of hematopoietic stem cells or progenitor cells caused by various factors is of central importance: • Activation of the immune system with primary or secondary (immunologically induced) bone marrow aplasia with activated cytotoxic T-cells, which cause destruction of CD34-positive progenitor cells via: – Direct T-cell-mediated cytotoxicity – Production of IFNγ and TNFβ – Induction of FAS receptor and antigen → apoptosis induction • Direct DNA damage (e.g., irradiation) • Cellular membrane damage and interference with the cellular metabolism (e.g., viral infec- tion) • Drug-induced: direct toxicity or hapten-mediated autoimmune reaction • Secondary clonal expansion of hematopoiesis • NK cells ↓ (as with other autoimmune diseases) Genetic Factors • Fanconi’s anemia: chromosomal instability based on multiple genetic defects (Fanconi anemia genes FANC A-L). Characteristics are: progressive bone marrow aplasia, increased incidence of malignancy, and abnormalities in skin, musculature, skeletal system, and urogenital system. In > 80% of cases, manifestation is during infancy. • Increased incidence of aplastic anemia in the presence of HLA A2, DR2, DR4, and DPw3. • PNH association (7 Chap. 6.4.3) • Mutations of telomerase reverse transcriptase (TERT) gene Drugs (in 25% of Cases) • Antibiotics (particularly sulfonamides, chloramphenicol), antimalarial drugs • yreostatics, antidiabetics • Antirheumatics, NSAIDs (e.g., phenylbutazone, gold) • Diuretics (furosemide), ticlopidine, nifedipine • Antiepileptics (e.g., carbamazepine, phenytoin) • Cytotoxic compounds (e.g., busulfan) Chemical Agents • Aromatic solvents (e.g., benzene) • Insecticides (lindane, DDT, etc.) Viral / Postinfectious (5% of Cases) • Parvovirus B19 (isolated erythropoietic aplasia, “pure red cell anemia”) • Hepatitis (non-A-B-C-G hepatitis, poor prognosis, mostly young men) • EBV (infectious mononucleosis, rare) • HIV Def: Def: ICD-10: ICD-10: Ep: Ep: Pphys: Pphys: Pg: Pg: and . Aplastic Anemia 6.1 Aplastic Anemia J. Finke, H. Bertz

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

���
Hematopoietic (bone marrow) failure with pancytopenia (bi- or tricytopenia) of the peripheral blood. Characteristics: hypocellular bone marrow with fatty substitution; no bone marrow stro-mal cell defects; no malignant cells.
D61
Incidence worldwide: 2–6 cases per 1,000,000 population/year; much higher in China, the Far East, and South East Asia.Age distribution: two peaks, around 20 years and 65 years.
Pathophysiological ModelDestruction / suppression of hematopoietic stem cells or progenitor cells caused by various factors is of central importance:• Activation of the immune system with primary or secondary (immunologically induced) bone
marrow aplasia with activated cytotoxic T-cells, which cause destruction of CD34-positive progenitor cells via:– Direct T-cell-mediated cytotoxicity– Production of IFNγ and TNFβ– Induction of FAS receptor and antigen → apoptosis induction
• Direct DNA damage (e.g., irradiation)• Cellular membrane damage and interference with the cellular metabolism (e.g., viral infec-
tion)• Drug-induced: direct toxicity or hapten-mediated autoimmune reaction• Secondary clonal expansion of hematopoiesis• NK cells ↓ (as with other autoimmune diseases)
Genetic Factors• Fanconi’s anemia: chromosomal instability based on multiple genetic defects (Fanconi anemia
genes FANC A-L). Characteristics are: progressive bone marrow aplasia, increased incidence of malignancy, and abnormalities in skin, musculature, skeletal system, and urogenital system. In > 80% of cases, manifestation is during infancy.
• Increased incidence of aplastic anemia in the presence of HLA A2, DR2, DR4, and DPw3.• PNH association (7 Chap. 6.4.3)• Mutations of telomerase reverse transcriptase (TERT) gene
Drugs (in 25% of Cases)• Antibiotics (particularly sulfonamides, chloramphenicol), antimalarial drugs• Thyreostatics, antidiabetics• Antirheumatics, NSAIDs (e.g., phenylbutazone, gold)• Diuretics (furosemide), ticlopidine, nifedipine• Antiepileptics (e.g., carbamazepine, phenytoin)• Cytotoxic compounds (e.g., busulfan)
Chemical Agents• Aromatic solvents (e.g., benzene)• Insecticides (lindane, DDT, etc.)
Viral / Postinfectious (5% of Cases)• Parvovirus B19 (isolated erythropoietic aplasia, “pure red cell anemia”)• Hepatitis (non-A-B-C-G hepatitis, poor prognosis, mostly young men)• EBV (infectious mononucleosis, rare)• HIV
Def:Def:
ICD-10:ICD-10:
Ep:Ep:
Pphys:Pphys:
Pg:Pg:
� Hematology and Hemostasis
�.� Aplastic Anemia
6.1 Aplastic Anemia
J.Finke,H.Bertz
���
Part � Hematology and Hemostasis
• CMV (bone marrow stromal cell invasion possible)• Flavivirus (dengue fever)
Radiation• Ionizing radiation• Thorotrast
Other Causes• Autoimmune diseases, associated with eosinophilic fasciitis• Pregnancy (estrogen-mediated?)• Thymoma• Idiopathic (70% of cases)
Classificationaccordingtonumberofgranulocytes,platelets,andreticulocytesa
Type Abbreviation Granulocytes Platelets Reticulocytes
Aplastic anemia AA < 1,500/µl < 50,000/µl < 20,000/µl
Severe AA SAA < 500/µl < 20,000/µl < 20,000/µl
Very severe AA VSAA < 200/µl < 20,000/µl < 20,000/µl
a At least 2 out of 3 criteria are necessary for diagnosis, hypocellular bone marrow
NOTE: Treatment-induced reversible hematopoietic insufficiency following chemo- or radio-therapy is not designated as aplastic anemia.
Symptoms are dominated by hematopoietic failure:• Symptoms of anemia: pallor, fatigue, reduced performance, dyspnea• Symptoms of neutropenia: oral ulcers, gingivitis, severe infections, pneumonia• Symptoms of thrombocytopenia: hemorrhage, petechiae (skin, mucous membranes), less
commonly hematomas
Medical History, Physical Examination• Medical history, including medication, infections• Physical examination (hemorrhage, mucous membranes, signs of infection, splenic status,
etc.)
Laboratory Tests• Complete blood count: bi- or trilineage cytopenia, generally without pathological morphol-
ogy, increased granulation, neutropenia, monocytopenia, and eosinopenia; reticulocytes ↓; in cases of thrombocytopenia: small platelets
• Ferritin, haptoglobin, Coombs’ test, blood group, coagulation parameters• ESR, total protein, electrophoresis, immunoglobulins, immunofixation, cold agglutinins,
rheumatoid factor, ANA• PNH exclusion (Ham’s test, sugar water test, GPI-linked proteins, CD55, CD59)• Vitamin B12, folic acid (exclusion of megaloblastic anemia)• Liver function (exclude past history of hepatitis)• Serology (EBV, CMV, HAV, HBV, HCV, HIV, HSV, parvovirus B19)
Bone Marrow (Aspiration, Histology, Immunohistochemistry, Iron Stain, Culture)• Hypocellular (cellularity < 25%) with predominance of fat cells• Lymphocytes, macrophages, and plasma cells present• CD34-positive progenitor cells ↓; in bone marrow cultures, reduced colony formation (CFU-
GM, colony-forming units – granulocytes / macrophages) and LTCIC (long-term culture-ini-tiating cells). Improved growth pattern in T-cell-depleted cultures (→ T-cell-mediated reac-tion?)
Class:Class:
Sy:Sy:
Dg:Dg:

���
Further Diagnostic Procedures• Chest x-ray, abdominal sonography• HLA typing (in cases of potential transplantation)• Cytogenetics, chromosome analysis (exclusion of MDS, Fanconi’s anemia)• Increased serum levels of hematopoietic growth factors: G-CSF (granulocyte colony-stimulat-
ing factor), TPO (thrombopoietin), M-CSF, and erythropoietin; SCF (stem cell factor) not increased
• Myelodysplasia with hypoplastic bone marrow (7 Chap. 7.2)• Primary Myelofibrosis (PM) (7 Chap. 7.3.4)• Vitamin B12 deficiency, folic acid deficiency (7 Chap. 6.4.2)• Paroxysmal nocturnal hemoglobinuria (PNH) (7 Chap. 6.4.3)• Leukemias, lymphomas, solid tumors with bone marrow infiltration
• Development of PNH in 7% of cases (7 Chap. 6.4.3)• Transformation into MDS or acute leukemia in 5–12% of cases (7 Chaps. 7.1.1, 7.1.2, 7.2)
IndicationsforTreatment
• Severe aplastic anemia (SAA or VSAA)• Patient at risk by complications arising from cytopenia (recurrent infections, hemorrhage, he-
mosiderosis)• Prevention of alloimmunization and subsequent transfusion refractoriness
Treatmentofaplasticanemia
MRD matched related donor (HLA-identical family bone marrow or stem cell donor)
TreatmentGuidelines
1. Aplastic anemia should always be treated in a hematological center.2. Patients under 55 years of age with HLA-identical siblings or relatives should be evaluated for
matched related allogeneic bone marrow or blood stem cell transplantation.3. In other patients, immunosuppression is carried out in the framework of clinical trials.
Dd:Dd:
Co:Co:
Th:Th:
�.� Aplastic Anemia

��0
Part � Hematology and Hemostasis
TreatmentModalities
Supportive Measures• Infection prophylaxis, antibiotics, amphotericin B prophylaxis• Oral hygiene• Prophylaxis / therapy of hemosiderosis (desferrioxamine mesylate)• Granulocyte transfusions (7 Chap. 5.4)• Suppress menses, avoid platelet aggregation inhibitors• Blood products (CMV negative, irradiated); erythrocyte transfusions according to symptoms,
platelet transfusions for counts below 5,000–10,000/µl• Growth factors: granulocyte colony-stimulating factor (G-CSF), erythropoietic factors
ATTENTION: use blood products as sparingly as possible until decision on BMT / PBSCT is made (danger of alloimmunization). Do not use blood products from relatives.
Transplantation Types (7 Chaps. 5.2, 5.3)• In patients under 55 years of age, allogeneic hematopoietic stem cell transplantation (HSCT)
from HLA-identical related (family) donors in conjunction with fludarabine / cyclophospha-mide-containing protocols
• Matched unrelated donor (MUD) transplantation recommended only in patients under 15 years of age
Immunosuppressive TherapyPatients > 55 years or without suitable stem cell donor. Effective compounds:• Antilymphocyte globulin (ALG) or antithymocyte globulin (ATG), since 1970• Cyclosporin A (CyA), since 1980• Methylprednisolone
Immunosuppressive therapy should only be carried out in clinical trials.
InnovativeTherapyApproachesandTreatmentofRelapse
If standard treatment fails:• Matched unrelated donor transplantation (MUD transplantation) in patients between 15 and
50 years of age• Hematopoietic growth factors• Treatment option without proven efficacy: androgens, used since 1954
ProgressionAplastic anemia can precede clonal hematological diseases (e.g., PNH). Incidence over 10 years: MDS 9%, leukemia 7%; after immunosuppressive therapy higher than after transplantation.
One-year Survival Rate with SAA• Untreated: 20%• Supportive treatment: 50%• Immunosuppressive treatment or allogeneic transplantation: 80%
Long-term Survival with Different Forms of Treatment• Patients < 25 years: 66–92%• Patients between 25 and 39 years: 69%• Patients > 39 years: 38%• With immunosuppressive treatment (ATG and CyA containing): 80%• Five-year survival after allogeneic matched related transplantation: 60–90%• Five-year survival after MUD transplantation: 29%• Relapse within an observation period of up to 14 years: 35%• Immunosuppression compared with BMT: no significant difference in terms of primary re-
sponse
Prg:Prg:

���
Relapse• After matched related allogeneic transplantation: 15–20%• After immunosuppressive medication (CyA + ATG containing): 30–50%
Bacigalupo A, Bruno B, Saracco P et al. Antilymphocyte globulin, cyclosporin, and granulocyte colony-stimulating factor for severe aplastic anemia: an update of the GITMO/EBMT Working Party. Blood 2000;95:1931–34Ball SE. The modern management of severe aplastic anaemia. Br J Haematol 2000;110:41–53Brodsky RA, Jones RJ. Aplastic anaemia. Lancet 2005;365:1647–56Davies JK, Guinan EC. An update on the management of severe idiopathic aplastic anaemia in children. Br J Haematol 2007;136:549–64Geroges GE, Storb R. Stem cell transplantation for aplastic anemia. Int J Hematol 2002;75:141–6Kojima S, Hibi S, Kosaka Y et al. Immunosuppressive therapy using antithymocyte globulin, cyclosporin, and danazol with or without human granulocyte colony-stimulating factor in children with acquired aplastic anemia. Blood 2000;96:2049–54Marsh JCW, Ball SE, Darbyshire P et al. British Committee for Standards in Hematology (BCSH). Guide-lines for the diagnosis and management of acquired aplastic anemia. Br J Haematol 2003;123:782–90Yamaguchi H, Calado RT, Ly H et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med 2005;352:1413–24Young NS. Immunosuppressive treatment of acquired aplastic anemia and immune-mediated bone mar-row failure syndromes. Int J Hematol 2002;75:129–40
1. http://www.aamds.org/ AA and MDS Foundation2. http://www.fanconi.org/ Fanconi Anemia Research Fund
1.
2.3.4.
5.6.
7.
8.
9.
Ref:Ref:
Web:Web:
�.� Aplastic Anemia

Hematology and HemostasisPart �
���
Neutropenia: Neutrophil count in the peripheral blood of adults < 1.5 × 109/l. Limit dependent on age and race: neonates show higher neutrophil levels, while certain African and Middle Eastern populations have physiologically lower numbers of neutrophils.
Agranulocytosis: Neutrophil count in the peripheral blood < 0.5 × 109/l. Usually symptomatic ac-quired disease with granulocytopenia and in severe cases, lymphocytopenia and monocytopenia. In adults, usually iatrogenic. Duration after discontinuation of the causative agent: 2–4 weeks.
D70
Neutropenia: common side effect of radio- / chemotherapy.
Agranulocytosis: rare occurrence, incidence of 3 cases per 1,000,000. Older patients are especially affected, male:female = 1:2. The incidence of specific forms of agranulocytosis depends on the causative agents and pathomechanisms.
Pathogenetic Mechanisms• Reduced production of neutrophils in the bone marrow• Redistribution from the circulating neutrophil pool to marginal areas (endothelium, tissues)• Peripheral destruction
Drug-induced Forms• Most common form: drug-induced toxic suppression of granulopoiesis or direct neutrophilic
damage (“delayed onset neutropenia,” e.g., after radio- or chemotherapy), usually with simul-taneous thrombocytopenia (7 Chap. 6.3)
• Drug-induced allergic reactions with destruction of neutrophils, often caused by metabolites• Usually, rapid granulocyte decrease within 1 week after exposure; in case of re-exposure,
within hours. Destruction of mature granulocytes (“abrupt onset neutropenia”), acute onset with fever and chills (DD: infection). Causative agent: e.g. phenylbutazone
• In rare cases, slow decrease, between 1 and 12 months after the beginning of treatment, due to destruction of hematopoietic progenitor cells. Causative agent: e.g., clozapine, in patients with HLA phenotype B38 and alleles DR4 and DQw3
Other Forms• Autoimmune diseases: T-cell-mediated inhibition of granulopoiesis (Felty’s syndrome, rheu-
matoid arthritis) or as a result of clonal T-cell expansion in patients with T-γ-lymphoprolifera-tive disease (“T-γ-disease”)
• Complement activation (e.g., with hemodialysis, sepsis): expression of adhesion molecules on the surface of neutrophils → neutrophilic aggregation, capillary occlusion (esp. pulmonary capillaries)
• Pseudoneutropenia (“shift neutropenia”): neutrophilic redistribution (shift) from the peripheral blood into the tissues, e.g., with infections
Neutropenia Caused by Congenital Granulopoietic Disorders• Congenital dysgenesis with familial pancytopenia• Reticular dysgenesis with congenital aleukocytosis: agranulocytosis + lymphoid hypoplasia +
thymic aplasia; unknown etiology• Periodic neutropenia: stem cell regulation defect; neutropenic phases in 10- to 35-day inter-
vals, compensatory monocytosis; autosomal dominant inheritance• Kostmann’s syndrome: severe agranulocytosis in children (abnormal differentiation in the
promyelocytic stage), reversible by administration of G-CSF (ATTENTION: possibly higher risk of MDS / AML development); autosomal dominant or recessive inheritance
• X-linked agammaglobulinemia
Def:Def:
ICD-10:ICD-10:
Ep:Ep:
Pg:Pg:
Class:Class:
6.2 Neutropenia and Agranulocytosis
J.Finke,H.Bertz

���
• Schwachman-Diamond-Oski syndrome: neutropenia + pancreatic insufficiency + metaphy-seal dysplasia; unknown etiology; autosomal recessive inheritance
• Neutropenia with bi- / tetraploid leukocytes: abnormal phagocytosis and chemotaxis as well as bi- and tetraploid granulocytes
• Chédiak-Higashi syndrome: albinism + neurological disorders + leukocytic granulation ab-normalities; unknown etiology
• Dyskeratosis congenita: neutropenia, skin abnormalities; X-linked inheritance• Lazy leukocyte syndrome: chemotaxis defect (actin defect); unknown etiology
Neutropenia Caused by Acquired Disorders of Granulopoiesis• Cytostatic treatment, immunosuppressives, azidothymidine (AZT), benzenes, ionizing radia-
tion• Idiosyncratic drug reactions (individual sensitivity) in 66% of cases: antibiotics (penicillin,
chloramphenicol, cephalosporins, sulfonamides), sulfasalazine, nonsteroidal antirheumatics (ibuprofen, indomethacin, phenylbutazone), phenothiazine, thyreostatics, quinidine, pro-cainamide, propafenone, ticlopidine, antihistamines, anticonvulsives, nifedipine, levamisole, tamoxifen, allopurinol, tranquilizers, neuroleptics (clozapine), gold, captopril + interferon
Neutropenia Caused by Increased Neutrophil Destruction• Hypersplenism• Autoimmune neutropenia: postinfectious (mononucleosis, viral infections), AIDS, Felty’s syn-
drome (rheumatoid arthritis + splenomegaly + neutropenia), systemic lupus erythematosus (SLE), Sjögren’s syndrome, malignant lymphoma
• Neonatal isoimmune neutropenia: transplacental passage of maternal antineutrophil antibod-ies
• Complement activation: hemodialysis, cardiopulmonary bypass, T-γ disease
Infections: Increased Margination / Consumption (Pseudoneutropenia)• Bacteria: typhus, paratyphus, brucellosis, tuberculosis, tularemia• Viruses: yellow fever, sandfly fever, infectious hepatitis, measles, influenza, chickenpox, Ger-
man measles, Colorado tick fever, dengue fever, HIV, EBV• Rickettsia: rickettsial pox, Rocky Mountain spotted fever• Protozoa: malaria, kala-azar, recurrent fever• Fungi: histoplasma
Other Causes• Bone marrow infiltration: leukemia (especially hairy cell leukemia), lymphomas, solid tumors• Malnutrition: vitamin B12 / folic acid deficiency, alcoholism• T-cell-associated neutropenia (T-γ disease), myelodysplasia (MDS)• DIDMOAD syndrome: diabetes insipidus + diabetes mellitus + optic nerve atrophy + deaf-
ness• Metabolic disorders: hepatic cirrhosis, ketoacidosis, Gaucher’s disease• Sepsis, hypothermia, acute anaphylaxis
• Initially usually asymptomatic• General symptoms: fatigue, decreased performance, anorexia, infections
Medical History, Physical Examination• Medical history: drug treatment, family history, infections, menstrual complaints• Physical examination: with lymph node status, liver / spleen, signs of infection, mucositis
Laboratory Tests• Blood count with differential, reticulocytes• Routine laboratory tests including vitamin B12 and folic acid, total protein, protein electropho-
resis, urinary protein (paraprotein diagnosis), copper• Immunology: immunoglobulin assay, immunoelectrophoresis, Coombs’ test, ANA, anti-DNA,
rheumatoid factor, granulocyte antibodies
Sy:Sy:
Dg:Dg:
�.� Neutropenia and Agranulocytosis

���
Part � Hematology and Hemostasis
• Differentiation of lymphocyte subpopulations (FACS): T-cell subpopulations, NK cells, exclu-sion of leukemia
• Infection monitoring: blood, fecal, and urine cultures, throat swab, viral serology (including HIV)
• Cytogenetics• Ham’s test, sugar water test (exclusion of PNH)
Histology• Bone marrow aspiration, biopsy and culture (CFU)
Imaging• Abdominal sonography (spleen), chest x-ray (exclusion of infection)
• Leukemia (7 Chaps. 7.1.1, 7.1.2)• Myelodysplasia (7 Chap. 7.2)• Primary Myelofibrosis (7 Chap. 7.3.4)• Aplastic anemia (7 Chap. 6.1)
• Susceptibility to infections, fever (7 Chap. 4.2)• Mucositis, gastroenteritis (“neutropenic enterocolitis”)
Supportive Therapy• Hygiene, anti-infectious environment, isolation• Mucositis prophylaxis• Selective intestinal decontamination• Oral antimycosis (e.g., fluconazole 200 mg/day p.o.)• Signs of infection: blood cultures, urine and stool cultures, swabs, immediate start of empirical
antibiotic treatment (7 Chap. 4.2)• With severe infections: granulocyte transfusion (7 Chap. 5.4)
Treatment of Acute Agranulocytosis• Discontinue all drugs administered within 4 weeks of onset of symptoms• G-CSF (filgrastim, lenograstim) 5–10 µg/kg daily s.c.
Treatment of Chronic NeutropeniaTreatment according to the assumed pathogenic causes, e.g.:• In patients with clinically relevant recurrent infections, G-CSF may be used as long-term treat-
ment• Use of other hematopoietic growth factors in clinical studies: GM-CSF, IL-3, stem cell factor
(SCF)• In cases of autoimmune neutropenia:
– Prednisolone 2 mg/kg daily p.o. (maximum 4 weeks)– Cyclosporin A (serum level target: 300–600 ng/ml): initial treatment over at least 4 weeks;
if successful, continue for at least 3 months– Azathioprine 2–4 mg/kg daily
• With hypersplenism: consider splenectomy (only after pneumococcus vaccination)• In cases of congenital neutropenia: consider allogeneic transplantation (7 Chap. 5.3)
ProphylaxisWith clozapine therapy and thyreostatic medication: regular weekly blood counts.
Berliner N, Horwitz M, Loughran TP Jr. Congenital and acquired neutropenia. Hematology (ASH Educ Program) 2004:63–79Boxer LA, Newburger PE. A molecular classification of congenital neutropenia syndromes. Pediatr Blood Cancer 2007;49:609–14Lakshman R, Finn A. Neutrophil disorders and their management. J Clin Pathol 2001;54:7–19Manny N, Zelig O. Laboratory diagnosis of autoimmune cytopenias. Curr Opin Hematol 2000;7:414–9
1.
2.
3.4.
Dd:Dd:
Co:Co:
Th:Th:
Ref:Ref:

