Yeast Yeast 2002; 19: 933–947. Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/yea.885 Research Article 6-phosphofructokinase from Pichia pastoris: purification, kinetic and molecular characterization of the enzyme J¨ urgen Kirchberger,* J ¨ org B¨ ar, Wolfgang Schellenberger, Hassan Dihazi and Gerhard Kopperschl¨ ager Institut f¨ ur Biochemie, Medizinische Fakult¨ at der Universit¨ at Leipzig, Liebigstrasse 16, D-04103 Leipzig, Germany *Correspondence to: J¨ urgen Kirchberger, Institut f¨ ur Biochemie, Medizinische Fakult¨ at der Universit¨ at Leipzig, Liebigstrasse 16, D-04103 Leipzig, Germany. E-mail: [email protected] Received: 8 February 2002 Accepted: 1 May 2002 Abstract 6-Phosphofructokinase from Pichia pastoris was purified for the first time to homo- geneity applying seven steps, including pseudo-affinity dye-ligand chromatography on Procion Blue H-5R-Sepharose. The specific activity of the purified enzyme was about 80 U/mg. It behaves as a typically allosteric 6-phosphofructokinase exhibiting activa- tion by AMP and fructose 2,6-bis(phosphate), inhibition by ATP and cooperativity to fructose 6-phosphate. However, in comparison with the enzymes from Saccharomyces cerevisiae and Kluyveromyces lactis, the activation ratio of 6-phosphofructokinase from Pichia pastoris by AMP is several times higher, the ATP inhibition is stronger and the apparent affinity to fructose 6-phosphate is significantly lower. Aqueous two- phase affinity partitioning with Cibacron Blue F3G-A did not reflect remarkable structural differences of the nucleotide binding sites of the Pfks from Pichia pastoris and Saccharomyces cerevisiae. The structural organisation of the active enzyme seems to be different in comparison with hetero-octameric 6-phosphofructokinases from other yeast species. The enzyme was found to be a hetero-oligomer with an molecular mass of 975 kDa (sedimentation equilibrium measurements) consisting of two distinct types of subunits in an equimolar ratio with molecular masses of 113 kDa and 98 kDa (SDS–PAGE), respectively, and a third non-covalently complexed protein component (34 kDa, SDS–PAGE). The latter seems to be necessary for the catalytic activity of the enzyme. Sequencing of the N-terminus (VTKDSIXRDLEXENXGXXFF) and of peptide fragments by applying MALDI–TOF PSD, m/z 1517.3 (DAMNVVNH) and m/z 2177.2 [AQNCNVC(L/I)SVHEAHTM] gave no relevant information about the identity of this protein. Copyright 2002 John Wiley & Sons, Ltd. Keywords: 6-phosphofructokinase; Pfk; Pichia pastoris ; purification; molecular and kinetic properties; methylotrophic yeast Introduction 6-phosphofructokinase (Pfk, phosphofructoki- nase-1, phosphohexokinase; EC 2.7.1.11) cataly- ses the transfer of phosphate from MgATP to the 1-OH group of fructose 6-phosphate, yielding fruc- tose 1,6-bis(phosphate) and MgADP. The catalytic activity is tightly regulated in a wide variety of organisms by diverse positive and negative effec- tors. The enzyme plays a major role in regulat- ing the glycolytic flux (Hofmann, 1976; Heinrich et al., 1977; Sols, 1981; Kotlarz and Buc, 1982; Kemp and Foe, 1983; Heinisch and Hollenberg, 1993; Larsson et al., 2000). In a few bacteria and most eukaryotic cells, ATP was found to function not only as a substrate but also as a potent inhibitor, whereas AMP and fructose 2,6- bis(phosphate) (Fru 2,6-P 2 ) are strong activators and are able to diminish ATP inhibition. In bacteria, Pfk is composed of four subunits of equal size, each of 36 kDa (Blangy, 1968; Hudson et al., 1979). In mammals, the enzyme consists of 80 kDa subunits that form a tetramer as the small- est catalytically active species, but aggregates of Copyright 2002 John Wiley & Sons, Ltd.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

YeastYeast 2002; 19: 933–947.Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/yea.885
Research Article
6-phosphofructokinase from Pichia pastoris:purification, kinetic and molecular characterizationof the enzymeJurgen Kirchberger,* Jorg Bar, Wolfgang Schellenberger, Hassan Dihazi and Gerhard KopperschlagerInstitut fur Biochemie, Medizinische Fakultat der Universitat Leipzig, Liebigstrasse 16, D-04103 Leipzig, Germany
*Correspondence to:Jurgen Kirchberger, Institut furBiochemie, Medizinische Fakultatder Universitat Leipzig,Liebigstrasse 16, D-04103Leipzig, Germany.E-mail:[email protected]
Received: 8 February 2002Accepted: 1 May 2002
Abstract6-Phosphofructokinase from Pichia pastoris was purified for the first time to homo-geneity applying seven steps, including pseudo-affinity dye-ligand chromatography onProcion Blue H-5R-Sepharose. The specific activity of the purified enzyme was about80 U/mg. It behaves as a typically allosteric 6-phosphofructokinase exhibiting activa-tion by AMP and fructose 2,6-bis(phosphate), inhibition by ATP and cooperativity tofructose 6-phosphate. However, in comparison with the enzymes from Saccharomycescerevisiae and Kluyveromyces lactis, the activation ratio of 6-phosphofructokinasefrom Pichia pastoris by AMP is several times higher, the ATP inhibition is strongerand the apparent affinity to fructose 6-phosphate is significantly lower. Aqueous two-phase affinity partitioning with Cibacron Blue F3G-A did not reflect remarkablestructural differences of the nucleotide binding sites of the Pfks from Pichia pastorisand Saccharomyces cerevisiae. The structural organisation of the active enzyme seemsto be different in comparison with hetero-octameric 6-phosphofructokinases fromother yeast species. The enzyme was found to be a hetero-oligomer with an molecularmass of 975 kDa (sedimentation equilibrium measurements) consisting of two distincttypes of subunits in an equimolar ratio with molecular masses of 113 kDa and 98 kDa(SDS–PAGE), respectively, and a third non-covalently complexed protein component(34 kDa, SDS–PAGE). The latter seems to be necessary for the catalytic activity ofthe enzyme. Sequencing of the N-terminus (VTKDSIXRDLEXENXGXXFF) and ofpeptide fragments by applying MALDI–TOF PSD, m/z 1517.3 (DAMNVVNH) andm/z 2177.2 [AQNCNVC(L/I)SVHEAHTM] gave no relevant information about theidentity of this protein. Copyright 2002 John Wiley & Sons, Ltd.
Keywords: 6-phosphofructokinase; Pfk; Pichia pastoris; purification; molecular andkinetic properties; methylotrophic yeast
Introduction
6-phosphofructokinase (Pfk, phosphofructoki-nase-1, phosphohexokinase; EC 2.7.1.11) cataly-ses the transfer of phosphate from MgATP to the1-OH group of fructose 6-phosphate, yielding fruc-tose 1,6-bis(phosphate) and MgADP. The catalyticactivity is tightly regulated in a wide variety oforganisms by diverse positive and negative effec-tors. The enzyme plays a major role in regulat-ing the glycolytic flux (Hofmann, 1976; Heinrichet al., 1977; Sols, 1981; Kotlarz and Buc, 1982;
Kemp and Foe, 1983; Heinisch and Hollenberg,1993; Larsson et al., 2000). In a few bacteriaand most eukaryotic cells, ATP was found tofunction not only as a substrate but also as apotent inhibitor, whereas AMP and fructose 2,6-bis(phosphate) (Fru 2,6-P2) are strong activatorsand are able to diminish ATP inhibition.