���
Palmblad JE, von dem Borne AE. Idiopathic, immune, infectious and idiosyncratic neutropenias. Semin Hematol 2002;39:113–20Welte G, Zeidler C, Dale DC. Severe congenital neutropenia. Semin Hematol 2006;43:189–95
1. http://www.rarediseases.org/ NORD, Rare Disorders2. http://www.nlm.nih.gov/medlineplus/ency/article/001295.htm Medline Plus article3. http://www.mascc.org MASCC, Supportive Care4. http://www.neutropenia.ca/ Neutropenia Support Assoc5. http://www.emedicine.com/med/topic82.htm E-medicine
5.
6.
Web:Web:
�.� Neutropenia and Agranulocytosis

Hematology and HemostasisPart �
���
Decreased platelet count (< 150,000/µl), most common cause of hemorrhagic diatheses.
D69.6
Platelet Kinetics• Thrombopoiesis: megakaryoblasts → megakaryocytes → platelets; regulated by thrombopoi-
etin and other cytokines (e.g., IL-3, IL-6, IL-11)• Directly after being released by the bone marrow, approximately one third of platelets are re-
versibly stored in the spleen (“pool”)• Two thirds of platelets circulate in the blood, life span 7–10 days, biological half-life 3–4 days;
15% of these platelets are spent daily to maintain hemostasis
The platelet count is influenced by:• Nutritional status: folic acid / vitamin B12 deficiency, alcohol abuse• Menstrual cycle: shortly after ovulation, platelet count ↑• Acute-phase reactions (infections, tumors) → platelet count ↑
Disorders of Thrombopoiesis• Infections (most common cause): e.g., CMV, EBV, HIV, mycoplasma, bacterial infection, para-
sites (malaria), sepsis (early symptom)• Hematopoietic (bone marrow) deficiency: aplastic anemia, primary myelofibrosis• Bone marrow infiltration: leukemia, lymphomas, solid tumors• Abnormal megakaryocytic maturation: myelodysplasia, folic acid / vitamin B12 deficiency• Drug-induced / toxic myelosuppression: cytostatics, thiazides, alcohol, estrogens, thiamazole,
gold, benzene, ionizing radiation• Hereditary platelet disorders (rare):
− Fanconi’s anemia− Wiskott-Aldrich syndrome (thrombocytopenia, eczema, and immunodeficiency)− von Willebrand’s disease type IIb− Thrombocytopenia with absent radii syndrome (TAR)− Bernard-Soulier syndrome (giant platelets and platelet dysfunction)− Thrombopoietin deficiency
Increased Splenic Platelet Sequestration (Hypersplenism)Splenomegaly (portal hypertension, splenic infiltration with hematological neoplasia).
Accelerated Peripheral Platelet Turnover• Heart valve and vascular prostheses• Extracorporeal circulation (surface activation)• Immune thrombocytopenia (ITP) (7 Chap. 6.3.1)• Microangiopathic disorders: hemolytic-uremic syndrome (HUS) , thrombotic-thrombocyto-
penic purpura (TTP) (7 Chap. 6.3.3)• Disseminated intravascular coagulation (DIC) (7 Chap. 6.5.5)• Disturbances in platelet and coagulation factor interaction: von Willebrand’s disease type IIb,
heparin-induced thrombocytopenia (HIT) (7 Chap. 6.3.2)• Evans’ syndrome: autoimmune hemolytic anemia and thrombocytopenia
Hemorrhage• Petechial type of hemorrhage with small pinpoint lesions on skin / mucous membranes, oc-
casionally epistaxis, menorrhagia• In rare cases: hematoma / bruising / diffuse hemorrhage
Def:Def:
ICD-10:ICD-10:
Phys:Phys:
Pg:Pg:
Sy:Sy:
6.3 Thrombocytopenia
A.K.Kaskel,J.Heinz

���
Clinical Diagnosis• Medical history (especially infections, drugs, hemorrhage)• Clinical findings: petechial bleeding (skin, mucous membranes), lymph nodes, spleen• In severe cases: signs of organ bleeding, anemia, hemorrhage
Laboratory Tests• Blood count with differential, reticulocytes, clotting studies (Quick, PTT, fibrinogen), hemoly-
sis parameters (LDH, haptoglobin), liver function tests, CRP• Exclusion of pseudothrombocytopenia by means of platelet count in citrated blood• Viral serology (HIV included)• With suspected vasculitis / SLE → immunology: antinuclear antibodies (ANA), rheumatoid
factor• With suspected HUS / TTP: screening for abnormal VWF multimers or VWF protease anti-
bodies (7 Chap. 6.3.3)• With suspected Evans’ syndrome (autoimmune hemolytic anemia and thrombocytopenia):
Coombs’ test• Blood group• Possibly detection of fixed thrombocytic antibodies (immune thrombocytopenia)
HistologyBone marrow aspiration and biopsy: megakaryocytes ↓ in case of dysfunctional thrombopoiesis, megakaryocytes normal or ↑ in cases of peripheral platelet loss. ATTENTION: if platelet count < 20,000/µl: risk of hemorrhage → iliac crest biopsy (no sternal puncture), apply careful pressure
ImagingChest x-ray (lymphomas, infections), abdominal sonography (lymphomas, spleen)
NOTE: if plasmatic coagulation and blood vessels are normal, there is only a low risk of hemor-rhage with a platelet count of > 10,000–20,000/µl.
“Pseudothrombocytopenia”: formation of platelet aggregates in EDTA blood: 0.1–2% of blood samples; cause: autoagglutinating IgG antibodies→ In vitro platelet aggregation in the presence of the anticoagulant agent EDTA→ False low count by automatic platelet counter→ Repeat platelet count with citrated or heparinized blood
Treatment of the Underlying Disease• In cases of drug-induced thrombocytopenia: avoid causative agent• Treatment of malignancies• Treatment of immunological disorders
Supportive Treatment• Prevention of menstrual bleeding (e.g., lynestrenol)• Avoid platelet aggregation inhibitors (acetyl salicylic acid)• Platelet transfusion at signs of bleeding / acute risk of hemorrhage (ATTENTION: HUS /
TTP)• With thrombopathy try DDAVP (desmopressin); dosage 0.3 µg/kg body weight in 0.9% saline
infusion every 8 h, maximum 3 days → repeat after 48 h
Platelet Transfusion (7 Chap. 4.9.1)• Therapeutic: at signs of bleeding or acute hemorrhage (e.g., petechiae, hemorrhage of mucous
membranes or epistaxis) with proven thrombocytopenia or thrombocyte dysfunction.• Prophylactic: platelet count < 10,000–20,000/µl. With concomitant diseases (especially acute
leukemia, fever, sepsis, splenomegaly) risk of hemorrhage with higher platelet counts (20,000–30,000/µl). With invasive interventions (e.g., catheter installation, punctures) the platelet count target is > 40,000–60,000/µl.
Dg:Dg:
Dd:Dd:
Th:Th:
�.� Thrombocytopenia

���
Part � Hematology and Hemostasis
Relative Contraindication• Allergy to human plasma protein• Post-transfusion purpura (PTP)• Idiopathic thrombocytopenic purpura (ITP)• Heparin-induced thrombocytopenia (HIT)• Thrombotic-thrombocytopenic purpura (TTP)
To avoid alloimmunization, transfusions should be avoided in patients scheduled for alloge-neic hematopoietic stem cell transplantation.
Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med 2007;357:580–7Bolton-Maggs PHB, Chalmers EA, Collins PW et al. A review of inherited platelet disorders with guide-lines for their management on behalf of the UKHCDO. Br J Haematol 2006;135:603–33Cines DB, Bussel JB, McMillan RB et al. Congenital and acquired thrombocytopenia. Hematology (ASH Educ Program) 2004:390–406Deutsch VR, Tomer A. Megakaryocyte development and platelet production: Br J Haematol 2006; 134:453–66Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 2004;103:390–8Geddis AE, Kaushansky K. Inherited thrombocytopenias: toward a molecular understanding of disorders of platelet production. Curr Opin Pediatr 2004;16:15–22George JN. Platelets. Lancet 2000;355:1531–9Jelic S, Radulovic S. Chemotherapy-associated thrombocytopenia. Ann J Cancer 2006;5:371–82
1. http://www.pdsa.org Platelet Disorder Support Assoc2. http://www.med.unc.edu/isth/ ISTH, Intl Soc Thromb Hemostasis3. http://www.nlm.nih.gov/medlineplus/ency/
article/000586.htm MedlinePlus4. http://www.emedicine.com/med/topic3480.htm E-medicine5. http://marrowfailure.cancer.gov/AMEGA.html NCI, Marrow Failure Disorders
1.2.
3.
4.
5.
6.
7.8.
Ref:Ref:
Web:Web:

���
Acquired thrombocytopenia, platelet count < 150,000/µl.
Classic definition: ITP = idiopathic thrombocytopenic purpura.Diagnosis by exclusion; acquired thrombocytopenia of unknown etiology with normal to in-creased megakaryocyte count in the bone marrow.
Alternative definition: ITP = immune thrombocytopenic purpura.Acquired thrombocytopenia caused by antithrombocytic antibodies.
D69.3
Incidence: 6–10 cases / 100,000 population / year. Distribution male:female = 1:2.
IgG-mediated immune reaction (rarely IgM) against platelet membrane antigens, e.g., GPIIb / GPIIIa (fibrinogen receptor), GPIb / IX (von Willebrand receptor), and GPIa / IIa (collagen recep-tor).• Specific platelet antibodies detectable in approximately 50–70% of cases• Macrophage binding via Fcγ I, II, and III receptors (in ITP patients: receptor polymorphism
with altered binding affinity for IgG)• Complement activation• Complement-mediated lysis and enhancement of phagocytosis
→ RES phagocytosis of IgG-coated platelets, esp. in spleen→ Biological half-life of platelets ↓↓ to a few hours
• Decreased thrombocytopoiesis (antibodies against megakaryocytes and thrombopoietic pro-genitor cells)
• Possibly T-cell-mediated process (in vitro, CD4+ T-cells can be activated by platelets)
Etiology• Without known causative disease (“primary ITP”)• In conjunction with an underlying disease (“secondary ITP”): lymphoproliferative diseases,
autoimmune diseases (systemic lupus erythematosus, etc.), viral diseases (e.g., HCV, HIV), bacterial infections (esp. in children), after bone marrow transplantation
Progression• Children: in > 90% of cases, “acute” course: severe thrombocytopenia, usually spontaneous
remission within 3 months• Adults: in > 90% of cases, “chronic” course (thrombocytopenia > 6 months): < 5% risk of fatal
hemorrhages (esp. intracranial), rarely spontaneous remission (5%), persists for more than 6 months despite adequate treatment in 35% of patients
Blood CountThrombocytopenia with normal differential and morphology.
Bone MarrowNormal or reactively increased megakaryocyte count, increased number of immature megakaryo-cytes. Otherwise, normal bone marrow, no abnormal cells.
Hemorrhage• Rare with platelet count > 30,000/µl
Def:Def:
ICD-10:ICD-10:
Ep:Ep:
Pp:Pp:
Pg:Pg:
Path:Path:
Sy:Sy:
�.� ITP
6.3.1 Immune (Idiopathic) Thrombocytopenic Purpura (ITP, Werlhof’s Disease)
A.K.Thomas,J.Heinz

��0
Part � Hematology and Hemostasis
• Petechial type of hemorrhage (skin, mucous membranes), with hematomas / bruising / epi-staxis
• Complication: intracerebral hemorrhage (rare), organ bleeding, retinal bleeding, gastrointesti-nal bleeds
The diagnosis of ITP is a diagnosis of exclusion. Therefore, the diagnostic strategies are aimed at identifying potential underlying causes of secondary thrombocytopenia.
Clinical Diagnosis• Medical history, family history, drug exposure, occupational hazards• Physical examination (petechiae, bruising, mucosal bleeds)
Laboratory Tests• Full blood count with differential• Virology: HCV / HIV serology in patients at risk• Screening for platelet antibodies (50% positive)
HistologyBone marrow biopsy and smear in accordance with recommendations of ASH (American Society of Hematology) and BCSH (British Committee for Standards in Hematology):• Patients over 60 years of age• Laboratory abnormalities (neutropenia, anemia)• Prior to splenectomy• Poor response to primary treatment.
Differential diagnosis of thrombocytopenia 7 Chap. 6.3
IndicationsforTreatment
Only a small number of randomized studies have been conducted in ITP. The life expectancy of ITP patients with a platelet count > 30,000/µl is equal to that of the normal population. With higher platelet counts (30,000/µl), treatment is therefore only indicated if blood loss is expected (perioperatively, before delivery) or in the case of active hemorrhage. Recommendations of the British Committee for Standards in Hematology (BCSH) with regard to safe platelet counts in adults:• Dental treatment: ≥ 10,000/µl• Tooth extraction: ≥ 30,000/µl• Minor operation: ≥ 50,000/µl• Major operation: ≥ 80,000/µl
TherapeuticconceptformanagementofITPinadults
Dg:Dg:
Dd:Dd:
Th:Th:

���
TreatmentOptions
Primary Treatment
Steroids• Initial response rate > 50%, long-term effect in 30% of patients, low-dose maintenance treat-
ment is required in most cases• Prednisolone 1–2 mg/kg daily, duration of treatment depending on response, or dexametha-
sone 40 mg/d for 4 days• With durable platelet response: dose reduction of prednisolone over 6–12 weeks, monitoring
of platelet counts• If no increment to > 30,000/µl within 2–4 weeks or required steroid dose markedly above the
threshold dose for Cushing’s disease → change treatment to immunoglobulins or alternative immunosuppressive drugs
Immunoglobulins (ivIG)• Initial response rate 75%, normalization of the platelet count in 50% of patients; however, only
transient (up to 4 weeks)• Standard dose: 0.4 g/kg daily i.v. days 1–5 or 1 g/kg daily i.v. day 1 + 2• Alternative: anti-D IgG in Rh-positive patients, 75 µg/kg body weight over 2–3 days. Disad-
vantage: i.v. product not available in all countries, high costs
In cases of severe or life-threatening hemorrhage: combined administration of methylprednis-olone 1 g daily i.v. over 3 days and immunoglobulins 0.4–1 g/kg daily over 2–4 days, platelet transfusion. Due to the short platelet half-life in ITP, the expected platelet need is approxi-mately 2–3 times higher than in other forms of thrombocytopenia. In patients with uncompli-cated ITP, platelet transfusions are, generally not indicated.
Secondary Treatment
RomiplostimThrombopoietic agent, binds to TPO receptor and stimulates platelet production of the bone mar-row. In Phase III studies in ITP, platelet responses in 80–90% of cases. Starting dose 1 µg/kg once weekly s.c., dose adjustment according to platelet counts.
Splenectomy• Approximately 60% response rate, no known predictors of response• Perioperatively, platelet count should be raised to > 50,000/µl (ivIG)• Preoperative vaccination against pneumococcus, Hemophilus influenzae, meningococcus• If no response, exclude accessory spleen, repeat steroids
Tertiary Treatment
Danazol• Mode of action: downregulation of Fc receptors on macrophages• Not effective in steroid-refractory cases, but may be useful in combination with prednisolone
to reduce steroid side effects
ImmunosuppressivesA number of smaller studies have provided limited data on efficacy and safety of various immuno-suppressives. In individual cases or in smaller groups of patients, the following substances have been used successfully: mycophenolate mofetil, azathioprine, cyclophosphamide, cyclosporin A.
Experimental Treatment• Immunoapheresis• Rituximab (CD20 antibody)
�.� ITP

���
Part � Hematology and Hemostasis
Prevention of hemorrhage / trauma• No intramuscular or intra-articular injections• No massages• No administration of platelet aggregation inhibitors (acetyl salicylic acid, ticlopidine, clopido-
grel)• No sports with high risk of hemorrhage• Emergency ID card
Andemariam B, Bussel J. New therapies for ITP. Curr Opin Hematol 2007;14:427–31British Committee for Standards in Hematology (BCSH). Guidelines for the investigation and man-agement of idiopathic thrombocytopenic purpura in adults, children and pregnancy. Br J Haematol 2003;120:574–96Bussel JB, Kuter DJ, George JN et al. AMG 531, a thrombopoiesis stimulating protein, for chronic ITP. N Engl J Med 2006;355:1672–81Cines DB, McMillan R. Management of adult idiopathic thrombocytopenic purpura. Annu Rev Med 2005;56:425–42George JN, Woolf SH, Raskob GE. Idiopathic thrombocytopenic purpura: a guideline for diagnosis and management of children and adults. American Society of Hematology. Ann Med 1998;30:38–44McMillan R, Durette C. Long-term outcomes in adults with chronic ITP after splenectomy failure. Blood 2004;104:956–60Portielle JEA, Westendorp RGJ, Kluin-Nelemans et al. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001;97:2549–54Stasi R, Stipa E, Masi M et al. Long-term observation of 208 adults with chronic idiopathic thrombocyto-penic purpura. Am J Med 1995;98:436–42
1. http://www.pdsa.org/ PDSA2. http://www.emedicine.com/EMERG/topic282.htm E-medicine3. http://www.emedicine.com/med/topic1151.htm E-medicine4. http://www.scripps.edu/itp/ Scripps Clinic5. http://www.itpsupport.org.uk ITP Support Assoc
1.2.
3.
4.
5.
6.
7.
8.
Px:Px:
Ref:Ref:
Web:Web:

���
Acquired heparin-induced thrombocytopenia
D69.5
Incidence of HIT type II (see below) with intravenous use of unfractioned heparin (UFH): 2–5%, with use of low-molecular-weight heparin (LMWH): < 0.5%.
Heparin-induced Thrombocytopenia (HIT) Type I• Dose-dependent mild early-onset thrombocytopenia (platelet count 100,000–150,000/µl) in
the initial 2–3 days of heparin treatment (UFH / LMWH)• Caused by minor heparin-induced platelet aggregation, no immunological genesis• Usually self-limiting (after 1–2 days) while heparin administration is ongoing• Frequency of up to 30%
Heparin-induced Thrombocytopenia (HIT) Type II• Dose-independent late-onset thrombocytopenia, 4–20 days after start of heparin treatment
(UFH / LMWH). In patients previously exposed to heparin (< 100 days), reoccurrence within hours
• Severe thrombocytopenia (platelets < 100,000/µl), median platelet count approximately 60,000/µl, rarely < 20,000/µl or decreased to < 50% of the initial count; worsening of thrombo-cytopenia if heparin treatment is continued
• Thromboembolic complications up to 40 days after heparin administration• IgG antibodies mostly against the platelet factor 4 (PF4)–heparin complex
→ Immune complex formation→ Platelet activation due to binding of the immune complex to the Fc receptor (Fcγ RIIA),
PF4 release→ Platelet aggregation, endothelial cell damage, thrombin activation→ Thromboembolic complications (“white clot syndrome”)
Clinical relevance: HIT type II:• Main symptom: thrombophilia, not hemorrhage• Warning signs: exanthema or necrosis at injection site• High incidence (up to 53%) of venous and arterial thrombosis, renal dysfunction, pulmonary
embolism, infarction (complications may occur weeks after discontinuation of heparin)
• Exclusion of other causes of thrombocytopenia (7 Chap. 6.3).• Combination of a functional test (e.g., heparin-induced platelet activation, HIPA) with ELISA
(detection of PF4–heparin complexes).• ATTENTION: if HIT II is clinically suspected, discontinue heparin immediately and use al-
ternatives, even without positive test. The diagnosis of HIT is based on clinical findings. Tests serve as confirmatory tools only.
Exclude other causes of thrombocytopenia (7 Chap. 6.3)
Therapeutic intervention (with HIT type II):• Discontinue heparin treatment (UFH / LMWH). ATTENTION: exclude exposure to “hidden”
heparin, e.g., coagulation factor products, “heparin lock” of central catheters• Anticoagulation must be continued for at least 4 weeks, using:
− Danaparoid sodium: heparin-free heparinoid, ATIII-mediated inhibition of factor Xa, half-life 24 h, renal elimination, monitoring via factor Xa levels, no antidote available
− Hirudin derivatives, e.g., lepirudin: bivalent direct thrombin inhibitor, half-life 1.5 h, renal elimination, monitoring via PTT, no antidote available
Def:Def:
ICD-10:ICD-10:
Ep:Ep:
Pg:Pg:
Sy:Sy:
Dg:Dg:
Dd:Dd:
Th:Th:
�.� HIT
6.3.2 Heparin-induced Thrombocytopenia (HIT)
A.K.Kaskel,J.Heinz

���
Part � Hematology and Hemostasis
– Argatroban: direct thrombin inhibitor, interacts with the active site of thrombin. Half-live 24 min., monitored by PTT. No dose adjustment in renal failure, due to hepatic elimina-tion.
• In cases of existing thrombosis: coumarin overlapping with danaparoid or hirudin. • Avoid using LMWH (cross-reaction)
Alving BM. How I treat heparin-induced thrombopenia and thrombosis. Blood 2003;101:31–7Arepally GM, Ortel TL. Heparin-induced thrombocytopenia. N Engl J Med 2006;355:809–17Keeling D, Davidson S, Watson H. British Committee for Standards in Haematology. The management of heparin-induced thrombocytopenia. Br J Haematol 2006;133:259–69Newman PM, Chong BH. Heparin-induced thrombocytopenia: new evidence for the dynamic binding of purified anti-PF4-heparin antibodies to platelets and the resultant platelet activation. Blood 2000;96:182–7Rice L, Attisha WK, Drexler A et al. Delayed-onset heparin-induced thrombocytopenia. Ann Intern Med 2002;136:210–5
1. http://www.med.unc.edu/isth/welcome Intl Soc Thrombosis Hemostasis2. http://www.tigc.org/eguidelines/hit05.htm TIGC, Guidelines
1.2.3.
4.
5.
Ref:Ref:
Web:Web:

���
Thrombocytopenic thrombotic microangiopathies with hemolytic anemia (microangiopathic he-molytic anemia, MAHA). Subtypes:• Thrombotic-thrombocytopenic purpura (TTP, Moschcowitz disease): main symptoms are mi-
croangiopathic hemolytic anemia, thrombocytopenia, and neurological symptoms; renal dys-function in 50% of cases
• Hemolytic-uremic syndrome (HUS, Gasser’s disease): main symptoms are acute renal failure (renal microangiopathy, glomeruli are particularly affected) and hemolytic anemia; thrombo-cytopenia and neurological symptoms are less pronounced than in TTP
• Toxic microangiopathic hemolytic anemia (toxic MAHA): after treatment with mitomycin C or high-dose chemotherapy
It is not yet clear whether TTP and HUS are separate diseases or whether they are different mani-festations of one syndrome. Due to the frequently overlapping symptoms, the more commonly used term is TTP-HUS (in adult patients). Exception: HUS in children after E. coli infection.
M31.1
TTP: age peak 30–50 years, distribution male:female = 1:2
HUS: incidence 3–5 cases/100,000 children/year, age peak 1–5 years, distribution male:female = 1:1
Thrombotic-Thrombocytopenic Purpura (TTP)• Acquired or congenital (total) dysfunction of the vWF-cleaving protease (= ADAMTS13;
a disintegrin and metalloprotease with thrombospondin type-1 motifs; cleaves vWF be-tween the amino acids 842 and 843), with unusually large von Willebrand factor multimers (UL-vWF-M), particularly in chronically recurrent TTP
• Acquired TTP: autoimmune disease with anti-vWF protease autoantibodies• Associated with infections (HIV), pregnancy, postpartum, after allogeneic bone marrow trans-
plantation, drugs (mitomycin C, cyclosporine, ticlopidine, clopidogrel, quinine), autoimmune diseases (SLE)
Hemolytic-Uremic Syndrome (HUS)• Normal vWF protease activity.• Commonly associated with gastrointestinal infections caused by Shiga toxin or verotoxin-pro-
ducing Escherichia coli (serotypes OH, particularly O157:H7, O103:HU, O103:H2), rarely shi-gella (Shigella dysenteriae serotype I).
• In the absence of gastrointestinal infections, HUS is probably complement-mediated and occurs in connection with autosomal recessively inherited factor H mutations. In sporadic forms, factor H autoantibodies are thought to be involved. In this case, association with glo-merulonephritis type II and involvement of autoantibodies against C3 convertase.
Under physiological conditions, vWF multimers are excreted by endothelial cells and deposited subendothelially. In the case of endothelial damage → complex formation of vWF multimers with thrombocytes → thrombocyte aggregation due to binding to platelet glycoproteins Ib, IX, and V as well as activated GP IIb/IIIa.
In cases of thrombotic microangiopathies, platelet aggregates or microthrombi are formed in cap-illaries and small vessels causing infarction, particularly in CNS and kidney.• Thrombocytopenia due to peripheral destruction• Anemia due to mechanical destruction of erythrocytes in partially thrombosed small vessels
(fragmentocytes, LDH ↑, haptoglobin ↓↓).
Def:Def:
ICD-10:ICD-10:
Ep:Ep:
Pg:Pg:
Path:Path:
�.� TTP-HUS
6.3.3 Thrombotic Microangiopathies (TTP-HUS)
A.K.Kaskel,J.Heinz

���
Part � Hematology and Hemostasis
Symptoms according to disease subtype:• Microangiopathic hemolytic anemia (MAHA): 100%; icterus, signs of acute hemolysis, pallor,
reduced performance• Thrombocytopenia (more common in TTP): 60–90%; petechiae, bruising, epistaxis, hemor-
rhage, bleeding• Neuropathy (more common in TTP): 70–90%; central neurological disorders, confusion,
cramps, headache, impaired vision, cerebellar ataxia, coma• Nephropathy (more common in HUS): 65%; hematuria, oliguria / anuria, renal failure• Fever: 30–50%• In infection-associated forms: preceding watery / bloody diarrhea caused by E. coli / shigella,
with abdominal pain, cramps• ARDS-like pulmonary complications
Clinical Diagnosis• Medical history (particularly infection)• Physical examination: type of hemorrhage, signs of infections, neuropathy, nephropathy (he-
maturia, oliguria, anuria), pulmonary symptoms
Laboratory Tests• Anemia, thrombocytopenia• Differential blood count / smear: reticulocytosis, fragmentocytes, anisocytosis, poikilocytosis• Signs of intravascular hemolysis: LDH ↑, haptoglobin ↓↓, bilirubin ↑• Coombs’ test negative (not antibody-mediated)• Renal dysfunction: creatinine ↑, urea ↑, electrolytes, uric acid ↑• Urine: proteinuria (1–2 g/24 h, up to 10 g/24 h), hematuria• Bleeding time ↑, fibrin monomers / fibrinogen cleavage products ↑• ELISA to detect Shiga toxin (EHEC)• Determination of the vWF protease activity (ADAMTS13)
• ITP → no hemolysis constellation• DIC / sepsis → lack of coagulation factors• Evans’ syndrome (autoimmunohemolysis and ITP) → positive direct Coombs’ test• Glomerulonephritis → hypertension, urine results, liver / kidney function ↓, kidney biopsy• Infections: malaria, leptospirosis, dengue fever, hantavirus infection
• Cardiac complications: ischemia, infarction, arrhythmia• Brain hemorrhage (rare)
Thrombotic microangiopathies constitute a hematological emergency → immediate specific treatment is of vital importance. Without adequate treatment, the mortality rate is 90%.
Plasmapheresis• Plasma exchange via pheresis with fresh frozen plasma (FFP) initially 40 ml/kg daily• Aim: depletion of vWF multimers and autoantibodies, substitution of vWF protease (t½ > 24 h)
through FFP or as cryoprecipitate• Success parameters: normalization of LDH and platelets, regression of neurological symptoms;
once laboratory parameters have normalized, lengthening of pheresis intervals• If symptoms persist: increase pheresis frequency to twice daily or raise volume to 80 ml/kg (in
individual cases, as much as 140 ml/kg/day may be indicated → however, twice daily phere-sis seems to be more effective); in addition, prednisone (1 mg/kg/day) or methylprednisolone (125 mg i.v. twice daily) and possibly vincristine or immunoglobulins
• Pheresis is often accompanied by moderate citrate toxicity (muscle cramps, tetany) → calcium replacement
• Even with adequate treatment, full reconstitution of renal function may be delayed
Sy:Sy:
Dg:Dg:
Dd:Dd:
Co:Co:
Th:Th:

���
Additional Treatment Options• With suspected acquired TTP: prednisolone 3 × 50 mg/day i.v. or p.o. over 1 week, withdraw
gradually over a period of at least 4 weeks• Patients with acquired antibody-mediated TTP who respond insufficiently to plasmapheresis
or have relapsed: additional immunosuppressive treatment, e.g., splenectomy, immunoad-sorption via protein A column, possibly azathioprine or other immunosuppressives (e.g., anti-CD20 antibody rituximab ± cyclophosphamide, cyclosporine).
• Congenital vWF protease deficiency: treatment according to symptoms: replacement of vWF protease ± plasmapheresis, prophylactic platelet aggregation inhibitors may be required with platelet recovery.
ATTENTION: Platelet transfusion only after careful benefit-risk assessment (e.g., life-threaten-ing hemorrhage) → possible deterioration of symptoms (increased intravascular thrombus formation).
Supportive Treatment• Hypovolemia: fluid replacement / hypovolemia control• Hypertension: antihypertensive treatment → in acute cases: nitrate / beta blockers, long-term
treatment: ACE inhibitors• Dialysis as required• Severe anemia: packed red cells
With adequate treatment (plasmapheresis, dialysis, supportive treatment), good prognosis:• Response rate: 80–90%, mortality 5–20%• Relapse rate: 15–20%• In 15–20% of cases, chronic disease-related effects: renal dysfunction, residual cerebral disor-
ders
Allford SL, Hunt BJ, Rose P et al. British Committee for Standards in Haematology. Guidelines on the diagnosis and management of the thrombotic microangiopathic purpura. Br J Haematol 2003;120:556–73George JN. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35Ho VT, Cutler C, Carter S et al. Blood and Marrow Transplant Clinical Trials Network Toxicity Commit-tee Consensus Summary: thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2005;11:571–5Levy GG, Motto DG, Ginsburg D. ADAMTS13 turns 3. Blood 2005;106:11–17Plaimauer B, Zimmermann K, Volkel D et al. Cloning, expression and functional characterization of the von Willebrand factor-cleaving protease (ADAMTS13). Blood 2002;100:3626–32Richards A, Goodship JA, Goodship THJ. The genetics and pathogenesis of HUS and TTP. Curr Opin Nephrol Hypertens 2002;11:431–5Sadler JE, Moake JL, Miyata T et al. Recent advances in thrombotic thrombocytopenic purpura. Hematol-ogy (ASH Educ Program) 2004:407–23Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uremic syn-drome. Lancet 2005;365:1073–86
1. http://moon.ouhsc.edu/jgeorge/TTP.html TTP-HUS Registry2. http://www.emedicine.com/emerg/topic579.htm TTP, E-medicine3. http://www.emedicine.com/emerg/topic238.htm HUS, E-medicine4. http://www.psbc.org/bulletins/bulletin_v7_n2.pdf Puget Sound Blood Center5. http://www.crttp.org/ TTP Foundation6. http://www.nlm.nih.gov/medlineplus/ency/article/000552.htm MedlinePlus7. http://www.nlm.nih.gov/medlineplus/ency/article/000510.htm MedlinePlus
1.
2.3.
4.5.
6.
7.
8.
Prg:Prg:
Ref:Ref:
Web:Web:
�.� TTP-HUS

Hematology and HemostasisPart �
���
Reduced hemoglobin concentration and hematocrit. Red blood cell (RBC) number below normal level.
Redbloodcell(RBC)parameters
Parameter Abbreviation Normal value
Hemoglobin Hb ♂ 14–18 g/dl, ♀ 12–16 g/dlHematocrit Hkt ♂ 40–52%, ♀ 37–48%Erythrocyte count Ery ♂ 4.3–5.7 × 106/µl, ♀ 3.9–5.3 × 106/µlMean corpuscular volume MCV 85–98 flMean corpuscular hemoglobin MCH, HbE 28–34 pgMCH concentration MCHC 32–37 g/dlErythrocyte diameter 6.8–7.3 µmReticulocyte count Reti 0.3–1.5%
NomenclatureofRedCellChanges
Size (Indices: Erythrocyte Diameter, MCV)• Macrocytosis: erythrocyte diameter ↑, MCV ↑• Microcytosis: erythrocyte diameter ↓, MCV ↓• Anisocytosis: pronounced variations in size of RBC
Shape• Poikilocytosis: different RBC shapes in blood smear• Elliptocytes: oval RBC• Spherocytes: spherical cells• Target cells: target-like appearance• Acanthocytes: irregularly spiculated cells, “spur cells”• Schistocytes: RBC fragments, fragmentocytes• Dacryocytes: drop-shaped cells, “teardrop” RBC• Drepanocytes: sickle cells (bipolar spiculated cells)
Staining (Indices: MCH, MCHC)• Hypochromic: RBC staining ↓, MCH ↓• Hyperchromic: RBC staining ↑, MCH ↑• Polychromatic: reddish-blue-gray staining
Cell Inclusions• Howell-Jolly bodies: basophilic inclusions (nuclear remnants)• Basophil stippling: punctuate basophilic inclusions (ribosomes)• Heinz bodies: denatured hemoglobin (special staining required)• Cabot’s rings: basophilic circular threadlike inclusions (nuclear remnants)
Def:Def:
Phys:Phys:
6.4 Anemia
D.P.Berger,R.Engelhardt

���
Erythropoiesisandclassificationofanemias
Symptoms of Anemia• Pallor of skin and mucous membranes, nail beds, conjunctivae• Weakness, tiredness, reduced performance• Lack of concentration, headache, vertigo• Dyspnea, tachycardia, palpitations (esp. with acute anemia)
History, Physical Examination• Risk factors, esp. infections, drugs, bleeding (menstruation history), nutritional habits• Physical examination including skin, mucous membranes, lymph node status, spleen / liver
findings, heart (tachycardia, particularly systolic murmur), rectal examination with fecal blood test, gynecological examination
Laboratory Tests• Hematology: blood count, with MCV, MCH, reticulocytes, differential blood count, blood
smear• Clinical chemistry: routine tests with bilirubin, renal function parameters, total protein, pro-
tein electrophoresis, iron status (iron, ferritin, transferrin-binding capacity), parameters of hemolysis (bilirubin, LDH, haptoglobin), CRP, BSG, vitamin B12 / folinic acid
• Coombs’ test (if hemolytic anemia is suspected)• Virus serology (including parvovirus B19)• Blood group• Erythropoietin level (if renal anemia is suspected)
HistologyBone marrow aspiration / biopsy, with iron stain (if stem cell/bone marrow disorder is suspected)
Pphys:Pphys:
Sy:Sy:
Dg:Dg:
�.� Anemia

��0
Part � Hematology and Hemostasis
Differentialdiagnosisofanemia
Hypochromic anemia Normochromic anemia Hyperchromic anemia
MCH ↓ MCH normal MCH ↑
Iron deficiency Tumor Inflammation, infection Thalassemia
Hemolysis Acute blood loss Aplastic anemia Renal anemia
Megaloblastic anemia (Vitamin B12 or folinic acid deficiency) Myelodysplastic syndromes
Supportive TreatmentSubstitution of packed red blood cells: restrictive indication (7 Chap. 4.9.1).
Guidelines for Transfusion Indication• Individual assessment of transfusion indication for each patient.• In acute blood loss, consider indication when hemoglobin < 8.0 g/dl.• With chronic anemia lower levels of hemoglobin (6–8 g/dl) are generally tolerated.• Patients with coronary heart disease or risk of cerebral ischemia: transfusion indication at
hemoglobin < 10 g/dl.• Specific conditions (surgery, thalassemia major, etc.) may require RBC transfusion support.
The indication for transfusion is based on clinical symptoms. Asymptomatic blood loss does not constitute an indication for transfusion.
Birgegard G, Aapro MS, Bokemeyer C et al. Cancer-related anemia: pathogenesis, prevalence and treat-ment. Oncology 2005;68(suppl 1):3–11Bokemeyer C, Aapro MS, Courdi A et al. EORTC guidelines for the use of erythropoietic proteins in anae-mic patients with cancer. Eur J Cancer 2004;40:2201–16British Committee for Standards in Hematology (BCSH). Guidelines for the clinical use of red cell transfu-sion. Br J Haematol 2001;113:24–31Littlewood TJ. The impact of hemoglobin levels on treatment outcomes in patients with cancer. Semin Oncol 2001;28(suppl 8):49–53Provan D, Weatherall D. Red cells I: inherited anaemias. Lancet 2000;355:1169–75Provan D, Weatherall D. Red cells II: acquired anemias and polycythaemia. Lancet 2000;355:1260–8Rizzo JD, Somerfield MP, Hagerty KL et al. ASH/ASCO 2007 clinical practice guideline update on the use of epoetin and darbepoetin. Blood 2007;111:x–y
1. http://www.anemiainstitute.org/ Anemia Institute2. http://www.guideline.gov/ Guideline Clearinghouse3. http://www.nlm.nih.gov/medlineplus/anemia.html MedlinePlus4. http://www.anemia.org Anemia Action Council
1.
2.
3.
4.
5.6.7.
Dd:Dd:
Th:Th:
Ref:Ref:
Web:Web:

���
Anemias with decreased corpuscular hemoglobin (MCH < 28 pg) and decreased corpuscular he-moglobin concentration (MCHC < 32%):• Iron deficiency anemia (> 90% of hypochromic anemias)• Anemia of chronic disease (inflammation- / infection- / tumor anemia)• Thalassemia (7 Chap. 6.4.3)• Rare causes: vitamin B6 deficiency, lead intoxication
Hypochromicanemia
Parameter Iron deficiency anemia
Inflammation- / tumor anemia
β-Thalassemia
Serum iron ↓ ↓ normal / ↑Transferrin ↑ ↑ normal / ↓Serum ferritin ↓ ↑ normal / ↑
IronDeficiencyAnemia
Most frequent form of anemia. Proportion male:female = 1:5. About 10–20% of women in child-bearing age demonstrate latent iron deficiency.
Ironmetabolism
Daily iron resorption required: men 1 mg, women and adolescents 2–3 mg, pregnant women 3–4 mg. About 60–70% of body iron store bound in hemoglobin, additional 10% in myoglobin. 1 g hemoglobin contains 3.4 mg of iron.
In iron deficiency the iron need is greater than the available iron supply, resulting in hemoglobin synthesis disorders → microcytic, hypochromic erythrocytes.
Parameters• Ferritinserum: correlates with total iron (↓ in iron deficiency)• Transferrinserum: correlates with circulating iron and need (↑ in iron deficiency)
Most important cause: loss of iron due to chronic bleeding → in manifest iron deficiency eval-uation of underlying cause is of central importance.
Def:Def:
Dd:Dd:
Ep:Ep:
Phys:Phys:
Pg:Pg:
�.� Hypochromic Anemia
6.4.1 Hypochromic Anemia
D.P.Berger,R.Engelhardt,T.Heinz

���
Part � Hematology and Hemostasis
Causes of Iron Deficiency• Poor iron uptake: infants, small children, vegetarians, alcoholics, nutritional disorders.• Recommended daily uptake: men 12 mg, women 15 mg, pregnancy 30 mg.• Decreased resorption: postoperative (gastrectomy), malassimilation• Increased need: growth, pregnancy, lactation period, during treatment of vitamin B12 defi-
ciency• Blood loss: urogenital / gastrointestinal bleeding, cystitis, angiodysplasia, esophagitis, hemor-
rhoids• Infection / parasites (worldwide most frequent cause of iron deficiency: hookworm infection)
Peripheral BloodMicrocytic, hypochromic erythrocytes, poikilocytosis, anisocytosis, anulocytes.
Bone MarrowIron stain (Prussian blue stain): storage iron not detectable (ferritin, hemosiderin).
Symptoms of Anemia• Pallor of skin and mucous membranes, nail beds, conjunctivae• Weakness, tiredness, reduced performance• Lack of concentration, headache• Exertional dyspnea, tachycardia, palpitations (DD: cardiac failure)
Symptoms of Iron Deficiency• Skin and nail changes: skin atrophy, spoon-shaped nails (koilonychia)• Oral rhagades, impairment of mucous membranes, in extreme cases painful mucous mem-
brane atrophy of tongue, pharynx, and esophagus with dysphagia (Plummer-Vinson syn-drome)
History, Clinical Findings• History, esp. infections, drugs, bleeding, nutritional habits• Physical examination: including skin, mucous membranes, lymph node status, spleen / liver,
heart (tachycardia, particularly systolic murmur), rectal examination with fecal blood test, urine dipstick
• Gynecological examination• Endoscopy: esophago-gastro- duodenoscopy, colonoscopy, rectoscopy
Laboratory Tests• Hematology: blood count, MCV ↓, MCH ↓, reticulocytes, differential blood count• Clinical chemistry: routine tests with bilirubin, renal function parameters, iron status (iron ↓,
ferritin ↓, transferrin-binding capacity ↑)• Blood group (if red cell substitution necessary)• Iron resorption test (if resorption deficiency is suspected)
HistologyIn inconclusive cases eventually bone marrow aspiration / biopsy, including iron staining, to ex-clude other causes of anemia.
• Anemia of chronic disease (iron ↓, ferritin normal or elevated, transferrin-binding capac-ity ↓)
• Thalassemia (MCV ↓↓, iron, ferritin, and transferrin-binding capacity normal)• Hemolytic anemia (bilirubin, LDH, haptoglobin, Coombs’ test)
Treatment of anemia with iron deficiency always requires a combined approach:1. Treatment of the underlying cause of iron deficiency (e.g., chronic blood loss)2. Iron substitution
Path:Path:
Sy:Sy:
Dg:Dg:
Dd:Dd:
Th:Th:Th:Th:

���
Oral Iron Substitution• Application of ferrous II preparation, e.g., Fe(II) sulfate, fumarate, gluconate, or succinate,
100–200 mg/day p.o., for 2–6 months.• PKIN: oral bioavailability, depending on preparation, 15–25%, better bioavailability when
taken prior to food.• SE: gastrointestinal tract symptoms (nausea, vomiting,), dark discoloration of stool (ATTEN-
TION misdiagnosis: upper gastrointestinal bleeding).• Treatment monitoring: after 5–7 days reticulocytes ↑, hemoglobin ↑. Most frequent cause of
a treatment failure is lack of compliance, followed by combined anemia (e.g., coexisting iron deficiency and lack of vitamin B12).
Parenteral Iron Substitution• Parenteral application of iron should be limited to individual cases (e.g., in malabsorption
syndrome), due to severity of side effects.• Strictly intravenous application of ferrous(III) preparations, consider premedication with ste-
roids and antihistaminics.• SE: thrombophlebitis, headache, flush, nausea, vomiting, fever, allergic reactions up to ana-
phylaxis. With paravenous injection local pain and visible iron deposits in tissue.
Red Cell SubstitutionApplication of packed red blood cells is generally not indicated in iron deficiency anemia. Excep-tions exist in patients with additional blood loss and clinical symptoms.
AnemiaDuetoInflammation,Infection,Tumor:AnemiaofChronicDisease(ACD)
Second most common form of anemia (after iron deficiency anemia).
Multifactorial anemia with chronic underlying disease (malignancy, inflammation, infection, col-lagen diseases). Pathogenetic factors:• Cytokine-mediated (TNFα, interleukin-1, interferon γ) → erythrocyte-survival time ↓, in-
terference with iron mobilization from reticuloendothelial iron stores (macrophages), iron uptake / utilization in normoblasts ↓, erythropoietin secretion and effect ↓, inhibition of ery-throid progenitor cells, etc.
• Treatment-associated (drugs, radiation therapy, etc.)• Consequence of underlying disease
Peripheral BloodNormochromic, normocytic or hypochromic, microcytic red blood cells, poikilocytosis, aniso-cytosis.
Symptoms of Anemia• Pallor of skin and mucous membranes, nail beds, conjunctivae• Weakness, tiredness, reduced performance, exertional dyspnea• Lack of concentration, headache
Symptoms of Underlying DiseaseDepending on disease, generally with• Tiredness, weakness, reduced performance• Fever, weight loss, night sweats (B symptoms)• Loss of appetite, myalgia, arthralgia, etc.
History, Clinical Findings• History: infections, drugs, exposition to hazardous substances, bleeding• Physical examination: skin, mucous membranes, lymph node status, spleen / liver, heart
(tachycardia, systolic murmur), rectal examination with fecal blood test
Ep:Ep:
Pg:Pg:
Path:Path:
Sy:Sy:
Dg:Dg:
�.� Hypochromic Anemia