In bacteria, Pfk is composed of four subunits ofequal size, each of 36 kDa (Blangy, 1968; Hudsonet al., 1979). In mammals, the enzyme consists of80 kDa subunits that form a tetramer as the small-est catalytically active species, but aggregates of
Copyright 2002 John Wiley & Sons, Ltd.

934 J. Kirchberger et al.
higher molecular weights have also been detected(for review, see Goldhammer and Paradies, 1979).Comparison of Pfk sequences from prokaryoticand eukaryotic cells led to the hypothesis that theenzyme of higher organisms has evolved by a geneduplication event of a prokaryotic ancestor (Poor-man et al., 1984; Heinisch et al., 1989).
In most yeasts investigated so far (e.g. Saccha-romyces cerevisiae, Kluyveromyces lactis, Candidaalbicans), Pfk is composed of two non-identicaltypes of subunits (α and β), which are encodedby PFK1 and PFK2, respectively (Herrmann et al.,1973; Heinisch, 1986; Heinisch et al., 1993; Lor-berg et al., 1999). Both genes exhibit high sim-ilarity in their middle parts and evolved likelyfrom a second gene duplication event. The N-terminal halves of both polypeptides account forthe catalytic function, while the C-terminal partsare mutated to regulatory sites for binding theallosteric effectors ATP, AMP and Fru 2,6-P2,respectively (Rodicio et al., 2000). In contrast toPfk from mammals, both kinds of yeast Pfk sub-units are elongated by two extra sequences at theC- and N-termini, which exhibit fewer similaritieswithin and between the different species. In mostyeasts containing two kinds of Pfk subunits, thenative enzyme is a hetero-octamer (α4β4), whichis also rather stable under in vitro conditions, i.e.in the cell-free extract. Single deletion mutants ofS. cerevisiae carrying only one type of the twogenes can apparently also generate Pfk activityin vivo, since they are able to grow on glucose(Klinder et al., 1998). However, the enzyme wasfound catalytically active only in the intact cell.After disruption of the cells, the enzyme inacti-vates rapidly and forms aggregates, as observed inextracts of single deletion mutants (Klinder et al.,1998; Arvanitidis and Heinisch, 1994).
Recently, we have purified and characterized aparticular form of Pfk from Schizosaccharomycespombe that is composed of only one type ofsubunit, forming a homo-octameric structure, andwas found to be stable in cell-free extract (Reuteret al., 2000).
Pfk activity was found also in cell-free extractof Pichia pastoris, a methylotrophic yeast, but theenzyme has not been purified and analysed untilnow. A sequence analysis of one gene (PFK1 )was carried out in respect to the observationthat the gene product of PFK1 has an impor-tant function in the process of glucose-induced
microautophagy of this yeast strain (Yuan et al.,1997). During glucose adaptation of the yeastgrown on methanol as carbon source, the perox-isomes are sequestered within finger-like exten-sions of the vacuole by a process analogous tomicroautophagy. The expression product of PFK1,which was identified as α-subunit of Pfk, modu-lates glucose-induced microautophagy independentof its ability to metabolize glucose intermediates.Mutants of Pichia pastoris, which are unable tosequester peroxisomes (gsa1 mutants), could becomplemented with PFK1. Furthermore, cellularlevels of both PFK1-mRNA and phosphofructok-inase activity were drastically reduced in gsa1mutants compared to the parental wild-type strain(Yuan et al., 1997).
In order to analyse Pichia pastoris Pfk in respectto its oligomeric structure and function in theglucose consumption, the enzyme was purified andcharacterized in terms of molecular and kineticproperties.
Materials and methods
Materials
The strain of Pichia pastoris (Collection No.70382) originates from the Deutsche Sammlungvon Mikroorganismen und Zellkulturen GmbHBraunschweig (DSMZ, Germany) and was a kindgift from Dr W. Babel (UmweltforschungszentrumLeipzig-Halle, Germany). All biochemicals for thePfk assay were from Roche Molecular Biochemi-cals (Mannheim, Germany). Fru 2,6-P2 and chem-icals for the SDS–PAGE and the immunoblot-ting procedure were from Bio-Rad Laboratories(Munchen, Germany). BACTO Peptone and yeastextract were obtained from BD Biosciences (Hei-delberg, Germany). Other chemicals used were ofanalytical reagent grade and were provided byMerck (Darmstadt, Germany) or Serva (Heidelberg,Germany). Procion Blue H-5R Sepharose 4B wasprepared according to Dean and Watson (1979) andCibacron Blue F3G-A was covalently coupled topolyethylene glycol (PEG) according to Johansson(1984).
Media and culture conditions
Yeast cells were grown at 30 ◦C in YPD-medium (1% yeast extract, 2% BACTO Peptone,
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

6-phosphofructokinase from Pichia pastoris 935
2% glucose) batchwise using a 15 l fermentor(Braun, Melsungen, Germany). Precultures wereprepared in 20 ml tubes which were placedon a rotary shaker overnight at 30 ◦C andthen transferred into a 500 ml Erlenmeyer flaskcontaining 200 ml YPD-medium. After 24 hshaking, the fermentor was inoculated with 200 mlof this preculture. Cells were harvested after24 h at an optical density (OD1 cm
580 nm) of about25, collected by centrifugation, washed twicewith deionized water and suspended in phosphatebuffer for disruption (see Purification protocol).Alternatively, the yeast was stored as a frozen pelletat −20 ◦C, but not longer than 1 month before use.
Enzyme and protein assay
Under standard conditions, the catalytic activ-ity of Pfk was measured spectrophotomet-rically at 25 ◦C, using a coupled enzymeassay containing 100 mM imidazole/HCl, pH7.2, 3 mM Fru 6-P, 0.6 mM ATP, 1 mM AMP,5 mM MgSO4, 5 mM (NH4)2SO4, 0.2 mM NADH,1 U/ml aldolase, 1.5 U/ml glycerol phosphatedehydrogenase, 5 U/ml triosephosphate isomerase.The reaction was started by the addition of10–50 µl enzyme sample appropriately dilutedwith 50 mM sodium phosphate buffer, pH 7.0.
For studying the kinetic properties of theenzyme, a coupled assay with regenerating sys-tems for ATP and Fru 6-P was composed inorder to allow measurements at low concentra-tions of the substrates. The assay mixture con-tained 100 mM imidazole/HCl, pH 6.6, 100 mM
KCl, 10 mM MgCl2, 20 mM potassium phosphate,0.2 mM NADH, 0.6 mM phosphoenolpyruvate,ATP, Fru 6-P, AMP and Fru 2,6-P2 as indicated,8.5 U/ml pyruvate kinase and 7 U/ml lactate dehy-drogenase. Fructose 1,6-bisphosphatase (1 U/ml)was added for continuous regeneration of Fru 6-P.The auxiliary enzymes were dialysed before use.The ATP velocity curves were determined at0.3 mM Fru 6-P and the Fru 6-P velocity curvesat 3.0 mM ATP. The effect of Fru 2,6-P2 and AMPwas measured at 0.3 mM Fru 6-P and at inhibitingconcentration of 3 mM ATP according to Nissleret al. (1983). The activity is given as internationalunits (1 U = 1 µmol/min at 25 ◦C).
Protein was determined according to Bradford(1976) using bovine serum albumin as standard.
Mathematical treatment
The dependence of the enzyme activity on theconcentration of Fru 6-P was described by the Hillequation (1):
v = V ∗ ([Fru − 6 − P]/KM)nH
1 + ([Fru − 6 − P]/KM)nH(1)
where V is the maximum velocity of the enzymewith respect to Fru 6-P, KM is the half-maximumrate constant and nH is the Hill coefficient.
For the description of the ATP velocity curves,the phenomenological equation (2) derived fromthe allosteric model of Monod et al. (1965)was used:
v = V ∗ [ATP]
([ATP] + K ATPS )
∗ 1
(1 + L)(2)
with
L = L0 ∗(
1 + [ATP]/K ATPT
1 + [ATP]/K ATPR
)8
where V is the maximum velocity of the enzymewith respect to ATP at the applied concentrationof Fru 6-P, K ATP
S abbreviates the affinity of ATPtowards its substrate binding site, K ATP
T and K ATPR
are the affinity constants for a ATP site of theinactive T and the active R state, respectively;L0 is the allosteric constant. For curve fitting, anoctameric enzyme structure (n = 8) was taken intoaccount.
For characterizing the influence of the effectorsAMP and Fru 2,6-P2, a generalized Hill equationwas used (equation 3):
v = V0 + (V − V0) ∗ ([X ]/KA)nH
1 + ([X ]/KA)nH(3)
where [X ] denotes the concentration of AMP orFru 2,6-P2; V0 is the activity of the enzyme in theabsence of the respective effector; V expresses theenzyme activity reached at infinitely high effectorconcentration [X ], KA is the effector concentrationcausing half-maximum activation and nH is the Hillcoefficient for the activation process.
SDS–PAGE and Western blot
Denaturation of protein and SDS–PAGE were car-ried out according to Laemmli in 7.5% acry-lamide running gels as described elsewhere (Kop-perschlager et al., 1993) using the Mini-Protean
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

936 J. Kirchberger et al.
II Dual Slab Cell system (Bio-Rad Laboratories).The mobility was related to the molecular massesof the premixed protein high-range molecularweight marker from Roche Molecular Biochemi-cals and purified native (21 S) as well as prote-olytically degraded Pfk (12 S), for which molecu-lar masses are well known (Kopperschlager et al.,1993). 1D-Gel analysis was done with the scannedSDS–PAGE images by using Labimage softwareversion 2.6 (Kapelan GmbH, Halle, Germany).
Western blotting was carried out according to Baret al. (1997). 0.5–5 µg protein per lane were sub-jected to electrophoretic runs followed by electro-transfer to a nitrocellulose membrane BioTrace NT(Pall, USA). Detection was carried out with poly-clonal rabbit anti-6-phosphofructokinase (baker’syeast) and anti-rabbit immunoglobulin/peroxidaseconjugate (Roche Molecular Biochemicals).
Amino acid sequence analysisThe N-terminal sequence of the peptide fragmentswas determined according to the Edman proce-dure, using an Applied Biosystems 473A sequenceraccording to Bar et al. (1997).
In-gel digestion and MALDI–TOF analysisProtein separation by SDS–PAGE (7.5%), stainingand destaining, and in-gel digestion by trypsin werecarried out according to Dihazi et al. (2001). Thematrix solutions were saturated with α-cyano-4-hydroxycinnamic acid and were prepared accordingto Asara and Allison (1999). The pellets obtainedafter protein digestion were dissolved in 50% ace-tonitrile containing 0.1% trifluoroacetic acid to afinal concentration of 50–80 ng/µl and desaltedwith ZipTip C18 (Millipore). The sample-matrixpreparation was carried out as two-layer samplepreparation (Dai et al., 1999). A stainless steeltarget was used. MALDI–TOF mass spectrome-try was performed on a Bruker Biflex III massspectrometer (Bruker Daltonik, Germany), usedin the reflector mode with internal calibrationas described by Dihazi et al. (2001). The pep-tide sequencing from protein digests was done byapplying MALDI–TOF PSD, according to Gevaertet al. (2001).
Analytical ultracentrifugationAnalytical ultracentrifugation experiments wereperformed with a Beckman XL-A ultracentrifuge
equipped with absorption optics at 20 ◦C. A doublesector cell of 12 mm light path, or the six-channelequilibrium centrepiece, and the rotor AN 60 Tiwere used. The sedimentation velocity runs wereperformed at 42 000 rpm. The molecular mass wasdetermined by the short column sedimentationequilibrium technique with a 2 h overspeed run at9000 rpm, following a reduced speed at 3500 rpmfor 18–20 h. The baseline offset was measured byoverspeeding after collecting the equilibrium dataset and reading the absorbance of the trailing gra-dient. The data of the sedimentation equilibrium,as well as of the sedimentation velocity runs, werecollected as a set of concentration measurements ofdifferent radial positions at a given time at 280 nm,using the Beckman user interface program. TheOptima XL-A Data analysis software version 2.0from Beckman Instruments was used for the calcu-lation of the apparent sedimentation coefficient andthe molar mass (Mapp). The residual plot servesas the most sensitive graphical representation forgoodness of fit. Moreover, systematic error char-acteristic of associating and non-ideal systems canbe seen in this residual plot (McRorie and Voelker,1993). The residuals are the difference betweeneach experimental data point and the correspond-ing point on the curve calculated from the modelequation.
Prior to ultracentrifugation, the enzyme wasexhaustively dialysed against 0.1 M sodium phos-phate buffer, pH 7.0, containing 5 mM 2-merca-ptoethanol, 1 mM EDTA and 0.5 mM phenyl-methylsulphonyl fluoride (PMSF).
Aqueous two-phase partitioning
2 g of a two-phase system containing 5% (w/w)PEG 6000, 7% (w/w) dextran and 50 mM sodiumphosphate buffer, pH 7.0, were prepared from stocksolutions of PEG 6000 (Serva; 20% w/w), dex-tran T70 (Amersham Pharmacia Biotech, Freiburg,Germany; 20% w/w) and 0.2 M sodium phos-phate buffer, pH 7.0, by weighing the respectiveamounts supplemented with effectors as indicatedin the legends of Table 3. The concentration of thedye-liganded PEG was determined spectrophoto-metrically using the molar absorption coefficient of13 600 l/mol/cm (λ = 610 nm). The two-phase sys-tems containing about 4 units of Pfk were cooledto 4 ◦C and equilibrated by gently mixing for 30 s.After centrifugation at 1500 × g for 5 min at 4 ◦C,
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

6-phosphofructokinase from Pichia pastoris 937
the systems were maintained for 10 min at 4 ◦C.Samples were withdrawn from both phases andappropriately diluted with 50 mM sodium phos-phate buffer, pH 7.0. An inhibition of the enzymein the assay mixture by the dye-PEG was avoidedby sufficient dilution of the samples.