���
Part � Hematology and Hemostasis
Laboratory Tests• Hematology: blood count, MCV (normal / ↓), MCH (normal / ↓), reticulocytes, differential
blood count• Clinical chemistry: renal function parameters, iron status (iron ↓, ferritin ↑, transferrin-bind-
ing capacity ↑), ESR ↑, fibrinogen ↑, CRP ↑, haptoglobin ↑ (acute-phase protein), possibly erythropoietin level
• Blood group (if red cell substitution necessary)
HistologyIn inconclusive cases consider bone marrow aspiration / biopsy, including iron staining, to ex-clude other causes of anemia.
• Iron deficiency anemia (iron ↓, ferritin ↓, transferrin- binding capacity ↑)• Thalassemia (MCV ↓↓, iron, ferritin, and transferrin-binding capacity normal)• Megaloblastic anemias (vitamin B12 / folinic acid)• Hemolytic anemia (bilirubin, LDH, haptoglobin, Coombs’ test)
Treatment of underlying disease
Donovan A, Andrews NC. The molecular regulation of iron metabolism. Hematol J 2004;5:373–80Goodnough LT, Skikne B, Brugnara C. Erythropoietin, iron, and erythropoiesis. Blood 2000;96:823–33Littlewood TJ. The impact of hemoglobin levels on treatment outcomes in patients with cancer. Semin Oncol 2001;28(2 suppl 8):49–53Means RT. Advances in the anemia of chronic disease. Int J Hematol 1999;70:7–12Thomas C, Thomas L. Anemia of chronic disease: pathophysiology and laboratory diagnosis. Lab Hematol 2005;11:14–23Umbreit J. Iron deficiency. Am J Hematol 2005;78:435–43Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med 2005;352:1011–23Zimmermann MB, Hurrell RF. Nutritional iron deficiency. Lancet 2007;370:511–20
1. http://www.nlm.nih.gov/medlineplus/ency/article/000565.htm Medline Plus2. http://www.nlm.nih.gov/medlineplus/ency/article/000584.htm Medline Plus3. http://www.umm.edu/blood/aneiron.htm Univ Maryland4. http://www.emedicine.com/med/topic1188.htm E-medicine
1.2.3.
4.5.
6.7.8.
Dd:Dd:
Th:Th:
Ref:Ref:
Web:Web:

���
Anemia with increased erythrocyte volume (MCV > 98 fl), usually caused by lack of vitamin B12 (cobalamin) and/or folic acid.
VitaminB12DeficiencyAnemia
Incidence 5–10 cases/100,000 population/year, distribution male:female = 3:2, age peak 60 years
VitaminB12metabolism
The reference nutrient intake (RNI) for vitamin B12 is 1 µg, with maximum daily absorption in the terminal ileum of 2–3 µg. “Intrinsic factor” (glycoprotein) is a prerequisite for vitamin B12 resorption.
Function of Vitamin B12 (Cobalamin)• Cofactor in the synthesis of succinyl CoA, methionine, and tetrahydrofolic acid• In case of vitamin B12 deficiency:
→ DNA synthesis and fatty acid metabolism impaired→ Delayed nuclear maturation, normal cytoplasmic development→ Ineffective myelopoiesis, large cells with altered nucleus: plasma ratio
Causes of Vitamin B12 Deficiency• Most frequent cause: pernicious anemia (80% of cases): autoimmune atrophic gastritis with
antibodies against gastric parietal cells (90% of cases) and/or antibodies against intrinsic fac-tor (50% of cases)→ Achlorhydria, intrinsic factor deficiency→ Decreased vitamin B12 resorption in the terminal ileum
• Insufficient vitamin B12 uptake (strict vegetarians, alcoholics)• Postoperatively (gastrectomy, resection of the terminal ileum, blind loop syndrome)• Vitamin B12 malabsorption, rare (Crohn’s disease, scleroderma, amyloidosis)• Infections / parasites (fish tapeworm, bacterial gastrointestinal infections)
Peripheral BloodMacrocytic hyperchromic erythrocytes, poikilocytosis, anisocytosis, hypersegmented granulo-cytes (right shift); in severe cases, granulocytopenia and thrombocytopenia.
Def:Def:
Ep:Ep:
Phys:Phys:
Pg:Pg:
Path:Path:
�.� Megaloblastic Anemia
6.4.2 Megaloblastic Anemia
D.P.Berger,R.Engelhardt,J.Heinz

���
Part � Hematology and Hemostasis
Bone MarrowMegaloblastic changes: ineffective left-shifted erythro-, thrombo-, and granulopoiesis, pronounced erythropoiesis with increased numbers of immature erythroid precursors (erythropoietic hyper-plasia with megaloblastic erythroblasts), giant band forms, immature megakaryocytes.
Anemia-related Symptoms• Pale skin and mucous membranes, icterus (due to intramedullary hemolysis)• Weakness, fatigue, reduced performance, dyspnea on exertion• Difficulty concentrating, headache
Neurological SymptomsIn advanced cases: funicular myelosis: neuropathy caused by symmetrical damage of the posterior columns of the spinal cord, the corticospinal tract and peripheral nerves; motor abnormalities mainly affecting the lower extremities; staggering gait, ataxia, spastic paresis, impaired vision, psy-chological disorders.
Gastrointestinal and Other Symptoms• Type A gastritis• Trophic disorders of the skin and mucous membranes: Hunter’s glossitis, etc.• Sterility (gonad dysfunction), reversible
Medical History, Physical Examination• Medical history: infections, drugs, hemorrhage, nutritional habits• Physical examination: skin, mucous membranes, lymph node status, spleen / liver, heart
(tachycardia, in some cases: systolic cardiac murmur), rectal examination and test for fecal blood, neurological examination
Laboratory Tests• Hematology: blood count with MCV (↑), MCH (↑), reticulocytes (↓), differential blood count• Clinical chemistry: liver and renal function tests, total protein, hemolysis parameters (biliru-
bin ↑, LDH ↑↑, haptoglobin ↓ due to intramedullary hemolysis)• Antibodies against gastric parietal cells and/or against intrinsic factor• Vitamin B12 serum level (normal: 200–900 pg/ml), folic acid serum level• Vitamin B12 absorption test (Schilling’s test): oral administration of radioactive B12 ± intrinsic
factor, determination of urinary vitamin B12, comparison of vitamin B12 absorption / excre-tion with and without intrinsic factor
• Blood group (if red cell transfusion is necessary)
Histology• Gastroscopy: detection of chronic atrophic gastritis, exclusion of gastric carcinoma (incidence
3 times higher with chronic atrophic gastritis)• Bone marrow aspiration / biopsy to confirm megaloblastic abnormalities
Other Causes of Macrocytosis• Alcoholism (most common cause of a macrocytic blood count)• Hepatic disorders, severe hypothyroidism• Reticulocytosis, myelodysplasia (7 Chap. 7.2), paraproteinemia• Cytostatic agents (antimetabolites, anthracyclines, anthracenediones, etc.)• Pregnancy, neonates
Other Forms of Anemia• Hypochromic anemia (iron deficiency anemia, anemia of chronic disease)• Hemolytic anemia (bilirubin, LDH, haptoglobin, Coombs’ test)• Parvovirus B19, renal anemia
Sy:Sy:
Dg:Dg:
Dd:Dd:

���
Vitamin B12 SubstitutionHydroxycobalamin 1 mg i.m. → initially: 6 injections within 2–3 weeks (to replenish vitamin B12 stores), then: one injection every 3 months. Additionally: application of ferrous II preparation and folic acid to cover increased erythropoesis during substitution phase.ATTENTION: close monitoring during the first days of treatment: critical increase in reticulo-cytes and platelets possible → increased risk of thrombosis, potassium and iron deficiency.
Gastroscopy at regular intervals due to increased risk of gastric cancer.
FolicAcidDeficiencyAnemia
Rare disorder
Folicacidmetabolism
Reference nutrient intake (RNI) for folic acid: 100–200 µg, during pregnancy 400 µg.
Function• Folic acid is a cofactor of thymidylate synthesis (C1 transfer), i.e., DNA synthesis• In case of folic acid deficiency:
→ Disorder of DNA synthesis→ Delayed nuclear maturation with normal cytoplasmic development→ Ineffective myelopoiesis, giant cells with an abnormal nucleus: plasma ratio
Causes of Folic Acid Deficiency• Insufficient folic acid intake: nutritional deficiency, alcoholism, anorexia nervosa• Malabsorption: gluten-induced enteropathy, tropical sprue, Crohn’s disease, scleroderma, amy-
loidosis, postoperatively (small bowel resection, gastrectomy)• Increased demand: pregnancy, chronic hemolytic anemia, chronic inflammatory disease, or
malignancies• Loss of folic acid: hemodialysis• Drug-induced (with folic acid antagonists): methotrexate, trimethoprim, pyrimethamine,
phenytoin, triamterene
Peripheral Blood and Bone MarrowSee Vitamin B12 Deficiency Anemia
Anemia-related Symptoms• Pale skin and mucous membranes, icterus (due to intramedullary hemolysis)• Weakness, fatigue, reduced performance, dyspnea on exertion• Difficulty concentrating, headache
Folic Acid Deficiency-related Symptoms• Folic acid deficiency during pregnancy: increased incidence of neural tube defects (spina bi-
fida, anencephaly)• Sterility (gonadal dysfunction), reversible
Th:Th:
Ep:Ep:
Phys:Phys:
Pg:Pg:
Path:Path:
Sy:Sy:
�.� Megaloblastic Anemia

���
Part � Hematology and Hemostasis
Medical History, Physical Examination• Case history including infections, drugs, hemorrhage• Physical examination: skin, mucous membranes, lymph node status, spleen / liver, heart
(tachycardia, in some cases: systolic cardiac murmur), rectal examination and test for fecal occult blood
Laboratory Tests• Hematology: blood count with MCV (↑), MCH (↑), reticulocytes (↓), differential blood count• Clinical chemistry: liver and renal function tests, total protein, hemolysis parameters (biliru-
bin ↑, LDH ↑, haptoglobin ↓ due to intramedullary hemolysis)• Vitamin B12 level, folic acid level (normal: 6–20 ng/ml)• Blood group (if red cell transfusion is necessary)
Histology• Esophago-gastro-duodenoscopy: exclusion of gluten-sensitive enteropathy (sprue)• Bone marrow aspiration / biopsy to confirm megaloblastic abnormalities
See Vitamin B12 Deficiency Anemia
Folic Acid SubstitutionFolic acid 5 mg daily p.o. for 4 months.
Dharmarajan TS, Norkus EP. Approaches to vitamin B12 deficiency. Early treatment may prevent devastat-ing complications. Postgrad Med 2001;110:99–105Fenech M. The role of folic acid and vitamin B12 in genomic stability of human cells. Mutat Res 2001;475:57–67Provan D, Weatherall D. Red cells II: acquired anaemias and polycythaemia. Lancet 2000;355:1260–8Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med 1997;337:1441–8Wickramashinghe SN. The wide spectrum and unresolved issues of megaloblastic anemia. Semin Hematol 1999;36:3–18Zittoun J, Zittoun R. Modern clinical testing strategies in cobalamin and folate deficiency. Semin Hematol 1999;36:35–46
1. http://www.nlm.nih.gov/medlineplus/ency/article/000567.htm Medline Plus2. http://web.indstate.edu/thcme/mwking/vitamins.html Introduction to Vitamins3. http://www.umm.edu/blood/aneper.htm Univ Maryland4. http://www.emedicine.com/MED/topic1420.htm E-medicine5. http://www.ashimagebank.org ASH Image Bank
1.
2.
3.4.5.
6.
Dg:Dg:
Dd:Dd:
Th:Th:
Ref:Ref:
Web:Web:

���
Anemia caused by erythrocyte destruction characterized by decreased erythrocyte survival (< 120 days)
Physiological Erythrocyte TurnoverIn the bone marrow, 2 × 1011 erythrocytes are produced per day; median erythrocyte survival: 120 days; erythrocyte destruction in spleen and liver (reticuloendothelial system, RES).
Hemoglobindegradation
Peripheral BloodGenerally, normochromic normocytic anemia with normal leukocytes and platelets; characteristic changes in cases of hereditary membrane defects (spherocytes, elliptocytes, etc.); anisocytosis, poikilocytosis, and, in some cases, fragmentocytes.
Def:Def:
Phys:Phys:
Path:Path:
�.� Hemolytic Anemia
6.4.3 Hemolytic Anemia
D.P.Berger,R.EngelhardtJ.Heinz

��0
Part � Hematology and Hemostasis
Bone MarrowErythropoietic hyperplasia, increase in erythroblasts.
Corpuscular Hemolytic Anemia (Erythrocyte Defects)
Hereditary Membrane Defects• Spherocytosis• Elliptocytosis• Stomatocytosis• Acanthocytosis
Hereditary Enzyme Defects• Glucose-6-phosphate dehydrogenase deficiency (G6PD deficiency)• Pyruvate kinase deficiency (PK deficiency)
Stem Cell Defects• Paroxysmal nocturnal hemoglobinuria (PNH)
Defects in Hemoglobin Synthesis• Sickle cell anemia and other hemoglobinopathies• Thalassemia
Extracorpuscular Hemolytic Anemia (Extraerythrocytic Defects)
Autoimmune Hemolytic Anemia• Warm antibody autoimmune hemolytic anemia (AIHA)• Cold antibody autoimmune hemolytic anemia (AIHA)• Isoimmune hemolytic anemia: transfusion reactions, rhesus incompatibility
Microangiopathic Hemolytic Anemia (MAHA)• Thrombotic-thrombocytopenic purpura (TTP)• Hemolytic-uremic syndrome (HUS)
Metabolic Disorders• Zieve’s syndrome: hemolytic anemia + alcohol-induced hepatic disease + hyperlipidemia
Hemolysis Due to Erythrocyte Damage• Traumatic hemolysis (after cardiac valve replacement, march hemoglobinuria)• Chemically induced hemolysis (snake poison)• Thermal hemolysis (burns)• Infection-associated hemolysis (malaria)• Drug-induced hemolysis
Anemia-related Symptoms• Pale skin / mucous membranes, icterus (hemolysis / bilirubin release)• Weakness, fatigue, reduced performance• Difficulty concentrating, headache• Dyspnea on exertion, tachycardia, palpitations (particularly with acute hemolysis)
Chronic HemolysisChronic hemolysis is usually associated with a lack of symptoms. Some patients can tolerate he-moglobin levels below 8 mg/dl without subjective restraints.• Low-grade icterus• Splenomegaly• Bilirubin gall stones
Class:Class:
Sy:Sy:

���
Acute Hemolysis (“Hemolytic Crisis”)• Fever, chills• Headache, back pain, abdominal pain• Icterus, hemoglobinuria
Medical History, Physical Examination• Medical history: infections, drugs, hemorrhage, family history• Physical examination: skin, mucous membranes, lymph node status, spleen / liver, heart
(tachycardia, in some cases systolic heart murmur), rectal examination and fecal occult blood test (exclusion of hemorrhagic anemia)
Laboratory Tests• Hematology: blood count with MCV, MCH, reticulocytes, differential blood count, blood
smear• Clinical chemistry: electrolytes (K+ ↑), liver and renal function tests, total protein, protein elec-
trophoresis, iron status (iron ↑, ferritin ↑), hemolysis parameters (indirect bilirubin ↑, LDH ↑, haptoglobin ↓), CRP
• Coombs’ test: direct (detection of erythrocyte-adherent antibodies) or indirect (detection of serum antibodies)
• Viral serology (including parvovirus B19), mycoplasma• Blood group
Hemolysis Parameters• LDH ↑, HBDH ↑, serum iron ↑• Indirect bilirubin ↑, urinary urobilinogen ↑• Haptoglobin ↓• Hemoglobin ↓, hematocrit ↓, erythrocyte count ↓• Reticulocytes ↑ (with chronic hemolysis)• Erythrocyte survival time ↓
HistologyConsider bone marrow aspiration / biopsy, including iron stain, to exclude other causes of anemia.
Therapeutic options depend on the anemia subtype. Treatment components are:• Supportive treatment: red cell transfusion (only in individual cases with symptomatic anemia,
controversial in cases of autoimmune hemolytic anemia)• Treatment of underlying disease• Immunosuppression (in cases of autoimmune hemolytic anemia)• Splenectomy → removal of the sequestration filter for damaged erythrocytes
ATTENTION Splenectomy• Splenectomy can correct the decrease in erythrocyte survival, but it is not a causal therapy in
the sense of a correction of the triggering hemolytic defect.• Prior to splenectomy, MANDATORY vaccination against Streptococcus pneumoniae, Neisseria
meningitidis, and Haemophilus influenzae because of the sepsis risk.• After splenectomy, prevention of thromboembolic events (platelets ↑) → low-dose heparin.
Membrane Defects
HereditarySpherocytosis
Most common hereditary hemolytic disease; prevalence 0.02%; in most cases hereditary disease (autosomal dominant), spontaneous mutation is rare.
Dg:Dg:
Th:Th:
Ep:Ep:
�.� Hemolytic Anemia

���
Part � Hematology and Hemostasis
Genetic modifications of erythrocyte membrane components: ankyrin (chromosome 8p), β-spec-trin (chromosome 14q), in rare cases α-spectrin or protein 4.2.→ Loss of membrane lipids→ Reduced membrane stability, osmotic resistance ↓, Na+ / H2O influx ↑→ Spherical erythrocytes→ Erythrocyte survival ↓, splenic sequestration ↑
• Anemia, icterus• Recurrent hemolytic crises (particularly after infections)• Splenomegaly (50–95%)• Bilirubin gall stones (20–60%)• Aplastic crises in cases of parvovirus B19 infection
Medical History, Physical Examination• Positive family history (icterus, gall stones, anemia)• Anemia symptoms
Laboratory Tests• Erythrocytes: blood smear with characteristic spherocytes, diameter < 7 µm• Normochromic microcytic anemia, MCV → ↑, MCHC ↑• Hemolysis parameters (LDH ↑, haptoglobin ↓, indirect bilirubin ↑)• Osmotic resistance ↓, negative Coombs’ test (exclusion of immune hemolytic anemia), reticu-
locytes ↑
In cases of severe anemia / hemolytic crises (10–15% of cases): splenectomy (not in patients under 5 years of age and preceded by splenic scintigraphy to exclude accessory spleens); consider subtotal splenectomy. Vaccination against S. pneumoniae, N. meningitidis and H. influenza mandatory.
HereditaryElliptocytosis
Rare, higher incidence in Mediterranean countries / Africa (increased malaria resistance of el-liptocytes)
Heterogenic disease group with > 25% of elliptic erythrocytes; protein defects of the erythrocytic cytoskeleton (spectrin, protein 4.1R)
Usually asymptomatic; only 10–30% of patients have varying degrees of anemia, icterus or hemo-lytic crises
• Positive family history• Blood smear with > 25% elliptocytes
In symptomatic patients: splenectomy
ParoxysmalNocturnalHemoglobinuria
Acquired clonal disorder of myeloid stem cells (i.e., of the erythrocytic, granulocytic, and throm-bocytic line) with somatic mutations of phosphatidylinositol-glycan A (PIG A) → defect of the “phosphatidylinositol-glycan anchor” (PIG anchor)
Rare
Pg:Pg:
Sy:Sy:
Dg:Dg:
Th:Th:
Ep:Ep:
Pg:Pg:
Sy:Sy:
Dg:Dg:
Th:Th:
Def:Def:
Ep:Ep:

���
The PIG anchor fixes various proteins to the cell membrane, including three complement-regulat-ing proteins: CD59 (membrane inhibitor of reactive lysis; MIRL), CD55 (decay accelerating fac-tor; DAF), and “C8 binding protein” (CBP).→ Changes in the PIG anchor lead to a decrease in the respective proteins in the cell membrane→ Reduced resistance against activated complement factors→ Complement-mediated lysis ↑, incidence of thromboembolic events ↑
• Chronic hemolytic anemia• Different severity levels of nocturnal hemolysis (even nocturnal hemolytic crises), with morn-
ing hemoglobinuria• Recurrent thrombosis, particularly portal vein, liver veins (Budd-Chiari syndrome), cerebral
vessels, splenic vein, skin veins (skin necrosis)• Iron deficiency anemia due to chronic loss of iron (renal)
Medical History, Physical Examination• Medical history: circadian occurrence of symptoms• Physical examination: anemia signs and symptoms, urinary discoloration
Laboratory Tests• Normochromic normocytic anemia, in some cases with granulocytopenia and thrombocyto-
penia• Hemolysis parameters (LDH ↑, haptoglobin ↓, indirect bilirubin ↑, hemoglobinuria)• Acid hemolysis test (Ham’s test) and sugar water test (sucrose test), pathological: complement-
mediated lysis after addition of sugar water or acid to the blood sample• Molecular genetic proof of the PIG defect
In rare cases development of aplastic anemia, myelodysplasia, or AML
Supportive Approach• Prophylactic anticoagulant therapy: phenprocoumon. ATTENTION: avoid heparin → possible
complement activation• Iron and folic acid supplementation• In cases of hemolytic crisis: corticosteroids (prednisolone 50–100 mg i.v.), supportive treat-
ment• Blood transfusion: only washed erythrocytes to avoid administration of additional comple-
ment• Eculizumab, antibody against complement c5, inhibits complement-mediated lysis of PNH
erythrocytes
Curative ApproachAllogeneic stem cell transplantation (7 Chap. 5.3): only in severe cases with repeated hemolytic crises or complications (thromboembolic events, etc.)
Enzyme Defects
Glucose-6-phosphateDehydrogenaseDeficiency(G6PDDeficiency,Favism)
Hereditary disease, genetic modification of the glucose-6-phosphate dehydrogenase (> 300 mu-tants worldwide)
One of the most common hereditary diseases worldwide, regional differences in incidence and prevalence. In Africa, Asia, and the Mediterranean region, as much as 20–60% of the population may be affected (patients are more resistant to malaria plasmodia). X-chromosomal recessive in-heritance → mainly males affected. Heterozygotics have two different populations of erythrocytes and usually have less pronounced symptoms.
Pg:Pg:
Sy:Sy:
Dg:Dg:
Co:Co:
Th:Th:
Def:Def:
Ep:Ep:
�.� Hemolytic Anemia