Purification protocolDisruption and fractionated ammonium sulphateprecipitation
About 150 g yeast (wet weight) were suspendedin 50 mM potassium phosphate buffer, pH 7.2,containing 1 mM EDTA, 5 mM 2-mercaptoethanoland 0.5 mM PMSF (buffer A), yielding a cyt-ocrit of 30%. Cell disruption was performedby vigorous shaking of the slurry with glassbeads (0.35 mm diameter) in a Vibrogen Cell Mill(Buhler, Tubingen, Germany) for 4 min at 4–6 ◦C.To reduce foam formation during disruption, smallamounts of 0.5% antifoam S 30 (Serva, Heidelberg,Germany) were added. Unless otherwise specified,the following steps were carried out at 4–6 ◦C. Thecell debris was centrifuged at 30 000 rpm (Beck-man L-80 ultracentrifuge, rotor type 35) for 45 minand the clear supernatant was subjected to fraction-ated precipitation with ammonium sulphate (33%and 50% saturation, respectively). The first pre-cipitate (centrifugation at 30 000 rpm for 30 min)was discarded, the second pellet was redissolved in70 ml buffer A.
Interfacial salting out
200 g Sepharose 4B CL, equilibrated withbuffer A, were added to the enzyme solution ofthe foregoing step and solid ammonium sulphatewas added up to 60% saturation. The slurry wasstirred for 15 min to allow the enzyme to adsorbon the gel. The gel was then stepwise washedon a Buchner funnel with a total volume of600 ml ice-cold buffer A containing ammoniumsulphate (50% saturation). For desorption ofthe enzyme from the gel, 200 ml of buffer Acontaining ammonium sulphate of 30% saturationwere stepwise loaded onto the funnel. Finally,the enzyme was precipitated by adding solidammonium sulphate to the eluate up to 75%saturation. The pellet was centrifuged (20 000 rpmfor 30 min) and dissolved in a small volume(20 ml) of buffer A.
Ion exchange chromatography on Resource Q
The enzyme solution was desalted using PD 10columns (Amersham Pharmacia Biotech) beforeloading onto the anion exchanger Resource Q(Amersham Pharmacia Biotech), which was equi-librated with buffer A. After washing the columnwith buffer A until the eluate was nearly free ofprotein, the enzyme was eluted by applying a lin-ear gradient of KCl (0–300 mM) in buffer A within180 min at a flow rate of 1 ml/min. The enzymeappeared at about 100 mM KCl as a sharp peak.
Affinity chromatography on Procion Blue H-5RSepharose 4B
The enzyme containing fractions of the forego-ing step were diluted five-fold with distilled waterand the Procion Blue H-5R Sepharose, equili-brated with buffer A, was added under stirring.For a quantitative binding of 100 enzyme unitsapproximately 10 g (wet weight) of the affinity gelwere necessary. After 30 min stirring, the gel waswashed exhaustively on a Buchner funnel (about600 ml buffer A) and then desorbed with 200 mlbuffer A containing 5 mM ATP and 10 mM MgCl2.Finally, the enzyme was concentrated by ultrafiltra-tion (Amicon, YM-30-membrane, Beverly, USA)and ammonium sulphate precipitation (85% satu-ration).
Gel permeation chromatography onBio-Sep-SEC-S 4000
The precipitate of the foregoing step was dissolvedin a small volume of 50 mM potassium phosphatebuffer, pH 7.0, containing 300 mM (NH4)2SO4 and0.5 mM PMSF, and chromatographed on a Biosep-SEC-S 4000 column (Phenomemex, Germany).The column was equilibrated with buffer A con-taining 300 mM ammonium sulphate and 0.5 mM
PMSF, and was run with a flow rate of 4 ml/min.The enzyme appeared as a sharp single peak, wasconcentrated by ultrafiltration (Amicon, YM-30-membrane) and stabilized by addition of glycerolto a final concentration of 30% (v/v). The enzymecould be stored in the presence of 0.5 mM PMSFat 4 ◦C for 4 weeks without loss of activity.
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.
938 J. Kirchberger et al.
Results
Purification
The yeast strain No. 70382 (DSMZ-Braunschweig)seems to be phenotypically similar to the strainGS115 as described by Cregg et al. (1985), becausewe found identical Western blot signals of Pfk andnearly the same specific activities of the enzyme inboth cell-free extracts (data not shown).
A typical purification protocol is depictedin Table 1, describing a combination of sevensteps. The interfacial salting-out step removedpredominantly contaminating lipoproteins. Both,ion-exchange and affinity chromatography onProcion Blue H-5R-Sepharose were most effective.By screening various dye-ligands coupled toSepharose 4B, Procion Blue H-5R was foundto be the optimum pseudo-biospecific ligandfor the affinity chromatography of the enzyme.Especially, the recovery was significantly higher incomparison to immobilized Cibacron Blue F3G-A.The specific activity of the purified enzyme wasabout 80 U/mg. The enzyme is sensitive againstammonium sulphate precipitation, particularly atlow protein concentrations in the highly purifiedstate, because a loss of activity by precipitationwas observed. The presence of 1 mM Fru 6-Pslightly stabilizes the enzyme against inactivation.The elution profile obtained after size exclusion
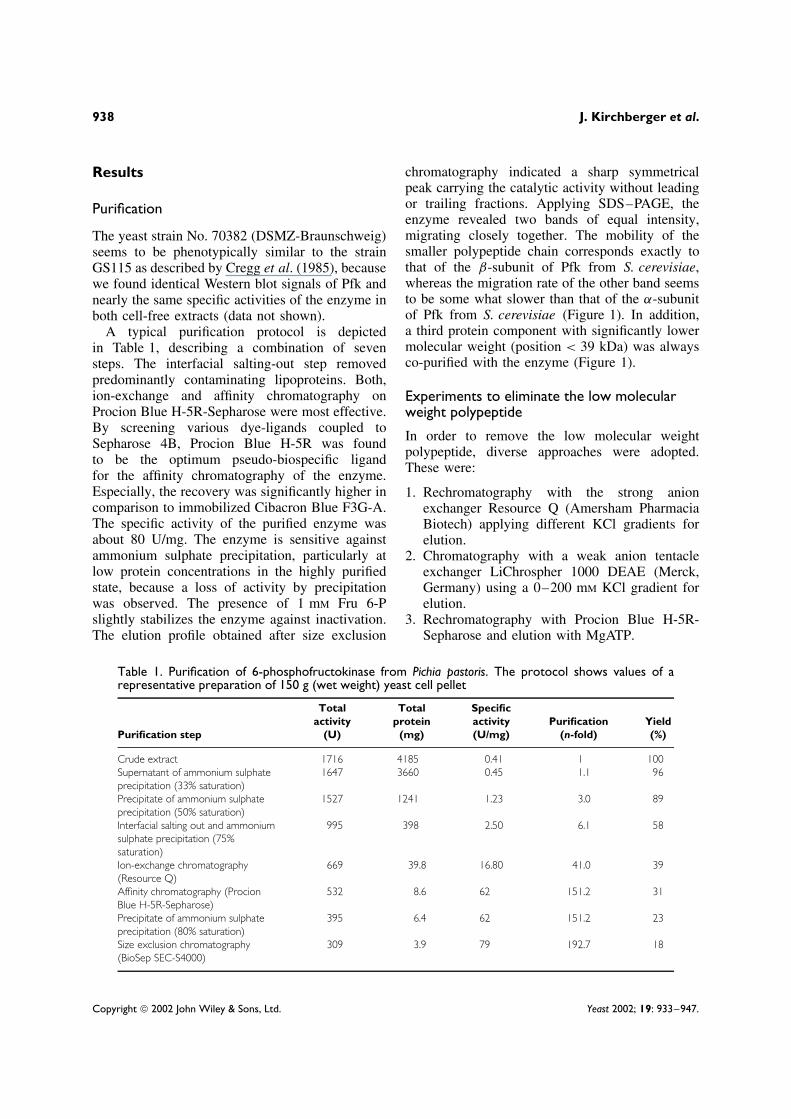
chromatography indicated a sharp symmetricalpeak carrying the catalytic activity without leadingor trailing fractions. Applying SDS–PAGE, theenzyme revealed two bands of equal intensity,migrating closely together. The mobility of thesmaller polypeptide chain corresponds exactly tothat of the β-subunit of Pfk from S. cerevisiae,whereas the migration rate of the other band seemsto be some what slower than that of the α-subunitof Pfk from S. cerevisiae (Figure 1). In addition,a third protein component with significantly lowermolecular weight (position < 39 kDa) was alwaysco-purified with the enzyme (Figure 1).
Experiments to eliminate the low molecularweight polypeptide
In order to remove the low molecular weightpolypeptide, diverse approaches were adopted.These were:
1. Rechromatography with the strong anionexchanger Resource Q (Amersham PharmaciaBiotech) applying different KCl gradients forelution.
2. Chromatography with a weak anion tentacleexchanger LiChrospher 1000 DEAE (Merck,Germany) using a 0–200 mM KCl gradient forelution.
3. Rechromatography with Procion Blue H-5R-Sepharose and elution with MgATP.
Table 1. Purification of 6-phosphofructokinase from Pichia pastoris. The protocol shows values of arepresentative preparation of 150 g (wet weight) yeast cell pellet
Purification step
Totalactivity
(U)
Totalprotein
(mg)
Specificactivity(U/mg)
Purification(n-fold)
Yield(%)
Crude extract 1716 4185 0.41 1 100Supernatant of ammonium sulphateprecipitation (33% saturation)
1647 3660 0.45 1.1 96
Precipitate of ammonium sulphateprecipitation (50% saturation)
1527 1241 1.23 3.0 89
Interfacial salting out and ammoniumsulphate precipitation (75%saturation)
995 398 2.50 6.1 58
Ion-exchange chromatography(Resource Q)
669 39.8 16.80 41.0 39
Affinity chromatography (ProcionBlue H-5R-Sepharose)
532 8.6 62 151.2 31
Precipitate of ammonium sulphateprecipitation (80% saturation)
395 6.4 62 151.2 23
Size exclusion chromatography(BioSep SEC-S4000)
309 3.9 79 192.7 18
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

6-phosphofructokinase from Pichia pastoris 939
200 kDa
116 kDa
97 kDa
66 kDa
39 kDa
A B C D E
Figure 1. SDS–PAGE and Western blot analysis of purified6-phosphofructokinase from Pichia pastoris. (A) Molecularweight standard. (B) 21 S Pfk from S. cerevisiae. (C) 12 SPfk from S. cerevisiae. (D) Pfk from P. pastoris. (E) Westernblot analysis of Pfk in cell-free extract of P. pastoris byusing polyclonal rabbit anti-Pfk-IgG and anti-rabbit IgG/PODconjugate for immunodetection. For Western blot analysis,5 µg protein/lane and for Coomassie blue staining 4 µgprotein/lane were applied
4. Isocratic size-exclusion HPLC on a Bio-SilectSEC 400-5 column (Bio-Rad Laboratories).
5. Reversed phase chromatography on a TSKODS-120T column (TosoHaas, Japan) inwater/trifluoroacetic acid solution and elutionwith increasing acetonitrile concentration.
6. Hydrophobic interaction chromatography onResource-Phenyl (Amersham Pharmacia Bio-tech, Germany) using a linear ammoniumsulphate gradient (40–0%).
7. Immunosorption on rabbit anti-yeast Pfk-Sepharose and elution by decreasing the pH.
Although the elution peaks were symmetrical in allcases, the eluate still contained the low molecularweight polypeptide in nearly the same ratio asbefore.
Because we found no difference in theSDS–PAGE pattern under reducing and non-reducing conditions, the low molecular weightpolypeptide must be non-covalently complexedwith the enzyme. In further experiments, theremoval of the third polypeptide was tried byapplying separation methods under denaturingconditions, i.e. in the presence of urea or SDS.
Because enzyme solutions of 2 mg/ml are stable in0.8 M urea or 0.0025% SDS at room temperaturefor several hours (data not shown), most of theexperiments were done within 1 h at 20 ◦C.
By applying anion exchange chromatography(MiniQ, Amersham Pharmacia Biotech) sucrosedensity gradient centrifugation (5–20% sucrosegradient) or SEC-HPLC (Bio-Silect SEC 400-5 col-umn), the low molecular weight polypeptide wasseparated in the presence of 0.8 M urea or 0.0025%SDS. But a complete loss of Pfk activity wasobserved in those fractions containing only the pairof bands. Experiments to regain the enzyme activ-ity by recombination of the separated componentsfailed. Reconstitution experiments with completelyunfolded subunits in the absence or presence ofthe third protein component according to Bar et al.(2000) were also unsuccessful (data not shown).
Molecular weight and subunit composition
An apparent sedimentation coefficient, s20,c =24 ± 0.2 S , was determined in the concentra-tion range 0.25–7.4 mg protein/ml. No significantdependence of the apparent s-value on the pro-tein concentration could be observed indicating astable enzyme structure. Sedimentation equilibriummeasurements of the enzyme revealed an averagemolecular mass of 975 ± 28 kDa by taking intoaccount a partial specific volume of 0.736 ml/g.For estimating the partial specific volume (Cohnand Edsall, 1943) the amino acid composition ofthe α-subunit was taken from the data bank (SWIS-SPROT Q92448) and the amino acid sequence ofthe ß-subunit was deduced from the PFK2 genesequence identified by Edelmann (2001; GenBankAccession No. AF 395078). The linearity of theln A vs. r2-plot, as well as the random distributionof points about the zero value in the residual plot(Figure 2) and the sedimentation of the purifiedP. pastoris Pfk with a single boundary (not shown),indicate a high homogeneity of the enzyme.
SDS–PAGE of the purified enzyme indicates anequimolar ratio of the α- and β-subunit of Pfk fromP. pastoris. Both subunits were designated accord-ing to the similarity of mobility of the Pfk sub-units from S. cerevisiae. Their masses, determinedby SDS–PAGE, are 112.9 ± 0.5 kDa and 98.0 ±0.7 kDa, respectively (Figure 1). The migrationrates of both bands in the purified state were foundto be equal to the migration of the bands detected
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

940 J. Kirchberger et al.
6.35 6.40 6.45 6.50 6.55 6.60 6.65
Res
idua
ls
-0.02
0.00
0.02B
Radius (cm)
41 42 43 44
ln A
-1.5
-1.0
-0.5
0.0A
r 2 (cm2)
Figure 2. Sedimentation equilibrium run of 6-phospho-fructokinase from Pichia pastoris. The run was performedapplying the short column technique with a six-channel cen-trepiece starting with an overspeed for 2 h at 9000 rpmfollowing a run at 3500 rpm for about 20 h. (A) ln A(absorbance) vs. r2-plot (°) Experimental data points. (B)Residual plot. The residuals (ž) are the difference betweeneach experimental point and the corresponding values cal-culated from the model referring to a system containingone ideal species
by Western blot analysis of the cell-free extract,indicating no proteolytic degradation of the sub-units in the course of preparation (Figure 1). Theß-subunit is a distinct polypeptide and is not gener-ated by proteolytic degradation, as shown by aminoacid sequence analysis (data not shown).