���
Part � Hematology and Hemostasis
G6PD Deficiency→ Defects in the erythrocytic pentose phosphate pathway → NADPH synthesis ↓ → decreased
glutathione (GSH)→ Lysis of erythrocytes due to oxidative stress, hemolytic crises
Triggers• Fava beans (Vica fava)• Infections• Drugs: primaquine, chloroquine, sulfonamides, acetylsalicylic acid, isosorbide dinitrate, an-
thracyclines, etc.• Chemicals: nitrates, nitrite compounds, phenylhydrazine
Hemolytic crisis with:• Fever, chills, icterus, hemoglobinuria• Headache, back pain, abdominal pain
• Positive family history• Decreased erythrocytic G6PD activity• Hemolysis parameters (bilirubin ↑, LDH ↑, haptoglobin ↓), blood smear with Heinz bodies
(denatured hemoglobin oxidation products)
Avoid exposure to triggering agents
PyruvateKinaseDeficiency(PKDeficiency)
Hereditary defect of the enzyme pyruvate kinase, i.e., the erythrocytic glycolysis.
Most common hereditary glycolytic defect (Embden-Meyerhof pathway), autosomal recessive in-heritance. Heterozygotic individuals are usually asymptomatic. Homozygosis (rare) leads to he-molytic anemia.
Pyruvate kinase deficiency results in abnormal glycolysis:→ ATP deficiency → abnormal Na+ / K+–ATPase activity in the erythrocyte membrane→ Membrane instability, hemolysis
Usually asymptomatic. In homozygotic individuals, hemolytic crises may occur.
• Blood smear with acanthocytes, anisocytosis, poikilocytosis• Hemolysis parameters, reduced erythrocytic pyruvate kinase activity
Symptomatic patients: splenectomy; in cases of iron overload: venesection therapy and adminis-tration of desferrioxamine.
Hemoglobinopathies
SickleCellAnemia
Qualitative changes in hemoglobin (hemoglobin S, HBS) with autosomal codominant inheritance and occurrence of sickle-shaped erythrocytes.
Most common hemoglobinopathy (HBS); occurs in particular in Mediterranean regions, Africa, Asia, and the USA (black population). HBS carriers are more resistant to malaria plasmodia.
Pg:Pg:
Sy:Sy:Sy:Sy:
Dg:Dg:
Th:Th:
Def:Def:
Ep:Ep:
Pg:Pg:
Sy:Sy:
Dg:Dg:
Th:Th:
Def:Def:
Ep:Ep:

���
Hemoglobin S (HBS): point mutation in the β-globin locus (chromosome 11) of the hemoglobin molecule in position 6: replacement of glutamic acid by valine (β6 Glu → Val).
HBS precipitates when deoxygenated (risk factors: lack of oxygen, dehydration, fever, increased serum osmolality, stasis):→ Sickle-shaped erythrocytes with reduced elasticity→ Hemolysis, disturbed microcirculation, capillary occlusion
Heterozygotic individuals (HBAS) are usually asymptomatic. In homozygotic cases (HBSS):• Hemolytic anemia and hemolytic crisis• Vaso-occlusive crises: organ infarction (particularly spleen, kidney, CNS), bone infarction,
pulmonary hypertension• Abdominal pain, bone pain, cerebral disorders, in some cases with fever, tachycardia, leucocy-
tosis• Hepatosplenomegaly, recurrent splenic infarction → “autosplenectomy,” functional asplenia
• Osteoporosis, growth defects due to recurrent bone infarction• Pure red cell aplasia / aplastic crisis with parvovirus B19 infections• Proliferative retinopathy → impaired vision• Bilirubin gall stones• Immunodeficiency (due to recurrent splenic infarction)
• Medical history (family history), clinical examination• Hemoglobin electrophoresis• Sickle cell test: erythrocytes show sickle shape after addition of sodium sulfide• Molecular genetic screening (PCR)
Other hemoglobinopathies: more than 450 hemoglobinopathies have been described. HB C, E, and D are the most common.
Sickle cell anemia is treated supportively:• Fluid replacement, at least 2,000 ml/day• Oxygen (via nasal tube, 3–4 l/min)• Treatment of infections, analgesia• Red cell transfusion, in case of severe complications: exchange transfusion• In cases of splenic infarction / hemorrhage / rupture: splenectomy• Prophylactic pneumococcus vaccination• Hydroxyurea
Prevention of lack of oxygen, dehydration, and infections.
β-Thalassemia
Quantitative disturbance of hemoglobin synthesis due to a genetic defect in globin chain forma-tion. Subtypes:• β-Thalassemia: abnormal β-chain synthesis• α-Thalassemia: abnormal α-chain synthesis (rare)
Regional differences in incidence: β-thalassemia in Mediterranean regions, Africa, and Asia; α-thalassemia in South East Asia and Africa.
Abnormal synthesis of the hemoglobin β-chain, i.e., no formation of normal adult HBA1 (αα/ββ).→ Compensatory formation of γ- or δ-chains (HBF = αα/γγ and HBA2 = αα/δδ)→ Ineffective erythropoiesis (free α-globin is toxic for erythroblasts) with intramedullary hemo-
lysis→ Hypochromic microcytic anemia, signs of hemolysis
Pg:Pg:
Sy:Sy:
Co:Co:
Dg:Dg:
Dd:Dd:
Th:Th:
Px:Px:
Def:Def:
Ep:Ep:
Pg:Pg:
�.� Hemolytic Anemia

���
Part � Hematology and Hemostasis
Heterozygotic Patients: Thalassemia MinorUsually, no clinical symptoms; in some cases minor chronic hemolysis, anemia, and splenomegaly.
Homozygotic Patients: Thalassemia Major (Cooley’s Anemia)• Chronic hemolysis, icterus• Hepatosplenomegaly• Cardiac insufficiency• Infections
• Microcytic hypochromic anemia (HB ↓, HCT ↓, MCV ↓, MCH ↓)• Ironserum ↑, ferritin ↑, transferrin iron-binding capacity ↓• Blood smear: microcytic hypochromic erythrocytes, target cells, polychromasia, isolated nor-
moblasts• Chronic erythropoietic bone marrow hyperplasia → expanded marrow, detectable in bone
marrow scan or skull x-ray (“hair-on-end” sign)• Hemoglobin electrophoresis: increase in HBF (αα/γγ) and HBA2 (αα/δδ)• Molecular genetic detection of the defective globin gene (via PCR)
Iron deficiency anemia (7 Chap. 6.4.1).
Supportive Approach• RBC transfusion• Hemosiderosis treatment: desferrioxamine 2,000 IU/day s.c.• Splenectomy• Prophylaxis of infections
Curative ApproachIn homozygotic cases / severe hemolysis: allogeneic stem cell transplantation during infancy.
Warm Antibody Autoimmune Hemolysis (AIHA)
Autoimmune hemolytic anemia caused by IgG incomplete “warm” autoantibodies (incomplete antibodies: antigen-antibody binding, but no lysis or agglutination).
Seventy-five percent of all autoimmune hemolytic anemias.
Formation of IgG Warm Autoantibodies• In non-Hodgkin’s lymphoma, particularly in low-malignant NHL (CLL)• With autoimmune diseases, e.g., systemic lupus erythematosus (SLE)• Following infections (viral infections, rarely bacterial infections)• Drug-induced hemolysis (various mechanisms): antibiotics, α-methyldopa, L-dopa, quinine,
quinidine, x-ray contrast agents, procainamide, diclofenac• Idiopathic (50% of cases)
Autoimmune Hemolysis• Binding of incomplete antibodies to erythrocytes• Destruction of antibody-coated erythrocytes in spleen and liver (extravascular non-comple-
ment-mediated lysis by cells of the reticuloendothelial system)
• Hemolysis and hemolytic crises, with icterus, hemoglobinuria, fever, etc.• Anemia symptoms (fatigue, weakness, reduced performance, pallor, headache, etc.)
Case History, Physical Examination• Case history including medication• Physical examination including signs and symptoms of anemia
Sy:Sy:
Dg:Dg:
Dd:Dd:
Th:Th:
Def:Def:
Ep:Ep:
Pg:Pg:
Sy:Sy:
Dg:Dg:

���
Laboratory Tests• Anemia (HB ↓, HCT ↓)• Signs of hemolysis (LDH ↑, indirect bilirubin ↑, haptoglobin ↓, etc.)• Blood group• Exclusion of potential underlying diseases
Coombs’ Test: Detection of Incomplete Antibodies• Direct Coombs’ test: detection of incomplete antibodies bound to erythrocytes• Indirect Coombs’ test: detection of incomplete serum antibodies• With warm antibody autoimmune hemolysis: direct Coombs’ test positive, indirect Coombs’
test positive or negative
Autoimmune hemolysis can show different degrees of severity, ranging from compensated chronic hemolysis to acute life-threatening hemolytic crisis. Every case of autoimmune hemo-lysis must initially be treated as a hematological emergency.
Causal TreatmentTreatment of underlying disease or discontinuation of causative drugs.
Symptomatic Treatment• Corticosteroids (prednisolone 100–500 mg/day i.v.), slowly taper dose after hemolysis para-
meters have normalized• In cases of chronic hemolysis and poor response to corticosteroids: use alternative immuno-
suppressive agents, e.g., azathioprine 80 mg/m2/day, cyclophosphamide 60 mg/m2/day p.o.• Splenectomy: in cases of treatment-refractory chronic hemolysis or refractory acute hemolytic
crisis• Transfusion of packed red cells only in cases of symptomatic anemia (e.g., cardiovascular
symptoms, dyspnea, cerebral ischemia)
Cold Agglutinin Autoimmune Hemolysis (AIHA)
Autoimmune hemolytic anemia caused by IgM complete “cold” autoantibodies, usually targeting the I-antigen of the erythrocyte membrane (complete antibodies: capable of agglutination and lysis induction after antigen-antibody binding).
Fifteen percent of all autoimmune hemolytic anemias.
Secondary Formation of Polyclonal Cold Autoantibodies (Cold Agglutinin Syndrome)• In low-malignant non-Hodgkin’s lymphoma or Hodgkin’s disease• After infection (viral infections, mononucleosis / EBV infection, mycoplasma pneumoniae) →
cold agglutinin titer up to 1:1,000
Primary Formation of Monoclonal Cold Autoantibodies (Cold Agglutinin Disease)Rare congenital disease→ Cold agglutinin titer up to 1:256,000
Autoimmune HemolysisWhen the intravascular temperature drops to < 20–25°C: antigen-antibody binding, agglutination and complement-mediated intravascular hemolysis.
• Exposure to cold leads to hemolysis and hemolytic crisis (with icterus, hemoglobinuria, fever, etc.)
• Anemia symptoms (fatigue, weakness, reduced performance, pallor, headache, etc.)• Acrocyanosis: painful / malperfused extremities (fingers / toes / nose)• Splenomegaly
Th:Th:
Def:Def:
Ep:Ep:
Pg:Pg:
Sy:Sy:
�.� Hemolytic Anemia

���
Part � Hematology and Hemostasis
• Medical history, physical examination• Diagnostic clues: erythrocyte agglutination when blood is drawn and during laboratory analy-
sis• Anemia (HB ↓, HCT ↓), signs of hemolysis (LDH ↑, indirect bilirubin ↑, haptoglobin ↓), de-
tection of cold autoantibodies• Exclusion of potential underlying diseases• Blood group
Causal ApproachTreatment of the underlying disease.
Symptomatic Approach• Protection against cold• With severe acute hemolysis: plasmapheresis (objective: removal of autoantibodies), often tech-
nically difficult (due to agglutination within the plasmapheresis system)• With chronic hemolysis: immunosuppressive drugs, e.g., azathioprine, cyclophosphamide, or
chlorambucil• With symptomatic anemia (cardiovascular symptoms, dyspnea, cerebral malperfusion, etc.):
transfusion of washed packed red cells (avoid complement administration in cases of comple-ment-mediated hemolysis)
• Corticosteroids and splenectomy are usually ineffective
Borgna-Pignatti C. Modern treatment of thalassaemia intermedia. Br J Haematol 2007;138:291–304British Committee for Standards in Haematology. Guidelines for the diagnosis and management of heredi-tary spherocytosis. Br J Haematol 2004;126:455–74Gallagher PG. Hereditary elliptocytosis: spectrin and protein 4.1R. Semin Hematol 2004;41:142–64Gertz A. Management of cold haemolytic syndrome. Br J Haematol 2007;138:422–9Hillmen P, Young NS, Schubert J et al. The complement inhibitor eculizumab in PNH. N Engl J Med 2006;355:1233–43King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol 2005;42:131–6Mehta A, Mason PJ, Vulliamy TJ. Glucose-6-phosphate dehydrogenase deficiency. Baillieres Best Pract Res Clin Haematol 2000;13:21–38Stuart MJ, Nagel RL. Sickle-cell disease. Lancet 2004;364:1343–60
1. http://www.nlm.nih.gov/medlineplus/ency/article/000571.htm Medline Plus2. http://www.nlm.nih.gov/medlineplus/ency/article/000534.htm Medline Plus3. http://www.umm.edu/blood/anehemol.htm Univ Maryland4. http://www.fpnotebook.com/HEM50.htm Family Practice5. http://www.emedicine.com/med/topic979.htm E-medicine
1.2.
3.4.5.
6.7.
8.
Dg:Dg:
Th:Th:
Ref:Ref:
Web:Web:

���
Anemia with normal corpuscular hemoglobin (MCH 27–34 pg) and normal corpuscular hemo-globin concentration (MCHC 31–36 g/dl).
• Hemolytic anemia (7 Chap. 6.4.3)• Aplastic anemia (7 Chap. 6.1)• Acute posthemorrhagic anemia• Renal anemia
RenalAnemia
Normochromic normocytic hyporegenerative anemia as a result of chronic renal failure.
Incidence: 50–60 cases/100,000 per year.
Chronic Renal Failure Anemia• Complex pathogenesis based on renal insufficiency• Renal erythropoietin synthesis ↓, the degree of anemia correlates with the severity of the un-
derlying disease• Myelosuppresion and intramedullary hemolysis due to accumulation of uremic toxins• Concurrent chronic blood loss due to hemodialysis
Anemia Symptoms• Pale skin and mucous membranes• Weakness, fatigue, reduced performance, dyspnea on exertion• Difficulty concentrating, headache
Uremia Symptoms• Uremic fetor• “Café au lait” complexion due to urochrome deposits and concurrent anemia, pruritus• Weakness, headache
Medical History, Physical Examination• Medical history: signs of chronic renal insufficiency• Physical examination: skin, mucous membranes, lymph node status, spleen / liver, heart
(tachycardia, systolic heart murmur), rectal examination and testing for fecal occult blood
Laboratory Tests• Hematology: blood count including MCV (normal), MCH (normal), reticulocytes (↓), differ-
ential blood count• Clinical chemistry: hepatic and renal function tests, total protein, hemolysis parameters (bili-
rubin, LDH, normal haptoglobin, low-grade hemolysis due to uremic toxins)• Vitamin B12 level, folic acid level• Serum iron, ferritin, transferrin; in cases of chronic blood loss due to hemodialysis, iron defi-
ciency may occur• Erythropoietin ↓ / normal (i.e., inadequate increase given the degree of anemia)• Blood group (if red cell transfusion is required)
Symptomatic Treatment• Erythropoiesis stimulation with darbepoetin 1.35 µg/kg body weight once weekly s.c. or i.v.,
adjust dose according to hemoglobin response• Alternatively, recombinant erythropoietin, 50 IU/kg body weight three times weekly s.c. or
i.v., adjust dose according to hemoglobin response
Def:Def:
Dd:Dd:
Def:Def:
Ep:Ep:
Pg:Pg:
Sy:Sy:
Dg:Dg:
Th:Th:
�.� Normochromic Anemia
6.4.4 Normochromic Anemia
R.Engelhardt,J.Heinz

��0
Part � Hematology and Hemostasis
• Target hemoglobin 10–12 g/dl• ATTENTION: blood pressure may rise as hematocrit increases, especially in cases of pre-exist-
ing hypertension• Hemodialysis• Additional iron supplementation with signs of iron deficiency (7 Chap 6.4.1)
Causal Treatment• Kidney transplantation
Eckardt KU. Pathophysiology of renal anemia. Clin Nephrol 2000;53(1 suppl):S2–8Eschbach JW. Current concepts of anemia management in chronic renal failure: impact of NKF-DOQI. Semin Nephrol 2000;20:320–9MacDougall IC. Novel erythropoiesis stimulating protein. Semin Nephrol 2000;20:375–81Ritz E, Schwenger V. The optimal target hemoglobin. Semin Nephrol 2000;20:382–6Valderrabano F. Quality of life benefits of early anaemia treatment. Nephrol Dial Transplant 2000;15(sup-pl 3):23–8
1. http://www.kidney.org/professionals/doqi/doqi/ doqianemia.html National Kidney Foundation Guidelines
2. http://www.anemiainstitute.org/ Anemia Institute3. http://www.asn-online.com/ American Society of Nephrology
1.2.
3.4.5.
Ref:Ref:
Web:Web:
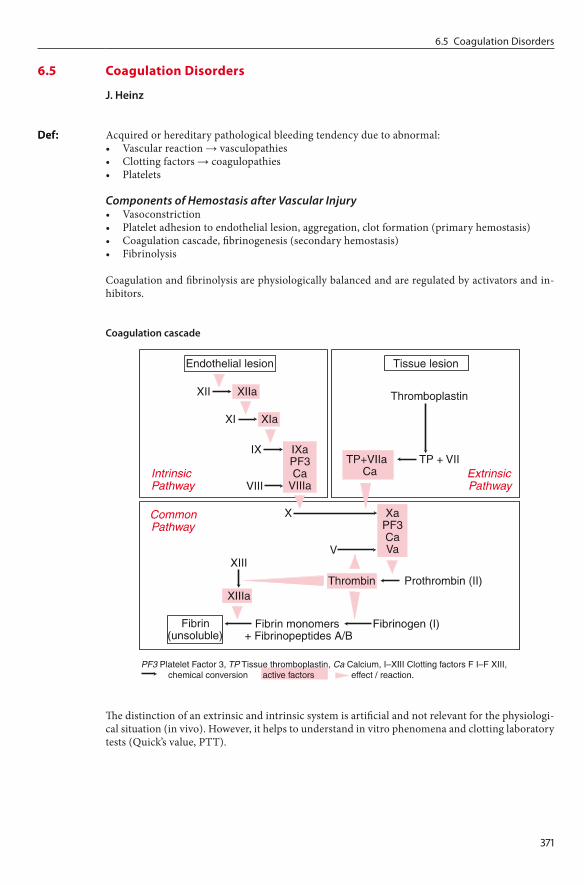
���
Acquired or hereditary pathological bleeding tendency due to abnormal:• Vascular reaction → vasculopathies• Clotting factors → coagulopathies• Platelets
Components of Hemostasis after Vascular Injury• Vasoconstriction• Platelet adhesion to endothelial lesion, aggregation, clot formation (primary hemostasis)• Coagulation cascade, fibrinogenesis (secondary hemostasis)• Fibrinolysis
Coagulation and fibrinolysis are physiologically balanced and are regulated by activators and in-hibitors.
Coagulationcascade
The distinction of an extrinsic and intrinsic system is artificial and not relevant for the physiologi-cal situation (in vivo). However, it helps to understand in vitro phenomena and clotting laboratory tests (Quick’s value, PTT).
Def:Def:
�.� Coagulation Disorders
6.5 Coagulation Disorders
J.Heinz

���
Part � Hematology and Hemostasis
Coagulationcascadeinhibitors
• Antithrombin (AT): prevention of excessive thrombin activation by formation of thrombin–AT complex, inhibition of IIa, IXa, Xa, XIa, XIIa; important physiological coagulation inhibitor; AT deficiency constitutes an increased risk of thrombosis (thrombophilia, 7 Chap. 6.6)
• Protein C: thrombin-induced conversion into active protein C (APC); APC inhibits FVa and FVIIIa and induces the release of tPA (plasminogenic activator); protein C deficiency consti-tutes an increased risk of thrombosis (7 Chap. 6.6)
• Protein S: cofactor of protein C• Heparin: activation of physiological AT → inhibition of thrombin generation; ineffective in
cases of AT deficiency. Unfractionated (UFH) and low molecular weight (LMWH) heparins• Hirudin: direct thrombin inactivation, effective in cases of AT deficiency• Coumarin: vitamin K antagonists; inhibition of the hepatic synthesis of the factors II, VII, IX,
and X as well as the proteins C and S
Fibrinolysiscascadeandfibrinolysisinhibitors
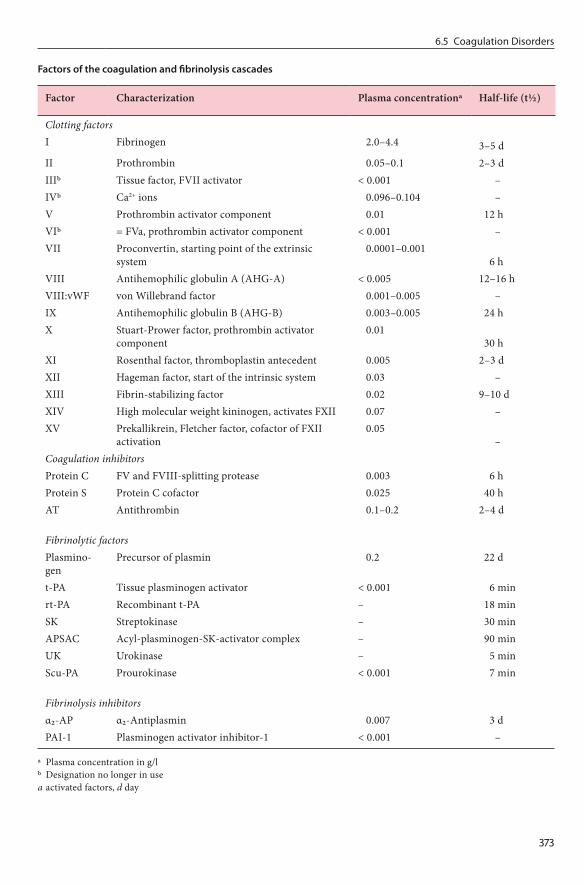
���
Factorsofthecoagulationandfibrinolysiscascades
Factor Characterization Plasma concentrationa Half-life (t½)
Clotting factorsI Fibrinogen 2.0–4.4 3–5 dII Prothrombin 0.05–0.1 2–3 dIIIb Tissue factor, FVII activator < 0.001 –IVb Ca2+ ions 0.096–0.104 –V Prothrombin activator component 0.01 12 hVIb = FVa, prothrombin activator component < 0.001 –VII Proconvertin, starting point of the extrinsic
system 0.0001–0.001
6 hVIII Antihemophilic globulin A (AHG-A) < 0.005 12–16 hVIII:vWF von Willebrand factor 0.001–0.005 –IX Antihemophilic globulin B (AHG-B) 0.003–0.005 24 hX Stuart-Prower factor, prothrombin activator
component 0.01
30 hXI Rosenthal factor, thromboplastin antecedent 0.005 2–3 dXII Hageman factor, start of the intrinsic system 0.03 –XIII Fibrin-stabilizing factor 0.02 9–10 dXIV High molecular weight kininogen, activates FXII 0.07 –XV Prekallikrein, Fletcher factor, cofactor of FXII
activation 0.05
–Coagulation inhibitorsProtein C FV and FVIII-splitting protease 0.003 6 hProtein S Protein C cofactor 0.025 40 hAT Antithrombin 0.1–0.2 2–4 d
Fibrinolytic factorsPlasmino-gen
Precursor of plasmin 0.2 22 d
t-PA Tissue plasminogen activator < 0.001 6 minrt-PA Recombinant t-PA – 18 minSK Streptokinase – 30 minAPSAC Acyl-plasminogen-SK-activator complex – 90 minUK Urokinase – 5 minScu-PA Prourokinase < 0.001 7 min
Fibrinolysis inhibitorsα2-AP α2-Antiplasmin 0.007 3 dPAI-1 Plasminogen activator inhibitor-1 < 0.001 –
a Plasma concentration in g/lb Designation no longer in usea activated factors, d day
�.� Coagulation Disorders