For the third component (low molecular weightpolypeptide), an average molecular weight of34 kDa (SDS–PAGE) was estimated. In order tocheck whether the polypeptide is generated by aproteolytic breakdown of Pfk subunits, the pro-tein band was subjected to the Edman degradationprocedure. The N-terminal amino acid sequence(VTKDSIXRDLEXENXGXXFF) did not matchany sequence of the α- or β-subunit of the enzymefrom Pichia pastoris. A similarity search in current
protein databases did also not report any hit withhigh homology.
After in-gel digestion of the 34 kDa pro-tein, the tryptic peptides were analysed byMALDI–TOF mass spectrometry (MS). Theresults are shown in Table 2. A search in the SWIS-SPROT/TrEMBL or NCBI’s nr (all non-redundantGenBank CDS translations + PDB + SwissProt +PIR + PRF) protein database, using different algo-rithms (Mascot, ProFound and PeptIdent), gaveno satisfying information about the identity ofthe protein. Additionally, two peptide fragments(m/z 1517.3 and m/z 2177.2; see Table 2) werechosen for sequence analysis with MALDI–TOFpost source decay (PSD). The large part of theamino acid sequences of both peptides could beidentified, for m/z 1517.3 (DAMNVVNH) andfor m/z 2177.2 [AQNCNVC(L/I)SVHEAHTM].However, a similarity search in current proteindatabases did also not report any hit with highhomology.
The fast moving protein band represents about14% of the total amount of protein prepared ascalculated by using a 1D-gel analysis software (seeMaterials and methods).
Kinetics
As shown in Figure 3, Pfk exhibits sigmoidal kinet-ics in dependence on Fru 6-P. In the presence of theactivators AMP or Fru 2,6-P2 the cooperativity iscompletely abolished as indicated by the decreaseof the Hill coefficients (see nH-values in the captionto Figure 3). Both AMP and Fru 2,6-P2 increase
Table 2. Masses of peptide fragmentsafter in-gel digestion of the 34 kDaprotein with trypsin and MALDI–TOFmass spectrometry
m/z
635.1 1517.3 2230.8691.0 1638.6 2301.8699.1 1813.0 2499.8786.8 1826.9 2847.9804.6 1516.8 2888.6918.5 1956.1 2905.3
1121.4 1980.6 3272.01127.3 2009.7 3786.31255.1 2028.4 4369.31423.7 2044.6 4697.81494.6 2177.2
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

6-phosphofructokinase from Pichia pastoris 941
Fru 6-P (mM)0 2 4 6 8 10 12 14
Enz
yme
activ
ity (
U/m
L)
0
1
2
3
4
5
6
Figure 3. Dependence of the activity of 6-phosphofruc-tokinase on the concentration of Fru 6-P. [ATP] = 3.0 mM.Kinetic parameters are calculated according to the Hill equa-tion (1). °, Without effector; ž, 1.0 mM AMP; �, 0.1 mMFru 2,6-P2
V (U/ml) KM (mM) nH
° 3.661 ± 0.092 6.971 ± 0.188 2.25 ± 0.08ž 5.650 ± 0.045 0.435 ± 0.015 1.13 ± 0.04� 5.482 ± 0.053 0.645 ± 0.019 1.12 ± 0.04
the maximum velocity (V ) and the affinity towardsFru 6-P (KM) significantly; the latter effect is littlemore pronounced by AMP.
Figure 4 shows the inhibiting effect of ATPon the enzyme activity. According to equa-tion 2 (Monod–Wyman–Changeux model), ATPis assumed to act as substrate and as effector,respectively. Both AMP and Fru 2,6-P2 diminishthe inhibition of the enzyme by ATP. Although,the maxima of the ATP-velocity curves arise atabout 0.1 mM ATP, the activating effect (ratio ofthe activities of the enzyme with and without theactivators) is most pronounced at ATP concentra-tions of about 1 mM. Under these conditions, theextent of activation by AMP significantly exceedsthe effect of Fru 2,6-P2. Although the applied con-centration of both activators was relatively high,the ATP inhibition could not be completely abol-ished. The observations can be explained on thebasis of the Monod–Wyman–Changeux model if itis assumed that both AMP and Fru 2,6-P2 act on theallosteric equilibrium and decrease the allostericconstant (Lo), but with different extents.
The lowest value of Lo was found for AMP.The affinity of ATP for the substrate binding site
0.01 0.1 1
Enz
yme
activ
ity (
U/m
L)
0
1
2
3
4
5
6
ATP (mM)
Figure 4. Dependence of the activity of 6-phosphofructok-inase on the concentration of ATP. [Fru 6-P] = 0.3 mM. °,Without effector; ž, 1.0 mM AMP; �, 0.1 mM Fru 2,6-P2.Data were described in terms of equation (2) with V = 6.40U/ml
L0 (U/ml) KATPS (mM) KATP
T (mM)
° 4.301 ± 0.987 0.012 ± 0.003 0.024 ± 0.007ž 0.032 ± 0.024 0.020 ± 0.001 0.029 ± 0.023� 0.057 ± 0.010 0.012 ± 0.001 0.055 ± 0.010
Inhibition ratio(Vmax :
KATPR (mM) V3mM ATP)
° 17.41 ± 3.09 12.2ž 38.11 ± 6.57 2.1� 33.00 ± 4.50 2.0
(K ATPs ) decreases slightly after addition of AMP,
but remains unchanged after addition of Fru 2,6-P2. The affinity of ATP for the effector bindingsite in the inactive state (K ATP
T ) of the enzyme isnot significantly affected by AMP but decreasesslightly in the presence of Fru 2,6-P2. The affinityof the enzyme towards ATP in the active state(K ATP
R ) is very low and both allosteric activatorsweaken the interaction of ATP at the effectorsite in the active state (K ATP
R ). Interestingly, whileFru 2,6-P2 does not affect the binding of ATP at thesubstrate binding site, a weak competitive action ofAMP and ATP at this site can not be excluded onthe basis of these data, as indicated by an increaseof (K ATP
S ). At low Fru 6-P concentration (0.3 mM)and in the presence of more than 10 µM ATP,both AMP and Fru 2,6-P2 increase the maximumactivity significantly and shift the maximum of theATP velocity curves to higher ATP concentrations.
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.