���
Part � Hematology and Hemostasis
Inhibitors of Platelet Aggregation• Acetylsalicylic acid: irreversible cyclooxygenase inhibition• Ticlopidine: inhibition of fibrinogen binding by interaction with GPIIb/IIIa• Tirofiban hydrochloride: GPIIb/IIIa receptor antagonist• Dipyridamole: increases the level of cellular cyclic AMP (cAMP)• Clopidogrel: selective inhibition of ADP binding, inhibition of ADP-mediated activation of the
GPIIb/IIIa receptor complex
Classification of Acquired and Congenital Coagulopathies
Vitamin K Deficiency or Abnormal Synthesis of Vitamin K-dependent Clotting Factors (7 Chap. 6.5.1)• Severe liver damage• Antibiotic treatment, malabsorption syndrome, abnormal fat absorption, alcoholism
Consumption Coagulopathy• Disseminated intravascular coagulation (DIC, 7 Chap. 6.5.5)
Immunocoagulopathies• Antibodies against clotting factors in conjunction with autoimmune diseases (e.g., lupus anti-
coagulant with systemic lupus erythematosus)
Microangiopathies• Thrombotic-thrombocytopenic purpura (TTP, 7 Chap. 6.3.3)• Hemolytic-uremic syndrome (HUS, 7 Chap. 6.3.3)
Hereditary Coagulopathies• Factor VIII deficiency (hemophilia A, 7 Chap. 6.5.2)• Factor IX deficiency (hemophilia B, 7 Chap. 6.5.3)• Von Willebrand’s disease (7 Chap. 6.5.4)• Other clotting factor deficiencies
Different coagulopathies are associated with different patterns of hemorrhage:• Thrombocytic abnormalities: pinpoint hemorrhages: petechiae, purpura• Vascular abnormalities: petechiae, purpura• Coagulopathies: soft tissue hemorrhage, hematomas, intra-articular hemorrhage
Medical History, Physical Examination• Medical history (including family history, bleeding, medication)• Physical examination: type of hemorrhage
Laboratory Tests• Blood count including platelet count, fibrinogen• Intrinsic pathway: PTT test (partial thromboplastin time)• Extrinsic pathway: Quick’s test (prothrombin time)• Vascular / platelet abnormalities: platelet count, platelet function tests, bleeding time (normal:
< 9 min), platelet function analysis• Coagulation activation assessment: fibrin monomers, prothrombin fragments 1 + 2• Assessment of fibrinolytic reactions: D-dimers (fibrin split products, signs of active fibrinoly-
sis)• Special tests: single factor analysis, platelet function tests, inhibitors
See respective coagulopathies (7 Chaps. 6.5.1–6.5.5)
Baglin T, Barrowcliffe TW, Cohen A et al. Guidelines on the use and monitoring of heparin. Br J Haematol 2006;133:19–34Dahlback B. Blood coagulation. Lancet 2000;355:1627–32Falanga A. Tumor cell prothrombotic properties. Haemostasis 2001;31(suppl 1):1–4
1.
2.3.
Class:Class:
Sy:Sy:
Dg:Dg:
Th:Th:
Ref:Ref:

���
Manucci PM, Duga S, Peyvandi F. Recessively inherited coagulation disorders. Blood 2004;104:1243–52Petralia GA, Lemoine NR, Kakkar AK. Mechanisms of disease: the impact of antithrombotic therapy in cancer patients. Nat Clin Pract Oncol 2005;2:356–63
1. http://www.med.unc.edu/isth/welcome ISTH2. http://tollefsen.wustl.edu/projects/coagulation/coagulation.html Washington Univ3. http://www.indstate.edu/thcme/mwking/blood-coagulation.html Indiana State Univ4. http://www.nlm.nih.gov/medlineplus/ency/article/001304.htm Medline Plus
4.5.
Web:Web:
�.� Coagulation Disorders

Hematology and HemostasisPart �
���
Coagulopathies which may occur spontaneously or as a result of an underlying disease which is not primarily related to the hemostatic system. In contrast to primary disorders, several compo-nents of the hemostatic system are usually affected. Impaired synthesis and metabolic defects can be distinguished.
D68
Causes of Acquired Bleeding Disorders• Vitamin K deficiency• Liver diseases• Uremia, nephrotic syndrome• Malignancies (lymphoma, leukemia, myeloproliferative syndromes, solid tumors)• Amyloidosis• Cardiovascular disorders• Autoimmune diseases• Drugs (asparaginase, penicillin, cephalosporin, interferon-α)• Pregnancy, post partum
VitaminKDeficiency
Most common bleeding disorder
• Vitamin K-free diet (rare)• Malabsorption syndrome, cholestasis, chronic pancreatitis, sprue• Iatrogenic: antibiotic treatment (e.g., cephalosporins), coumarin treatment, parenteral nutri-
tion without vitamin K substitution
Vitamin K is a fat-soluble vitamin and a cofactor for synthesis of the clotting factors II, VII, IX, and X as well as protein C and S. Vitamin K deficiency:→ Synthesis of prothrombin complex ↓ → bleeding tendency ↑
Higher incidence of hemorrhages / hematomas
• Quick’s test ↓, aPTT normal or ↑• Determination of each individual factor is not usually required
• Exogenous deficiency without hemorrhage: usually single oral dose of phytomenadione 10–20 mg; in cases of abnormal absorption: i.v. administration. The efficacy of vitamin K substitu-tion can differ considerably between individuals. Coagulation parameters (Quick’s value) will increase 6–12 h after treatment at the earliest.
• With coumarin treatment without hemorrhage: phytomenadione: dose according to INR (In-ternational Normalized Ratio). INR within target range and no hemorrhage: dosage 2–5 mg, INR too high and no hemorrhage: dosage 10–20 mg (possibly repeated).
• Obvious hemorrhage / before emergency surgery: administration of prothrombin complex con-centrates (PPSB) 25–50 IU/kg body weight plus vitamin K 10 mg; in cases of uncontrollable bleeding: administration of activated factor VIIa.
CoagulationDisordersinHepaticDiseases
The liver is the primary site of formation and elimination of coagulation factors and their inhibi-tors as well as filtration → hepatic dysfunction can lead to complex hemostatic disorders:
Def:Def:
ICD-10:ICD-10:
Et:Et:
Ep:Ep:
Et:Et:
Pphys:Pphys:
Sy:Sy:
Dg:Dg:
Th:Th:
Pphys:Pphys:
6.5.1 Acquired Coagulation Disorders
J.Heinz

���
• Impaired synthesis of clotting factors → factor deficiency (esp. factor II, V, VII, IX, X, XIII, fibrinogen, plasminogen, α2-antiplasmin, antithrombin, protein C, protein S)
• Impaired elimination of clotting factors → factor excess (e.g., VIII, von Willebrand-factor)• Impaired thrombopoiesis / platelet function (hypersplenism, bone marrow defect due to toxic
effects of alcohol, vitamin B12 / folic acid deficiency, thrombopoietin deficiency)• Hyperfibrinolysis• Ascites → loss of coagulation factors (loss of coagulation factors via ascites)
• Signs of hepatic failure• Bleeding signs and symptoms: hematomas, mucous membrane hemorrhage, epistaxis• Esophageal variceal bleeding (life-threatening)
• Quick’s test ↓ (earliest indication of hepatic coagulation defects: includes factor VII which is the first to decrease due to its short half life of 6 h); suitable parameter for monitoring hepatic disorders
• aPTT: may be normal or increased in advanced hepatic disorders• Platelets ↓, fibrinogen ↓, factor V ↓, protein C ↓, protein S ↓, antithrombin ↓ (may be increased
in case of cholestasis), D-dimers ↑• Determination of separate factors usually not required
Acute Hemorrhage• Initial treatment with fresh frozen plasma (FFP) 10 ml/kg.• If insufficient: antithrombin supplementation, fibrinogen supplementation (for levels below
1.0 g/l), administration of cryoprecipitate.• Administration of platelet concentrates, desmopressin (DDAVP; 0.4 µg/kg) and antifibrinolyt-
ics (e.g., aprotinin 250,000 IU in 30 min, 2 million IU/day i.v.) may be considered. ATTEN-TION: DIC (7 Chap. 6.5.5).
• If factor XIII concentration < 50% and FFP is without effect: administration of factor XIII concentrate.
• If initial values are unknown / emergency situations: empirical treatment with antithrombin 50 IU/kg, fibrinogen 3 g, and PPSB 50 IU/kg. In cases of severe hemorrhage, treatment with activated factor VIIa may be considered.
TargetValuesintheTreatmentofHepaticCoagulationDisorders
Parameter Target value range
Antithrombin > 40%Quick’s value > 40%Fibrinogen > 50–100 mg/dlPlatelet count > 50,000/µl
Prophylaxis of HemorrhageAdministration of antifibrinolytics (e.g., tranexamic acid 1 g three times daily), vitamin K
BleedinginUremicConditions
Acute or chronic renal insufficiency
• Platelet function disorder due to accumulation of urinary toxins• Fibrin polymerization ↓• vWF (von Willebrand factor)-mediated platelet adhesion ↓
• Signs of renal failure• Mucous membrane hemorrhage, hematomas, gastrointestinal hemorrhage
Sy:Sy:
Dg:Dg:
Th:Th:
Et:Et:
Pphys:Pphys:
Sy:Sy:
�.� Acquired Coagulation Disorders

���
Part � Hematology and Hemostasis
• Creatinine ↑, urea ↑, creatinine clearance ↓• Bleeding time ↑, platelet dysfunction, in severe cases with thrombocytopenia• vWF antigen level ↑
• Hemodialysis• DDAVP (0.3 µg/kg body weight)• In emergency situations: administration of platelet concentrates and von Willebrand factor-
enriched factor VIII concentrate (dose: 25 IU/kg)
CoagulationDisordersinMalignantDiseases
Most frequent malignant diseases associated with bleeding disorders:• Malignant lymphomas• Myeloproliferative syndromes• Multiple myeloma, monoclonal gammopathy (MGUS)• Solid tumors (esp. prostate, ovarian, and colon carcinoma)
• Thrombocytopenia due to bone marrow infiltration• Hepatic metastases → impaired synthesis of coagulation factors• Functional impairment of platelets / coagulation factors• Hyperfibrinolysis triggered by procoagulatory activity (esp. solid tumors)• Paraprotein → increased viscosity, platelet dysfunction, inhibition of coagulation factors, and
fibrin polymerization
Hemorrhages of all degrees and in all locations (hematomas, mucous membrane hemorrhage, postoperative bleeding)
• Platelet count usually normal• Prolonged clinical or in vitro bleeding time (PFA 100 test)• Platelet dysfunction after stimulation with epinephrine and / or ADP• Quick’s and aPTT usually normal (exception: inhibitor formation, severe hepatic dysfunc-
tion)• D-dimers ↑, fibrinogen ↓ as a sign of hyperfibrinolysis
• Treatment of the underlying disease• In case of thrombocytopenia and hemorrhage: administration of platelet concentrates• In case of paraprotein-associated hemorrhage (Waldenström’s macroglobulinemia or multiple
myeloma): administration of DDAVP, in case of severe hyperviscosity: apheresis treatment• Hyperfibrinolysis: antifibrinolytics (e.g., aprotinin)
AcquiredFactorVIIIInhibitor(Antigen-inducedHemophilia)
Antibodies to clotting factors, occurring as primary (spontaneous) or secondary (due to underly-ing disease) antibodies:• Autoantibodies
− Inhibitors to individual clotting factors (most commonly to factor VIII, antigen-induced hemophilia)
− Antiphospholipid antibodies (7 Chap. 6.6)− Monoclonal immunoglobulins, heparin-like antibodies
• Alloantibodies (inhibitors in hemophilia A / B replacement therapy)
Incidence of factor VIII inhibitors 1:1,000,000
Acquired factor VIII inhibitors: 50% of cases occur spontaneously, secondary inhibitors in con-junction with:
Dg:Dg:
Th:Th:
Et:Et:
Pphys:Pphys:
Sy:Sy:
Dg:Dg:
Th:Th:
Def:Def:
Ep:Ep:
Et:Et:

���
• Autoimmune diseases, SLE (systemic lupus erythematodes), asthma• Malignancies• Drug-induced: penicillin, ampicillin, interferon-α• Others: post partum, skin diseases, sarcoidosis, amyloidosis, GVHD
Spontaneous bleeding with large hematomas, mucous membrane hemorrhages, vaginal bleeding, in severe cases uncontrollable bleeding after minor injuries; high mortality of up to 25%
• aPTT ↑, factor VIII ↓• Determination of inhibitor levels according to the Bethesda method
Acute Hemorrhage• Administration of recombinant factor VIIa• In cases of low titer inhibitors (< 5 B.U.): administration of high-dose factor VIII• In severe cases additional administration of activated prothrombin complex concentrate, im-
munoadsorption therapy
Causal Treatment• Immunosuppressive treatment: steroids (prednisone 1 mg/kg daily for at least 4 weeks), pos-
sibly in combination with cyclophosphamide (2 mg/kg daily)• Alternatively: i.v. immunoglobulins 1 g/kg daily for 2 days, cyclosporine, anti-CD20 antibody
(rituximab)• In cases of high titer inhibitors > 5 B.U. or severe bleeding: immunoadsorption
AcquiredvonWillebrand’sSyndrome
Occurrence of von Willebrand factor inhibitors in conjunction with:• Malignancies: lymphomas, leukemias, myeloproliferative diseases, solid tumors• Cardiovascular diseases• Autoimmune diseases, drugs
• Autoantibodies against von Willebrand factor• Binding of von Willebrand factor to the surface of malignant cells• Proteolysis of von Willebrand factor (e.g., with acute promyelocytic leukemia 7 Chap. 7.1.2)• High shear stress → destruction of vWF multimers (e.g., in cases of aortic stenosis)• Impaired synthesis
Skin / mucous membrane hemorrhage, postoperative bleeding
See congenital von Willebrand’s syndrome (7 Chap. 6.5.4)
• Treatment of the underlying disease• Replacement therapy with von Willebrand concentrate, inhibitor elimination
HemorrhagicDisorderswithAsparaginaseTreatment
Asparaginase therapy of acute leukemias
• Impaired synthesis of clotting factors (esp. fibrinogen, antithrombin, protein C and S, fac-tors II, IX, and XIII)
• Potential complication: DIC (7 Chap. 6.5.5)
Hemorrhages of all degrees and in all locations (hematomas, mucous membrane hemorrhage, postoperative bleeding)
Levels of fibrinogen, antithrombin, D-dimers
Sy:Sy:
Dg:Dg:
Th:Th:
Et:Et:
Pphys:Pphys:
Sy:Sy:
Dg:Dg:
Th:Th:
Et:Et:
Pphys:Pphys:
Sy:Sy:
Dg:Dg:
�.� Acquired Coagulation Disorders

��0
Part � Hematology and Hemostasis
• FFP 10 ml/kg (coagulation factor increase by 10–20%)• Antithrombin concentrate: 20 IU/kg → increase by approximately 20–40%• Fibrinogen 3 g → increase by 1 g/l
Dahlback B. Blood coagulation and its regulation by anticoagulant pathways: genetic pathogenesis of bleeding and thrombotic diseases. J Intern Med 2005;257:209–23Delgado J, Jimenez-Yuste V, Hernandez-Navarro F et al. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol 2003;121:21–35Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002;346:752–63Li T, Chang CY, Jin DY et al. Identification of the gene for vitamin K epoxide reductase. Nature 2004;427:541–4Wiestner A, Cho HJ, Asch AS et al. Rituximab in the treatment of acquired factor VIII inhibitors. Blood 2002;100:3426–8Zeitler H, Ulrich-Merzenich G, Hess L et al. Treatment of acquired hemophilia by the Bonn-Malmö proto-col. Blood 2005;105:2287–93
1. http://www.med.unc.edu/isth/welcome Intl Soc Thrombosis Hemostasis2. http://www.ctds.info/vitamink.html Vitamin K Deficiency3. http://www.emedicine.com/med/topic2385.htm E-Medicine4. http://www.nlm.nih.gov/medlineplus/ency/
article/002407.htm Medline Plus
1.
2.
3.4.
5.
6.
Th:Th:
Ref:Ref:
Web:Web:

���
Hereditary bleeding disorder caused by deficiency (90% of cases) or inactivity (10%) of coagula-tion factor VIII (FVIII, AHG-A, antihemophilic globulin A).
D66
Most common hereditary coagulopathy, incidence 1 case/5,000 men/year. Women are heterozy-gotic carriers of the gene. Clinically apparent hemophilia in women is rare. Ratio between hemo-philia A and B approximately 5:1.
• Factor VIII coding gene located on the X chromosome → mainly men are affected, X-linked recessive inheritance (70% of cases) or spontaneous mutations (30%)
• Synthesis in liver, 265 kDa protein, no vitamin K dependence; half-life: 8–12 h• Factor VIII circulates in the plasma bound to von Willebrand factor (vWF) → protection from
proteolytic degradation
SeverityofFVIIIdeficiency
Severity FVIII activity Symptoms
Normal > 70% NoneSubhemophilia 15–40% No symptoms in normal lifeMild 5–20% Hematoma following trauma, discrete tendency to bleedModerate 1–5% Hematoma following mild trauma, tendency to bleedSevere < 1% Spontaneous bleeding, bleeding into joints, hematuria
Increased tendency to bleed (manifestation during infancy and early childhood)• Extensive bleeding, hematomas• Soft tissue bleeding, bleeding into joints (hemarthrosis)• Gastrointestinal bleeding, hematuria
Medical History, Physical Examination• Medical history: including family history• Physical examination: including type of bleeding, complications
Laboratory Tests• Coagulation parameters: factor VIII ↓↓, aPTT ↑, Quick’s test normal (extrinsic system), nor-
mal bleeding time (verified by platelet function test)• Genetic diagnosis: RFLP (analysis of the restriction fragment length polymorphism); most
common genetic defect: intron 22 inversion
• Von Willebrand’s disease• Other coagulation factor deficiencies• Acquired factor VIII antibodies (7 Chap. 6.5.1)
• Arthropathy → joint destruction, arthrosis, stiffening• Retroperitoneal bleeding, psoas hemorrhage, cerebral bleeding (rare)• Hepatitis / HIV infection due to transfusion and administration of FVIII products (especially
before 1984). HIV patients on protease inhibitor treatment: bleeding risk ↑• Pseudotumor formation / liquefaction of tissue at the hemorrhage site → surgical excision
Def:Def:
ICD-10:ICD-10:
Ep:Ep:
Pg:Pg:
Class:Class:
Sy:Sy:
Dg:Dg:
Dd:Dd:
Co:Co:
�.� Hemophilia A
6.5.2 Factor VIII Deficiency (Hemophilia A)
O.Schmah,J.Heinz

���
Part � Hematology and Hemostasis
Detailed recommendations see guidelines of the International Society for Thrombosis and Hemo-stasis. The following types of treatment can be distinguished:• Treatment on demand (spontaneous / traumatic bleeding)• Continuous prophylactic treatment (esp. children and teenagers)• Prophylactic treatment (before surgery, physical stress, etc.)
ATTENTION: Treatment must be provided as early as possible and must be sufficient with re-spect to dosage and treatment period.
Mild Bleeding and FVIII > 15–40%DDAVP, nasal spray, or intravenous administration (0.3–0.4 µg/kg in 100 ml saline over 30 min, every 12–24 h); effect occurs within 30–60 min: transient FVIII increase by factor 2–3 for up to 4 days; may also be given prior to minor surgery (e.g., tooth extraction), possibly with antifibri-nolytics.
Severe Bleeding and/or Patients with FVIII < 15%Administration of recombinant factor VIII or plasma factor VIII. Administration of recombinant factor products excludes the risk of viral contamination (HBV, HCV, HIV, HSV, EBV, CMV, etc.). Coagulation factors are applied i.v. as a slow bolus injection or via continuous infusion (2–4 IU/kg/h) with reduced factor content.
Required Amount: Dose (IU) = Desired Factor Increase (%) × 0.5 × Body Weight (kg)Rule of Thumb: Administration of Factor VIII 1 IU/kg → Plasma FVIII ↑ by 1%
DosageguidelinesforFVIII
Type of bleeding Target FVIII activitya Duration of therapy
Joint bleeding 15–50% 1–7 daysExtensive soft tissue bleeding, muscular bleeding
30–50% 2–7 days
Complicated bleeding (tongue, neck, forearm, calf)
40–70% Several weeks
Intracranial / gastrointestinal bleeding
70–100% Several weeks
Minor surgery 25–40% 3–5 daysMajor surgery, tonsillectomy 80–150% 14–21 daysb
a Therapeutic factor VIII activity in plasmab Or until wound healing is complete
Monitoring of FVIII ReplacementaPTT monitoring is not sufficient, specific measurement of plasma factor VIII should be per-formed.• Determine FVIII level 30–60 min after bolus was administered = confirmation of increase;
when biological half-life is reached, and prior to administration of the next dose• 15–35% of patients develop antibodies against infused factor VIII → alloantibodies may form
within the first 50–100 days of exposure, and may lead to treatment resistance → monitoring via FVII inhibitor assay
Patients with Factor VIII Antibodies• Hemorrhage and low antibody titer (Bethesda titer < 5): increase dose and frequency of FVIII
products, close monitoring; use of porcine FVIII may be considered (no cross-reaction)• Hemorrhage and high antibody titer (Bethesda titer > 5): give recombinant factor VIIa 90 µg/
kg body weight (= 4.5 KIU/kg body weight) every 2–4 h or Factor Eight Bypassing Activity
Th:Th:

���
(FEIBA) 20–100 IU/kg body weight every 8–12 h. In emergency situations: plasmapheresis or immunoadsorption
Normal life expectancy
Patient information and instruction are the best and most important bleeding prophylaxis• Early detection of signs of bleeding• Controlled exercise and sports program to prevent bleeding into joints and to maintain mobil-
ity• Avoid platelet aggregation inhibitors (ASS, etc.), no intramuscular injections• Caries prophylaxis, meticulous local hemostasis during surgical procedures; no surgery with-
out prophylactic administration of FVIII• Hepatitis A/B vaccination is recommended• X-linked inheritance → examine coagulation status of patient’s relatives
Special Attention when Caring for InfantsBleeding-related arthropathy often goes unnoticed → close monitoring, permanent FVIII treat-ment in cases of severe hemophilia: 25–40 IU/kg 1–3 times weekly → rate of complications / ar-throplasty significantly decreased.
Berntorp E. Immune tolerance induction: recombinant vs. human-derived product. Haemophilia 2001;7:109–13Bolton-Maggs P, Pasi KJ. Haemophilia A and B. Lancet 2003;361:1801–9Evatt BL, Farrugia A, Shapiro AD et al. Haemophilia 2002: emerging risks of treatment. Haemophilia 2002;8:221–9Graw J, Brackmann HH, Oldenburg J et al. Haemophilia A: from mutation analysis to new therapies. Nat Rev Genet 2005;6:488–501Kubisz P, Stasko J. Recombinant activated factor VII in patients at high risk of bleeding. Hematology 2004;9:317–32Srivastava A. Dose and response in haemophilia: optimization of factor replacement therapy. Br J Haema-tol 2004;127:12–25Van den Berg HM, Fischer K, van der Bom JG. Comparing outcomes of different treatment regimens for severe haemophilia. Haemophilia 2003;9:27–31
1. http://www.haemophilia-forum.org Haemophilia Forum2. http://www.hemophilia.org Natl Hemophilia Foundation3. http://www.haemophilia.org.uk Haemophilia Society4. http://www.wfh.org World Fed Hemophilia5. http://www.nlm.nih.gov/medlineplus/hemophilia.html Medline Plus6. http://med.unc.edu/isth/welcome International Society of Thrombosis and
Hemostasis
1.
2.3.
4.
5.
6.
7.
Prg:Prg:
Px:Px:
Ref:Ref:
Web:Web:
�.� Hemophilia A
Hematology and HemostasisPart �
���
Hereditary coagulopathy due to deficiency or inactivity of coagulation factor IX (FIX, Christmas factor, antihemophilic globulin B, AHG-B).
D67
Rare hereditary coagulopathy, incidence 1 case/25–30,000 men/year. Women are heterozygotic carriers of the gene.
• Factor IX coding gene is located on the X chromosome → mainly men are affected, X-linked recessive inheritance; hereditary forms (80% of cases) and spontaneous mutations (20%)
• Hepatic synthesis, 55 kDa protein, vitamin K-dependent, half-life 24 h
SeverityofFIXdeficiency
Severity FIX activity Symptoms
Normal > 50% No symptomsSubhemophilia 15–40% No symptoms in normal lifeMild 5–15% Hematoma following trauma, discrete tendency to bleedModerate 1–5% Hematoma following mild trauma, tendency to bleedSevere < 1% Spontaneous bleeding, bleeding into joints, hematuria
Increased tendency to bleed, similar to hemophilia A (clinically indistinguishable):• Hematomas, soft tissue bleeding, bleeding into joints (hemarthrosis)• Hematuria, gastrointestinal bleeding
Medical History, Physical Examination• Medical history: including family history• Physical examination: including type of bleeding, complications
Laboratory TestsFactor IX ↓↓, PTT ↑, Quick’s test normal (extrinsic system), normal bleeding time (verified by platelet function test).
• Von Willebrand’s disease• Other coagulation factor deficiencies
Arthropathy, severe bleeding, infections.
Detailed recommendations on aspects of hemophilia treatment see guidelines of the International Society for Thrombosis and Hemostasis.
Bleeding ManagementAdministration of factor IX products, half-life 12–24 h.
Required Amount: Dose (IU) = Desired Factor Increase (%) × Body Weight (kg)Rule of Thumb: Administration of Factor IX 1 IU / kg → Plasma IX ↑ by 2%
Dosing Guidelines (7 Chap. 6.5.1)• Mild bleeding: increase factor IX for 1–2 days by 10–30%• Moderate bleeding: increase factor IX for 5–7 days to 30–50% of normal activity
Def:Def:
ICD-10:ICD-10:
Ep:Ep:
Pg:Pg:
Sy:Sy:
Dg:Dg:
Dd:Dd:
Co:Co:
Th:Th:
6.5.3 Factor IX Deficiency (Hemophilia B)
J.Heinz

���
• Severe bleeding / planned operation: increase factor IX for 3 days to > 70% , then keep at > 50% for 7 days
• In cases of emergency, fresh frozen plasma (FFP) may be used, if recombinant FIX concentrate is not available
Monitoring of FIX Replacement• aPTT monitoring is not sufficient, plasma factor IX should be determined (shortly after re-
placement and before administration of the next dose)• 1–4% of patients develop antibodies against infused factor, with treatment resistance → moni-
toring via FVII inhibitor assay
Normal life expectancy
Patient information and instruction (7 Chap. 6.5.2)
Special Attention when Caring for InfantsBleeding-related arthropathy often goes unnoticed → close monitoring, prophylactic factor IX treatment in cases of severe hemophilia in children: 25–40 IU/kg 2 times weekly → significant decrease of complication / arthropathy rate.
Berntorp E, Astermark J, Björkman S et al. Consensus perspectives on prophylactic therapy for haemo-philia: summary statement. Haemophilia 2003;9(suppl 1):1–4Bolton-Maggs P, Pasi KJ. Haemophilia A and B. Lancet 2003;361:1801–9Di Michele D. Inhibitor development in haemophilia B. Br J Haematol 2007;138:305–15Kubisz P, Stasko J. Recombinant activated factor VII in patients at high risk of bleeding. Hematology 2004;9:317–32Srivastava A. Dose and response in haemophilia: optimization of factor replacement therapy. Br J Haema-tol 2004;127:12–25Stobart K, Iorio A, Wu JK. Clotting factor concentrates given to prevent bleeding and bleeding-related complications in people with hemophilia A or B. Cochrane Database Syst Rev 2005;CD003429Van den Berg HM, Fischer K, van der Bom JG. Comparing outcomes of different treatment regimens for severe haemophilia. Haemophilia 2003;9:27–31
1. http://www.kcl.ac.uk/ip/petergreen/haemBdatabase.html Hemophilia B Database2. http://www.haemophilia-forum.org Haemophilia Forum3. http://www.hemophilia.org Natl Hemophilia Foundation4. http://www.haemophilia.org.uk Haemophilia Society5. http://www.wfh.org World Fed Hemophilia6. http://www.nlm.nih.gov/medlineplus/hemophilia.html Medline Plus7. http://med.unc.edu/isth/welcome International Society of Thrombosis and
Hemostasis
1.
2.3.4.
5.
6.
7.
Prg:Prg:
Px:Px:
Ref:Ref:
Web:Web:
�.� Hemophilia B

Hematology and HemostasisPart �
���
Hereditary coagulopathy due to qualitative or quantitative deficiencies of the von Willebrand fac-tor (vWF).
D68.0
Most common hereditary coagulopathy, heterozygotic gene carriers 1:100 to 1:1,000; incidence of symptomatic cases: 125 cases/1,000,000 population.
Von Willebrand factor is a heterogenic multimeric plasma glycoprotein (normal serum level: 10 mg/l). The vWF precursor is synthesized as a monomer in the endothelium and megakaryocytes. Active forms (vWF multimers) are found in the endothelium, platelets, and plasma. Functions:• Mediation of platelet adhesion to vascular wall (collagen) via high-molecular vWF multimers
and binding to platelet glycoprotein Ib (GPIb)• Factor VIII carrier in plasma
Hereditary defect caused by mutation in the vWF gene (chromosome 12); autosomal-dominant (subtype 1 and 2) or autosomal-recessive (subtype 2 and 3) inheritance. Consequences:• Impaired platelet adhesion• Reduced FVIII activity
Rare: acquired cases due to vWF antibodies in connection with autoimmune diseases, lymphop-roliferative diseases, or after multiple transfusions (“von Willebrand syndrome,” VWS). Defects in the vWF-binding glycoprotein GPIb can mimic von Willebrand’s disease (“Pseudo-VWD”).
VWDClassificationoftheInternationalSocietyonThrombosisandHemostasisISTH(1993)
Type Frequency Definition
1 70% Partial quantitative vWF deficiency, vWF plasma level 10–50%2 25–30% Qualitative vWF deficiency, atypical binding of vWF to platelets
2A 10–15% vWF multimers ↓↓, impaired platelet function2B 5% Increased affinity of vWF to platelet GPIb2M 5–10% Platelet function ↓↓, normal vWF multimer2N Up to 3% Reduced FVIII binding capacity3 < 10% Complete vWF deficiency, vWF level < 1%
• Type 1: mild form, bleeding time ↑, discrete tendency to bleed, epistaxis, gum bleeding, in-creased menstruation, bleeding after minor surgery
• Type 2: different characteristics depending on subtype; increased soft tissue bleeding, mucous membrane bleeding, gastrointestinal bleeding, hematuria; bleeding into joints less common than with hemophilia; rarely intracerebral bleeding
• Type 3: most severe form with pronounced bleeding (soft tissue bleeding, bleeding into joints, and petechial type bleeding)
• ATTENTION: in all types life-threatening bleeding may occur up to 14 days after surgery.
Medical History, Physical Examination• Medical history (including family history)• Physical examination including type of bleeding
Def:Def:
ICD-10:ICD-10:
Ep:Ep:
Phys:Phys:
Pg:Pg:
Class:Class:
Sy:Sy:
Dg:Dg:
6.5.4 Von Willebrand’s Disease (VWD)
O.Schmah,J.Heinz

���
Laboratory Tests• vWF antigen, FVIII function (FVIII:C), vWF multimers• Ristocetin cofactor (RiCof) ↓↓, ristocetin-induced platelet aggregation (RIPA)• Bleeding time or platelet function analysis• Platelet count, collagen-binding assay (CBA)• Blood group (patients with blood group O have a 25% lower vWF concentration)
Differential Diagnosis of VWD Types
LaboratoryfindingsforVWDsubtypes
Parameter Type 1 Type 2A Type 2B Type 2N Type 3
PTT n n n n / ↑ ↑Bleeding time n / ↑ n / ↑ n / ↑ n ↑Platelet count n n n / ↓ n nFVIII:C n n / ↓ n ↓ ↓vWF antigen n / ↓ n / ↓ n / ↓ n –Multimers n – – n –RiCof n / ↓ n / ↓ n / ↓ n –RIPA n n ↑ n –CBA ↓ ↓ ↓ n –
n normal, ↑ increased, ↓ decreased, – absent, RiCof ristocetin cofactor, RIPA ristocetin-induced platelet ag-gregation, CBA collagen-binding activity
ATTENTION: vWF is an acute-phase protein with high intraindividual variability → findings can often be inconclusive, determination during pregnancy and acute infections is not mean-ingful. Retesting may be required.
Bleeding in VWD Subtypes 1, 2A, or 2M• Mild bleeding: vasopressin analog desmopressin (DDAVP), nasal spray or intravenously, e.g.,
every 12–24 h 0.3 µg/kg body weight i.v. in 100 ml saline 0.9% over 30 min → release of vWF in endothelium, increase of the vWF level by factor 3–5. Response within 30–60 min in > 80% of patients; duration of effect 8–10 h. Since not all patients respond, conduct provocation test prior to treatment; treatment must be interrupted after 3–5 days due to depletion of endog-enous vWF stores.
• With menstruation, single doses of DDAVP prior to menstruation are usually sufficient; sup-portive estrogen therapy with subtype 1.
• Severe bleeding: similar strategy to type 2B, 2N, and 3.
Bleeding in VWD Subtypes 2B, 2N, and 3• Administration of high-vWF plasma products (e.g., 20–70 U/kg 2–4 times daily or 3–5 U/kg/
h per infusor) until ristocetin cofactor activity > 60% for at least 72 h. ATTENTION: Recom-binant FVIII products contain no vWF and are ineffective in von Willebrand’s disease → use special high-vWF plasma or FVIII products.
• Platelet concentrates• If surgery is planned: vWF antigen as well as ristocetin cofactor activity should be 60%, pre- as
well as postoperatively.• ATTENTION: With subtype 2B, DDAVP did not demonstrate a clear benefit (risk of throm-
bocytopenia). With subtype III, it is ineffective.
Monitoring of Substitution• Monitoring of vWF antigen, FVIII function (FVIII:C), ristocetin cofactor (RiCof) according
to disease subtype.
Th:Th:
�.� Von Willebrand’s Disease

���
Part � Hematology and Hemostasis
• Development of vWF alloantibodies in 10–15% of cases, risk of anaphylactic reactions with repeated exposure. With neutralizing antibodies and bleeding complications factor VIIa may be given.
Adjuvant Treatment / Preparation for Surgery• If DDAVP has proven to be effective, give 30 min prior to surgery• High risk of hemorrhage (e.g., tonsillectomy): raise vWF antigen and ristocetin cofactor activ-
ity up to 60%; administer high-vWF FVIII concentrate• Intraoperative use of fibrin glue and fibrinolysis inhibitors (e.g., tranexamic acid mouthwash
with dental surgery)
During Pregnancy• During pregnancy, hormone-induced increase in vWF and FVIII:C → with subtype 1 and 2
no further treatment required• Peripartum: keep vWF antigen and Ristocetin cofactor activity above 50%; with cesarean sec-
tion, aim for 100% pre- and postoperatively
Battle J, Noya MS, Giangrande P et al. Advances in the therapy of von Willebrand disease. Haemophilia 2002;8:301–7Ginsburg D. Molecular genetics of von Willebrand disease. Thromb Haemost 1999;82:585–91Manucci PM. Treatment of von Willebrand’s disease. N Engl J Med 2004;351:683–94Rodeghiero F, Castaman G. Treatment of von Willebrand disease. Semin Hematol 2005;42:29–35Ruggeri ZM. Developing basic and clinical research on von Willebrand factor and von Willebrand disease. Thromb Haemost 2000;84:147–9Sadler JE, Mannucci PM, Berntorp E et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 2000;84:160–74Schneppenheim R, Budde U. Phenotypic and genotypic diagnosis of von Willebrand disease: a 2004 update. Semin Hematol 2005;42:15–28
1. http://www.shef.ac.uk/vwf/ ISTH VWF Database2. http://www.allaboutbleeding.com/ VWD Resource3. http://www.nlm.nih.gov/medlineplus/ency/
article/000544.htm VWD, Medline Plus4. http://www.hemophilia.org/bdi/bdi_types3.htm Hemophilia Foundation5. www.wfh.org World Fed Hemophilia6. www.hemophilia.ca/en/2.2.php Canadian Hemophilia Soc7. www.emedicine.com/med/topic2392.htm E-Medicine
1.
2.3.4.5.
6.
7.
Ref:Ref:
Web:Web:

���
Systemic consumption coagulopathy due to release of coagulation activators, with intracapillary coagulation, microthrombus formation, subsequent ischemic organ damage (kidney, liver, lung) and organ failure. Diffuse tendency to bleed due to collapsed hemostasis with secondary hyperfi-brinolysis.
Acute DIC is a severe life-threatening disease. Chronic DIC with continuous coagulation may occur in patients with malignant diseases.
D65
Excessive Release of Activators of the Coagulation Cascade• Infections: sepsis (gram-negative / gram-positive), malaria, rickettsia, chlamydia, mycobac-
teria, meningococcus (Waterhouse-Friderichsen syndrome: consumption coagulopathy with adrenocortical bleeding), viral infections
• Solid tumors: carcinomas of the lung, pancreas, stomach, colon, prostate, Kasabach-Merritt syndrome (hemangiomas)
• Hematological neoplasia: acute promyelocytic leukemia (FAB M3)• Obstetric complications: placenta abruptio, amniotic fluid embolism, septic abortion, eclamp-
sia, postpartal hemolytic-uremic syndrome• Hypoxia and shock: traumatic, hemorrhagic, cardiac, septic• Hemolysis: transfusion errors, toxins, paroxysmal nocturnal hemoglobinuria• Operations on organs with a high thrombokinase content (prostate, pancreas, lung), extracor-
poreal circulation (contact activation of the endogenous coagulation system)• Trauma: head injury, soft tissue damage, fat embolism• Others: snake bites, heat stroke (endothelial damage), abdominal aortic aneurysms
Pathophysiology: Four PhasesExcessive thrombin synthesis leads to fibrin formation and subsequent intravascular coagulation with consumption of platelets and clotting factors. Inhibitory mechanisms (e.g., inhibition of FVa and FVIIIa via thrombomodulin-activated protein C) cannot compensate the thrombin forma-tion.
Phase Characteristics
I Hypercoagulability, microthrombus formation, microembolismsII Lack of coagulation factors, incipient fibrinolysisIII Hemostasis collapse, severe reactive fibrinolysisIV Reconstitution
Def:Def:
ICD-10:ICD-10:
Pg:Pg:
Pphys:Pphys:
�.� DIC
6.5.5 Disseminated Intravascular Coagulation (DIC)
J.Heinz

��0
Part � Hematology and Hemostasis
Initially (phase I and II), pathological laboratory parameters only. Only with severe consumption coagulopathy (phase III) clinically detectable symptoms:• Hemorrhagic diathesis with ubiquitous bleeding, 75% of cases: skin / mucous membrane bleed-
ing, hematomas, secondary bleeding after venipuncture / from puncture sites, pulmonary hemorrhage, gastrointestinal bleeding, renal bleeding, hematuria, adrenal bleeding / insuffi-ciency, intracerebral bleeding
• Multiple microthromboses with impaired organ function, 70%: acute renal failure, impaired liver function, acute respiratory insufficiency (ARDS, “acute respiratory distress syndrome”), intra-dermal microvascular thrombosis → “purpura fulminans” (skin bleeding with central necro-sis), cerebral small vessel ischemia (coma, epileptic seizures)
• Shock: tachycardia, decrease in blood pressure, edemas, organ failure• Chronic course: coagulation factor synthesis ↑, thrombosis ↑ → malperfusion of larger blood
vessels (embolisms, cerebral ischemia, etc.)
Case History, Physical Examination• Case history including risk factors• Physical examination
Laboratory Tests
Phase Description Parameters
I Activation Rapid decrease of platelets, platelet count n / ↓, antithrombin n / ↓, FV n / ↓, FVIII n / ↓, coagulation products ↑ (soluble fibrin, prothrombin fragment F1+2, thrombin–antithrombin complex TAT)
II Early consumption Platelets ↓, antithrombin ↓, Quick ↓, coagulation factors (fibrinogen, FV) ↓, PTT ↑, TAT ↑, protein C ↓
III Late consumption Platelets ↓↓ (< 30,000/µl), antithrombin ↓↓, Quick ↓↓, coagulation factors ↓↓ (fibrinogen, FV, and FVIII), PTT ↑↑, thrombin time ↑↑, fibrinogen degrading products / fibrin monomers +, D-dimers +++, detectable fragmentocytes
IV Recovery Decrease in coagulation products (soluble fibrin, prothrom-bin fragment F1+2, TAT), increase of clotting factors (fibrino-gen, FV, FVIII), normalization of global clotting tests
n normal, F factor, TAT thrombin-antithrombin complex, PTT partial thromboplastin time
Diagnostic Key Parameters• Basic diagnosis: platelets (platelet decrease often first symptom), antithrombin, D-dimers, fi-
brinogen, Quick’s test, PTT• Advanced diagnosis: fibrin monomers (soluble fibrin), prothrombin fragments F1+2, throm-
bin–antithrombin complex (TAT), plasmin-plasmin inhibitor complex, factor V, protein C, possibly protein S (in cases of purpura fulminans)
DICscoringsystemoftheISTH(InternationalSocietyonThrombosisandHemostasis)
Basic screening test Score
0 1 2 3
Platelets > 100/nl 50–100/nl <50/nl –Quick’s test INR < 1.4 INR 1.5–2.0 INR > 2.0 –Fibrinogen > 100 mg/dl ≤ 100 mg/dl – –Fibrinogen degradation products Normal – + ++
INR international normalized ratio
Sy:Sy:
Dg:Dg:

���
A score ≥ 5 indicates ongoing DIC; with scores < 5 diagnostics should be repeated every 12–24 h depending on the individual clinical condition.
ATTENTION:• With tumors / infections / pregnancy, the platelet count is often increased (“reactive throm-
bocytosis”) → normal platelet counts may already indicate DIC.• Fibrinogen is an acute-phase protein → “normal” fibrinogen levels may already be patho-
logically decreased (e.g., with infections).• With DIC, frequent monitoring is required to determine the dynamics and course of dis-
ease.
• Primary hyperfibrinolysis: normal platelet count, normal ATIII, no fibrin monomers
Treatment of DIC requires a combined approach, with treatment of the underlying disease and cor-rection of the coagulation disorder. Early diagnosis improves survival and long-term outcomes.
Principles of DIC TreatmentBasic therapy• Antithrombin (AT) replacement if AT level < 70% (initially 1,000 IU, then 500 IU every 6 h),
check level (target: 80–100%)• Heparin: 100–300 IU/kg/day (not with AML type M3 or patients with high bleeding risk),
depending on platelet countOrgan dysfunction, bleeding• Fresh frozen plasma (FFP, 10 ml/kg)• Antithrombin (AT), 500–1000 IU every 6 h• Fibrinogen replacement with fibrinogen levels of < 100 mg/dl• Platelet transfusion (target: > 50,000/μl)• Red cell transfusion according to hemoglobin level, compensation of acidosis• Heparin is contraindicated, no intramuscular injections• Patients should be treated in intensive care unit• Severe uncontrollable bleeding: administration of activated FVIIa. ATTENTION: potential
risk of thromboembolic complications
ATTENTION: administration of coagulation factors can increase DIC and should be avoided. For replacement therapy, use fresh frozen plasma (FFP).
Special Cases• Severe sepsis: activated protein C (drotrecogin) 24 µg/kg/h over 96 h → decreases mortality
from 31% to 25%. ATTENTION: with thrombocytopenia higher risk of hemorrhagic compli-cations. Contraindicated after brain hemorrhage, epidural catheter, etc.
• In DIC, avoid fibrinolysis inhibitiors: → in cases of uncontrollable bleeding: use aprotinin (e.g., 250,000 units in the first half hour, followed by 2 million units/day), platelets, fibrinogen, and PPSB. In cases of life-threatening bleeding or lack of success: administer recombinant FVIIa.
• Fibrinolysis inhibitors (antifibrinolytics, e.g., tranexamic acid) may be indicated with hyperfi-brinolytic conditions (prostate carcinoma, AML M3) in phase I of a DIC only.
• Low-dose heparinization is indicated in cases of: purpura fulminans, acral ischemia, venous thrombosis. For phase I, some studies favor low-molecular weight heparin (100–200 units/kg/day).
Monitoring During Treatment• Clinical monitoring: close monitoring of neurological, cardiovascular, respiratory and renal
parameters• Monitoring of bleeding: tachycardia, hemoglobin decrease, retroperitoneal bleeding (→ so-
nography), neurology• Laboratory tests: coagulation parameters, blood count, hepatic and renal function parameters,
electrolytes
Dd:Dd:
Th:Th:
�.� DIC

���
Part � Hematology and Hemostasis
In cases of manifest severe DIC: 50–80% mortality
Administration of heparin 10,000–15,000 IU/24 h with all predisposing conditions
Bernard GR, Vincent JH, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001;344:699–709De Cicco M. The prothrombotic state in cancer: pathogenetic mechanisms. Crit Rev Hematol Oncol 2004;50:187–96Franchini M, Manzato F. Update on the treatment of disseminated intravascular coagulation. Hematology 2004;9:81–5Levi M. Disseminated intravascular coagulation: what’s new? Crit Care Clin 2005;21:449–67Toh CH, Dennis M. DIC 2002: a review of disseminated intravascular coagulation. Hematology 2003;8:65–71Warren BL, Reid A, Singer P et al. High dose antithrombin III in severe sepsis. JAMA 2001;286:1869–78Yanada M, Matsushita T, Suzuki M et al. DIC in acute leukemia: clinical and laboratory features at presen-tation. Eur J Haematol 2006;77:282–7
1. http://www.med.unc.edu/isth/ Intl Soc Thromb Haemost2. http://www.nlm.nih.gov/medlineplus/ency/article/000573.htm Medline Plus3. http://www.emedicine.com/med/topic577.htm E-medicine
1.
2.
3.
4.5.
6.7.
Prg:Prg:
Px:Px:
Ref:Ref:
Web:Web:

���
Thrombosis: localized intravascular aggregation of blood components → thrombus (clot) forma-tion with consecutive vascular occlusion. Clinical symptoms differ depending on the blood vessels affected: venous or arterial thrombosis.
Embolism: migration of detached thrombus (clot) elements in the blood stream with consecutive vascular occlusion. Triggers: thrombotic material, tumor particles or leukemic cell thrombi, scle-rotic material, fat droplets, amniotic fluid, air.
Thrombophilia: increased risk of occurrence of thromboembolic events, hereditary or acquired.
I82.9
Thrombosis incidence: 3 cases/1,000 population/year; location: > 90% in inferior vena cava or leg / pelvic veins; male:female = 1:1; particularly in patients > 50 years of age; pulmonary embolisms occur in 1–2% of all hospitalized patients.
Thromboembolic Events in Patients with Malignancies••
•
•
10–30% of tumor patients experience thrombosis in the course of their diseaseOccurrence depends on tumor location and histology (common with lung cancer, gastro-intestinal adenocarcinomas, pancreatic and ovarian carcinoma)“Idiopathic” deep vein thrombosis or pulmonary embolism in clinically “healthy” adults is due to an underlying malignancy in 6–35% of casesPulmonary embolisms or venous thromboses are found in up to 50% of cancer patients at autopsy
VirchowTriad:Major Pathomechanisms of Thrombogenesis
•••
Endothelial alterations: vascular sclerosis, inflammation, trauma, etc.Circulation disorders: intravascular stasis, vortex formation, etc.Hypercoagulability: thrombophilia, thrombocytosis, etc.
Endothelial Alterations (esp. with Arterial Thrombosis)• Arteriosclerosis• Vascular puncture, vascular prostheses, trauma• Vasculitis (arteritis nodosa, thromboangiitis obliterans, etc.), phlebitis
Circulation Disorders• Immobilization, bed rest• Intravascular stasis due to vascular constriction or compression: e.g., after extended periods of
travel (“economy class syndrome”), varicosis, obesity, pregnancy, solid tumors, or lymphomas• Altered blood flow due to cardiac disorders (“low output failure”: cardiac insufficiency, infarc-
tion, cardiogenic shock)
Hypercoagulability
ThrombocytosisE.g., myeloproliferative syndromes (7 Chap. 7.3)
Def:Def:
ICD-10:ICD-10:
Ep:Ep:
Pg:Pg:
�.� Thromboembolism and Thrombophilia
6.6 Thromboembolism and Thrombophilia
J.Heinz

���
Part � Hematology and Hemostasis
Thrombophilia•
•
•
•
•
•
APC (activated protein C) resistance: most common cause of thrombophilia; in 95% of cases due to factor V mutations → abnormal APC binding site (mainly point mutations in the factor V gene, G1691A, “factor V Leiden”) → insufficient inactivation of factor V by mutated APC. Prevalence: heterozygotic carriers 3–9% of normal population, 20–40% among patients with thrombosis. Relative thrombosis risk in heterozygotic cases approxi-mately 3- to 7-fold, in homozygotic cases 50- to 100-fold increased. Other causes (< 5%) for APC resistance: antiphospholipid antibodies, oral contraceptives, pregnancy.Factor II mutation: prothrombin mutation G20120A, often associated with increased prothrombin levels. Prevalence: heterozygotic carriers 2–4% of normal population; among patients with thrombosis 5–7%. Relative risk of thromboembolisms: 2- to 4-fold increased.Factor VIII increase: 25% of thrombosis patients show persistent FVIII increase of un-known etiology; with significant risk for relapse thrombosis. ATTENTION: FVIII levels not meaningful in acute situations (acute phase protein).Antithrombin / protein C / protein S defects: rare hereditary disorders; AT deficiency espe-cially is associated with a high risk of thrombosis. DD: hepatic diseases.Hyperhomocysteinemia: hereditary defect of cystathionine β synthetase; or acquired due to vitamin B6, B12, or folic acid deficiency.Antiphospholipid syndrome: most common acquired form of thrombophilia; occurs as primary or secondary (as a result of systemic lupus erythematosus SLE, collagenosis, ma-lignancy, medication, infections) subtype. Typical triad: thrombosis (venous or arterial) + recurrent miscarriages + thrombocytopenia.
Other Causes of Abnormal Coagulation• Abnormal fibrinolysis: plasminogen deficiency, tPA deficiency (rare)• Hyperviscosity syndrome: hypergammaglobulinemia with plasmocytomas, hyperglobulinemia,
dehydration, leukemia• Trauma, burns• Estrogens: pregnancy, postpartum, contraceptives. Contraceptives increase the thrombosis risk
by factor 5, simultaneous smoking by factor 20.
ReasonsforIncreasedThrombogenesisinPatientswithMalignancies
• Vessel wall defects: endothelial alterations due to intravascular catheters / lines, antineoplastic treatment, direct invasion of tumor tissue; cytokine-mediated activation of the endothelium → enhanced expression of tissue factor / adhesion molecules / PAI and decreased endothe-lial thrombomodulin expression → thrombophilic surface
• Changes in blood flow: immobilization, tumor-related vascular compression, stasis, hypervis-cosity
• Changes in coagulation system: fibrinogen ↑, factor V ↑, FVIII ↑, von Willebrand factor ↑, FXII ↑, AT ↓
• Release of procoagulating substances (“cancer coagulants,” e.g.,tissue factor = TF, FX activa-tors) with activation of the extrinsic system via factor VII or direct FX activation; high levels of TF in promyelocytes of acute leukemia type FAB M3
• Decrease in coagulation inhibitors (antithrombin, protein C and S) due to chemotherapy (asparaginase)
Thrombus Types• White thrombus / blood platelet thrombus: platelet aggregates occurring with endothelial de-
fects; adherent to vascular wall• Coagulation thrombus: intravascular coagulation due to stasis or decreased blood flow rate,
platelet / erythrocyte / coagulation factor aggregates, only moderate adherence to vessel wall, high risk of embolism
• Hyaline thrombus: capillary thrombus in disseminated intravascular coagulation (DIC, 7 Chap. 6.5.5), high content of coagulation factors
Path:Path:

���
• Tumor cell / leukemia cell thrombus: solid tumor particles or aggregates of malignant cells in patients with leukemia (usually with cell counts of > 100,000/µl)
Locations• Arterial thrombi: aorta, coronary vessels, carotids, cerebral arteries, extremities• Venous thrombi: predominantly in lower extremity veins (50%), pelvic veins (30%), vena cava
inferior (10%); with iatrogenic damage of the subclavian or jugular veins (catheters)
Embolism• Detachment of venous thrombi results in pulmonary embolism in > 90% of cases; paradoxical
arterial embolism in cases of patent foramen ovale / pulmonary hypertension• Displacement of thrombi from the left side of the heart → arterial embolism of cerebral vessels
(60%), extremity arteries (30%), renal arteries, mesenteric arteries
Deep Vein Thrombosis (DVT) of the LegOnly < 30% of patients show classic clinical signs:• Calf pain at dorsal flexion of the ankle (Homans’ sign) or ballottement of the calf• Tenderness when pressure is applied to the sole of the foot (Payr’s sign)• Tenderness when pressure is applied along the deep veins of the leg• Dilation of epifascial veins• Local swelling, hyperthermia, tightness, pain, fever
Phlebothrombosis of the Arm (Paget-von Schroetter Syndrome)• Swelling of the arm, hyperthermia, livid discoloration, tightness• Pain in forearm, upper arm and/or shoulder, fever
Phlegmasia Cerulea Dolens• Combined venous and arterial occlusion• Rapidly increasing swelling of the leg• Pulseless extremity, swelling, livid discoloration, tenderness• Severe pain
Pulmonary Embolism• Dyspnea, tachypnea, cough 80% of patients• Chest pain, infradiaphragmatic pain 80%• Tachycardia 60–70%• Anxiety, sweating, vegetative symptoms 30–50%• Syncope, shock 10–20%
Arterial Occlusion: Ischemia Syndrome (“6 Ps”)Pain, pallor, pulselessness, paresthesia, paralysis (motor dysfunction), prostration
Medical History, Physical Examination• Medical history including risk factors• Physical examination including local signs and symptoms, cardiopulmonary signs, circulatory
status
Laboratory Tests• Routine laboratory analyses including urea + electrolytes, serum creatinine, LDH• D-dimer: decomposition product of cross-linked fibrin; high sensitivity with thrombosis
(95–100%) but low specificity (positive D-dimers after surgery, with trauma, hemorrhage, in-flammation, malignancy, and pregnancy)
• ATTENTION: if thrombosis or pulmonary embolism is suspected, further diagnostic proce-dures should be implemented even if D-dimers are negative.
• Capillary / arterial blood gas analysis: pO2 ↓, pCO2 ↑ (with pulmonary embolism)• Thrombophilia work up:
− Indication: thrombosis of unknown etiology in patients < 40 years of age; hereditary dis-position; recurrence of thrombosis or embolism; unusual thrombosis location (e.g., sinus
Sy:Sy:
Dg:Dg:
�.� Thromboembolism and Thrombophilia

���
Part � Hematology and Hemostasis
veins, mesenteric veins); thromboembolic event despite effective anticoagulation; throm-bosis during pregnancy and tendency to miscarriage and stillbirth
− Analysis of: fibrinogen, antithrombin, protein C, protein S, prothrombin (FII), FVIII, APC resistance, factor II mutation, antiphospholipid antibodies (lupus anticoagulants, anticar-diolipin antibodies), plasminogen deficiency
− Extended diagnostics: homocysteine, methyltetrahydrofolate reductase (MTHFR) muta-tion G77T, FIX, and FXII
− In most cases, repeated tests are required. With suspected thrombophilia, patients should be referred to specialized hematology centers.
Imaging in Cases of Suspected Thrombosis• Sonography: CW Doppler, B-mode, duplex scan• Only in uncertain cases: phlebography• Possibly CT scan or MRI (esp. abdomen and pelvis)
Imaging with Suspected Embolism• Spiral CT• Ventilation / perfusion lung scan• Echocardiography revealing signs of right heart failure• Angiography / arteriography• ECG: signs of pulmonary embolism: sinus tachycardia, SI QIII type, incomplete right bundle
branch block, P pulmonale, newly developed arrhythmia / extrasystoles
Tumor Screening: Young Patients, Thrombosis of Unknown Etiology, Recurrent Thrombosis• Thorough clinical examination (including lymph node status, rectal examination, fecal occult
blood test, gynecological / urological examination)• Blood count, LDH, PSA• Chest x-ray, abdominal sonography, possibly CT abdomen / pelvis
DVT of the lower extremity• Vascular compression by tumors, aneurysms, hematomas, Baker’s cyst, retroperitoneal fibro-
sis, vena cava compression, etc.• Erysipelas• Edema of different etiology (pulmonary edema, cardiac edema)• Superficial thrombophlebitis
Thrombosis-related Complications• Venous thrombi → pulmonary embolism (in > 95% of cases due to phlebothrombosis, ap-
proximately 50% of patients with phlebothrombosis develop pulmonary embolism)• Arterial / cardiac thrombi → cerebral malperfusion, renal infarction, extremities
Long-term Sequelae of Lower Extremity DVT• Post-thrombotic syndrome (after 10–15 years, in 40–60% of conventionally treated patients)• Chronic leg ulcer (in 10% of patients)
Anticoagulation
InitialTreatmentwithHeparin
Low Molecular Weight Heparin (LMWH)• Effect: factor Xa inhibition, half-life: 100–180 min• Dosage: enoxaparin 1 mg/kg twice daily s.c., dalteparin 100 IU/kg twice daily, or tinzaparin
175 IU/kg once daily• Advantages of treatment with LMWH compared to UFH:
DD:DD:
Co:Co:
Th:Th:

���
− Rapid onset of the anticoagulation effect; minimal laboratory monitoring required (plate-let count during first 3 weeks, antifactor Xa levels in patients with renal failure, cachectic, or overweight patients)
− No i.v. access, outpatient treatment possible− Low molecular weight heparin has a favorable side effect profile, hemorrhagic complica-
tions, osteoporosis, and heparin-induced thrombocytopenia are less common• Target antifactor Xa activity: if given twice daily, 0.5–1.0 IU/ml (3–4 h postinjection); with
single daily dose, 1.0–2.0 IU/ml (3–4 h postinjection); levels should be determined after steady state has been reached
Unfractioned Heparin (UFH)• Effect: inhibition of thrombin, factor Xa, IXa, half-life: 60 min• Intravenous bolus: 5,000 IU i.v. in patients < 70 kg, 10,000 IU bolus > 70 kg; then continuous
intravenous treatment: 30,000 IU/24 h, maximum 50,000 IU/24 h; dosage according to PTT: target PTT > 60–90 s, first PTT test after 6 h, then every 12 h; once stable, it is sufficient to check PTT once daily
• Alternatively, subcutaneous administration: 7,500–10,000 IU s.c. 3 times daily; studies have shown that with identical PTTs, intravenous and subcutaneous administration are equally ef-fective
• Side effects: hemorrhage (in up to 10% of patients), hypersensitivity (urticaria, bronchospasm, fever, even shock), alopecia (rare), vasospasm (rare), osteoporosis (with long-term use), hepa-rin-induced thrombocytopenia (in 2–10% of patients, 7 Chap. 6.3.2)
Indications• Deep vein thrombosis of the lower extremity, thrombosis of major veins (arm veins, cerebral
veins, etc.)• Pulmonary embolism (stable circulation)• Arterial occlusion (extremity arteries, acute myocardial infarction, etc.)
Discontinuation of Heparin Treatment• Adoption of coumarin: discontinuation of heparin treatment once target INR is reached (usu-
ally after 5 days); exception: in cases of extensive thrombosis (calf to pelvis), continue heparin treatment for 10–14 days, do not give coumarin before day 5.
• Absence of contraindications and patient compliance provided, LMWH treatment may be possible in an outpatient setting.
SecondaryProphylaxis:Coumarin(Phenprocoumon)
EffectVitamin K antagonism → inhibition of the hepatic synthesis of coagulation factors F II, VII, IX, X (prothrombin complex) as well as protein C and protein S.
IndicationsIntroduce on first or second day of heparin treatment (exception: in case of extensive thrombosis (calf to pelvis), give oral anticoagulants only after 5 days of heparin treatment).
Contraindications• Patients > 65 years of age, hemorrhagic diathesis, sepsis• Uncompensated hypertension, liver or renal insufficiency• Surgery within last 7–10 days, arterial puncture, intramuscular injections• CNS surgery within the last 3 months, cerebral bleeding, cerebral sclerosis, CSF puncture
within the last 10 days• Pancreatitis, endocarditis lenta, diabetic retinopathy, nephrolithiasis• Pulmonary / gastrointestinal diseases with high risk of bleeding (tuberculosis, bronchiectasis,
ulcers, colitis, esophageal varices, neoplasia)• Pregnancy, in particular during the first 3 months
�.� Thromboembolism and Thrombophilia

���
Part � Hematology and Hemostasis
DosageAccording to INR (“international normalized ratio”).
INR =Patient Plasma Prothrombin Time (s)Normal Plasma Prothrombin Time (s)
In cases of uncomplicated deep vein thrombosis and/or pulmonary embolism, the recommended INR is 2.0–3.0. A higher target INR (3.0–4.0) is recommended in cases of recurrent venous throm-bosis despite adequate anticoagulation.
Treatment InitiationIn normal weight patients (≈ 70 kg), the following rule of thumb applies: current Quick value divided by 10 is the number of coumarin tablets to be given in the first 4 days. On day 1, give 3 tablets, on days 2 and 3 give 2 (or 1) tablets (slow initiation to reduce the risk of coumarin necrosis). The INR should be checked on day 4. The result determines the dosage of subsequent treatment. Heparin treatment may be discontinued once the target INR has been reached (usually after 5–6 days).
Duration of Treatment: Depending on Thrombosis TypeThe duration of anticoagulation treatment has to be determined individually for each patient, based on thrombosis type, location, risk factors and comorbidities. Guideline:
Thrombosis type and location Duration of treatment
Deep vein thrombosis of the lower leg (> 2 veins, trifurcation) 1.5–3 monthsArm vein thrombosis 3 monthsThrombosis of the popliteal vein and/or the femoral vein 6 monthsInvolvement of the iliac vein (pelvic vein thrombosis) (6)–12 monthsPulmonary embolism with/without deep vein thrombosis of the leg (6)–12 monthsRecurring thrombosis 3–4 yearsDVT due to severe thrombophilia (AT deficiency, homozygotic FV mutation, malignancy, etc.)
Possibly life-long
Life-threatening pulmonary embolism or thrombosis (mesenteric, sinus veins, cerebral)
Possibly life-long
Secondary Prophylaxis AlternativePatients with contraindications against coumarin may receive low molecular weight heparin as secondary prophylaxis on a long-term basis. Half the therapeutic LMWH dose is usually recom-mended (comparable to an INR of 2–3); start after 10–14 days of “full dose” therapeutic LMWH treatment. Cancer patients in particular benefit from treatment with low molecular weight hepa-rin.
Thrombolytic Therapy (Fibrinolysis Treatment)
Due to frequent occurrence of hemorrhagic complications (10–15%), increased mortality (1–2%) and limited long-term benefit (no reduction in occurrence of post-thrombotic syndrome), fibri-nolysis now only plays a secondary role. An indication for treatment with fibronolytics (e.g., strep-tokinase, urokinase) may exist in young patients with extensive fresh thrombosis.

���
Surgical Treatment
Surgical ThrombectomySurgical thrombectomy allows immediate perfusion of the blood vessel. However, endothelial in-jury and incomplete thrombus removal often lead to rapid reformation of thrombi. Indications:• Phlegmasia cerulea dolens• Fresh isolated descending pelvic vein thrombosis (not older than 1–2 days)• Acute arterial occlusion
Cava FilterPlacement of a filter in the V. cava reduces the risk of severe pulmonary embolism in patients with recurrent thromboses. Indications are:• Recurrent pulmonary embolism despite effective anticoagulation• Contraindication against anticoagulants
Supportive Treatment
• Immobilization: studies did not confirm a role for immobilization in the prevention of pulmo-nary embolisms.
• In patients with severe pain or edema: elevation and immobilization of the leg for a limited number of days.
• Compression therapy: compression dressings with bandages or compression stockings; contra-indicated with peripheral arterial occlusive disease and phlegmasia cerulea dolens. Compres-sion stockings should be worn for at least 2 years as secondary prophylaxis after DVT of the lower extremity. In most cases, calf compression stockings on the affected leg are sufficient.
• Anticoagulants• Platelet aggregation inhibitors, acetylsalicylic acid 100 mg daily p.o. (protective effect in par-
ticular with arterial occlusion and coronary heart disease)• Elimination of risk factors (see above), early postoperative mobilization, physiotherapy, com-
pression stockings
Baglin IP, Cousins D, Keeling DM et al. Recommendations from the British Committee for Standards in Haematology and National Patient Safety Agency. Br J Haematol 2006;136:26–9British Committee for Standards in Haematology (BCSH). Guideline. Investigation and management of heritable thrombophilia. Br J Haematol 2001;114:512–28Francis CW. Prophylaxis for thromboembolism in hospitalized medical patients. N Engl Med 2007;356:1438–44Klerk CPW, Smorenburg SM, Otten HM et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol 2005;23:2130–5Kyrle PA, Eichinger S. Deep vein thrombosis. Lancet 2005;365:1163–74Lee AYY, Rickles FR, Julian JA et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol 2005;23:2123–9Lopez JA, Kearon C, Lee AY. Deep venous thrombosis. Hematology (ASH Educ Program) 2004;439–56Lyman GH, Khorana AA, Falanga A et al. ASCO Guideline: recommendations for venous thromboembo-lism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007;25:5490–505
1. http://www.nlm.nih.gov/medlineplus/ency/article/000156.htm Medline Plus2. http://www.emedicine.com/emerg/topic122.htm E-Medicine3. http://www.med.unc.edu/isth ISTH4. http://www.tri-london.ac.uk/ Thrombosis Res Inst5. http://www.tigc.org TIGC
1.
2.
3.
4.
5.6.
7.8.
Px:Px:
Ref:Ref:
Web:Web:
�.� Thromboembolism and Thrombophilia
Related Documents