942 J. Kirchberger et al.
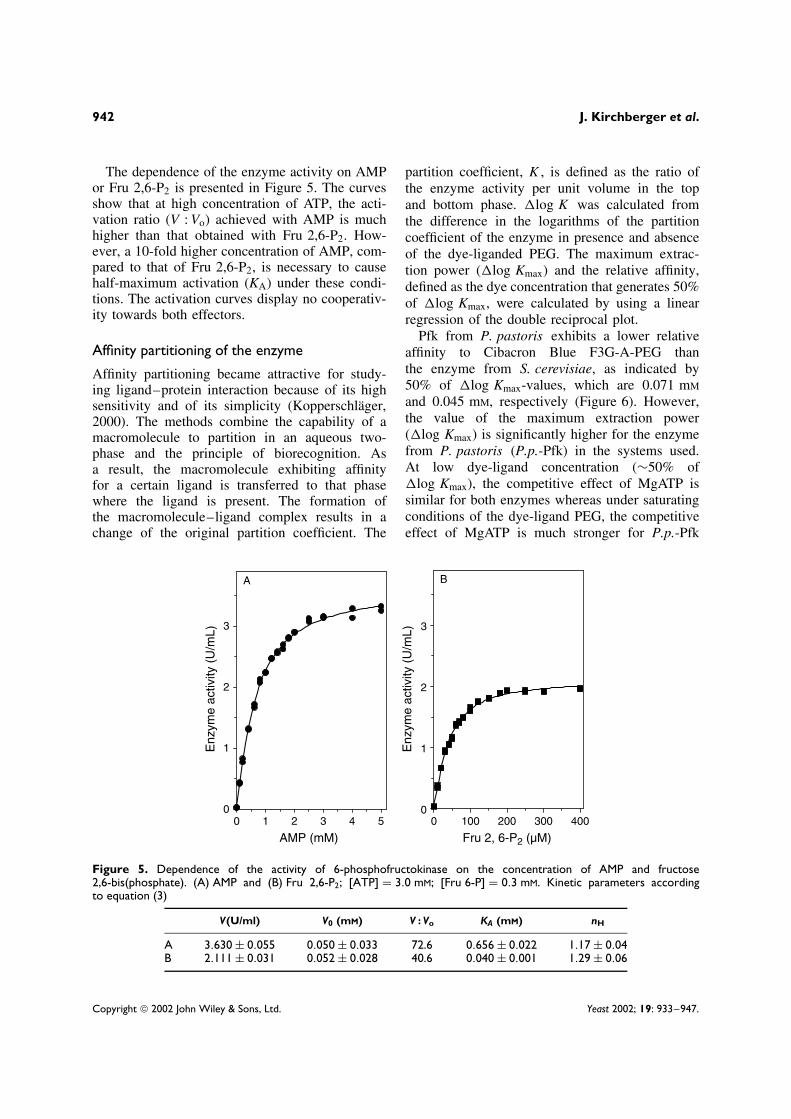
The dependence of the enzyme activity on AMPor Fru 2,6-P2 is presented in Figure 5. The curvesshow that at high concentration of ATP, the acti-vation ratio (V : Vo) achieved with AMP is muchhigher than that obtained with Fru 2,6-P2. How-ever, a 10-fold higher concentration of AMP, com-pared to that of Fru 2,6-P2, is necessary to causehalf-maximum activation (KA) under these condi-tions. The activation curves display no cooperativ-ity towards both effectors.
Affinity partitioning of the enzyme
Affinity partitioning became attractive for study-ing ligand–protein interaction because of its highsensitivity and of its simplicity (Kopperschlager,2000). The methods combine the capability of amacromolecule to partition in an aqueous two-phase and the principle of biorecognition. Asa result, the macromolecule exhibiting affinityfor a certain ligand is transferred to that phasewhere the ligand is present. The formation ofthe macromolecule–ligand complex results in achange of the original partition coefficient. The
partition coefficient, K , is defined as the ratio ofthe enzyme activity per unit volume in the topand bottom phase. �log K was calculated fromthe difference in the logarithms of the partitioncoefficient of the enzyme in presence and absenceof the dye-liganded PEG. The maximum extrac-tion power (�log Kmax) and the relative affinity,defined as the dye concentration that generates 50%of �log Kmax, were calculated by using a linearregression of the double reciprocal plot.
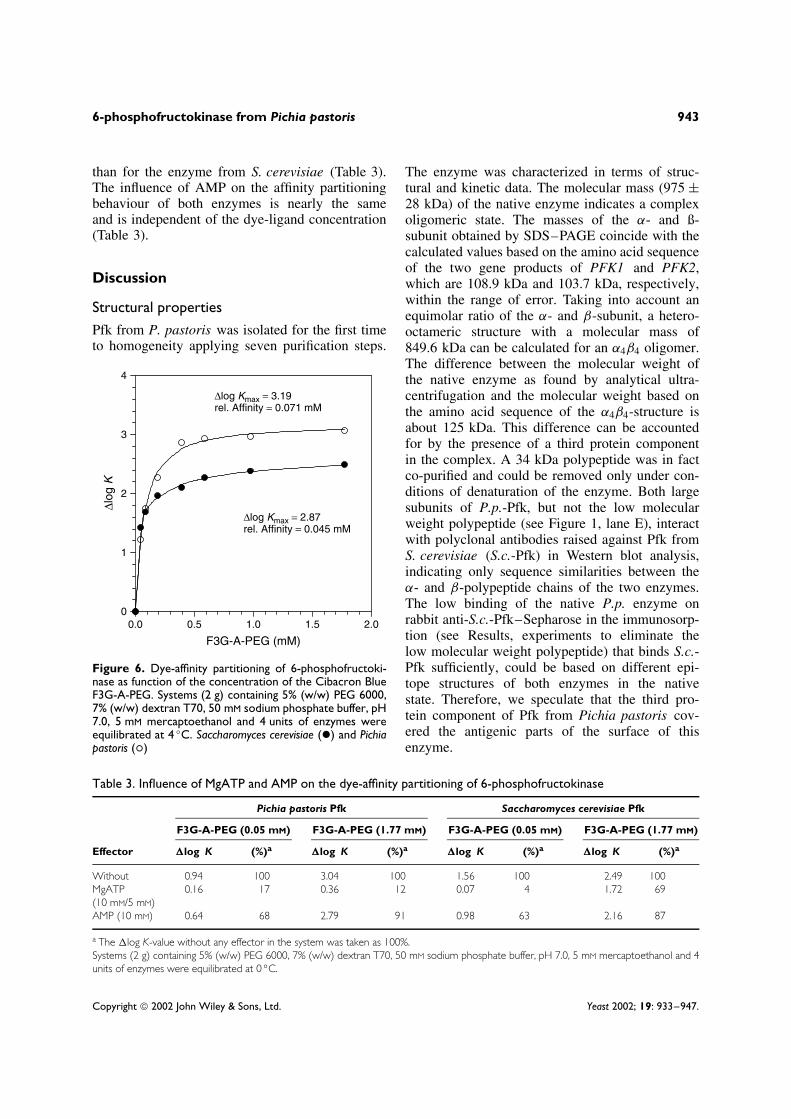
Pfk from P. pastoris exhibits a lower relativeaffinity to Cibacron Blue F3G-A-PEG thanthe enzyme from S. cerevisiae, as indicated by50% of �log Kmax-values, which are 0.071 mM
and 0.045 mM, respectively (Figure 6). However,the value of the maximum extraction power(�log Kmax) is significantly higher for the enzymefrom P. pastoris (P.p.-Pfk) in the systems used.At low dye-ligand concentration (∼50% of�log Kmax), the competitive effect of MgATP issimilar for both enzymes whereas under saturatingconditions of the dye-ligand PEG, the competitiveeffect of MgATP is much stronger for P.p.-Pfk
0 100 200 300 400
Enz
yme
activ
ity (
U/m
L)
0
1
2
3
0 1 2 3 4 5
Enz
yme
activ
ity (
U/m
L)
0
1
2
3
A B
AMP (mM) Fru 2, 6-P2 (µM)
Figure 5. Dependence of the activity of 6-phosphofructokinase on the concentration of AMP and fructose2,6-bis(phosphate). (A) AMP and (B) Fru 2,6-P2; [ATP] = 3.0 mM; [Fru 6-P] = 0.3 mM. Kinetic parameters accordingto equation (3)
V(U/ml) V0 (mM) V : Vo KA (mM) nH
A 3.630 ± 0.055 0.050 ± 0.033 72.6 0.656 ± 0.022 1.17 ± 0.04B 2.111 ± 0.031 0.052 ± 0.028 40.6 0.040 ± 0.001 1.29 ± 0.06
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.
6-phosphofructokinase from Pichia pastoris 943
than for the enzyme from S. cerevisiae (Table 3).The influence of AMP on the affinity partitioningbehaviour of both enzymes is nearly the sameand is independent of the dye-ligand concentration(Table 3).
Discussion
Structural properties
Pfk from P. pastoris was isolated for the first timeto homogeneity applying seven purification steps.
0.0 0.5 1.0 1.5 2.0
∆log
K
0
1
2
3
4
F3G-A-PEG (mM)
∆log Kmax = 2.87rel. Affinity = 0.045 mM
∆log Kmax = 3.19rel. Affinity = 0.071 mM
Figure 6. Dye-affinity partitioning of 6-phosphofructoki-nase as function of the concentration of the Cibacron BlueF3G-A-PEG. Systems (2 g) containing 5% (w/w) PEG 6000,7% (w/w) dextran T70, 50 mM sodium phosphate buffer, pH7.0, 5 mM mercaptoethanol and 4 units of enzymes wereequilibrated at 4 ◦C. Saccharomyces cerevisiae (ž) and Pichiapastoris (°)
The enzyme was characterized in terms of struc-tural and kinetic data. The molecular mass (975 ±28 kDa) of the native enzyme indicates a complexoligomeric state. The masses of the α- and ß-subunit obtained by SDS–PAGE coincide with thecalculated values based on the amino acid sequenceof the two gene products of PFK1 and PFK2,which are 108.9 kDa and 103.7 kDa, respectively,within the range of error. Taking into account anequimolar ratio of the α- and β-subunit, a hetero-octameric structure with a molecular mass of849.6 kDa can be calculated for an α4β4 oligomer.The difference between the molecular weight ofthe native enzyme as found by analytical ultra-centrifugation and the molecular weight based onthe amino acid sequence of the α4β4-structure isabout 125 kDa. This difference can be accountedfor by the presence of a third protein componentin the complex. A 34 kDa polypeptide was in factco-purified and could be removed only under con-ditions of denaturation of the enzyme. Both largesubunits of P.p.-Pfk, but not the low molecularweight polypeptide (see Figure 1, lane E), interactwith polyclonal antibodies raised against Pfk fromS. cerevisiae (S.c.-Pfk) in Western blot analysis,indicating only sequence similarities between theα- and β-polypeptide chains of the two enzymes.The low binding of the native P.p. enzyme onrabbit anti-S.c.-Pfk–Sepharose in the immunosorp-tion (see Results, experiments to eliminate thelow molecular weight polypeptide) that binds S.c.-Pfk sufficiently, could be based on different epi-tope structures of both enzymes in the nativestate. Therefore, we speculate that the third pro-tein component of Pfk from Pichia pastoris cov-ered the antigenic parts of the surface of thisenzyme.
Table 3. Influence of MgATP and AMP on the dye-affinity partitioning of 6-phosphofructokinase
Pichia pastoris Pfk Saccharomyces cerevisiae Pfk
F3G-A-PEG (0.05 mM) F3G-A-PEG (1.77 mM) F3G-A-PEG (0.05 mM) F3G-A-PEG (1.77 mM)
Effector �log K (%)a �log K (%)a �log K (%)a �log K (%)a
Without 0.94 100 3.04 100 1.56 100 2.49 100MgATP(10 mM/5 mM)
0.16 17 0.36 12 0.07 4 1.72 69
AMP (10 mM) 0.64 68 2.79 91 0.98 63 2.16 87
a The �log K-value without any effector in the system was taken as 100%.Systems (2 g) containing 5% (w/w) PEG 6000, 7% (w/w) dextran T70, 50 mM sodium phosphate buffer, pH 7.0, 5 mM mercaptoethanol and 4units of enzymes were equilibrated at 0 ◦C.
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

944 J. Kirchberger et al.
Based on the protein partition in SDS–PAGE andthe difference in the molecular weights a ratio ofone polypeptide (34 kDa) per enzyme dimer (i.e.four per octamer) can be assumed. We suggest forthe native P.p.-Pfk a hetero-oligomeric structureconsisting of four α-, four ß- and four 34 kDa-polypeptide chains. That means that Pfk fromP. pastoris is more complex than the enzymes fromother yeasts such as C. albicans (Lorberg et al.,1999), K. lactis (Bar et al., 1997) and S. cerevisiae(Kopperschlager et al., 1977).
Because we were not able to purify an activeenzyme without the third protein component, wesuggest that this protein is an important non-covalently bound part of active P.p.-Pfk. However,we do not know whether the removal of the thirdprotein component per se, or the denaturation ofthe enzyme in the presence of urea, or SDS by anunavoidable decrease of the protein concentrationduring the separation processes, is responsible forthe loss of activity.
Kinetic properties
In Table 4, the kinetic effects of the enzyme fromP. pastoris are compared with those obtained fromS. cerevisiae and from K. lactis (K.l.-Pfk). Theymay be discussed as follows.
Effects of fructose 6-phosphate
In the absence of AMP or Fru 2,6-P2, the affinity ofP.p.-Pfk towards Fru 6-P is significantly lower thanthose from S. cerevisiae and K. lactis. WhereasP.p.-Pfk and S.c.-Pfk generate sigmoidal curves,the K.l.-Pfk shows no cooperativity towards Fru 6-P. After addition of AMP the KM-values aresimilar and significantly decreased in all cases.The cooperativity of S.c.-Pfk towards Fru 6-P isnot diminished by the addition of AMP, whilethe activating effect of AMP decreases the Hillcoefficient of P.p.-Pfk significantly. In the presenceof Fru 2,6-P2, the parameters KM and nH of P.p.-Pfk and K.l.-Pfk are similar and nearly in the
Table 4. Comparison of kinetic data of 6-phosphofructokinase from different yeast strains
Conditions Effector ParameterPichia
pastorisSaccharomyces
cerevisiae1Kluyveromyces
lactis2
[Fru 6-P] Without KM (mM) 6.97 1.65 1.58nH 2.3 2.8 1.2
AMP KM (mM) 0.44 0.51 0.37nH 1.1 2.7 1.2
Fru 2,6-P2 KM (mM) 0.65 0.11 0.29nH 1.1 1.7 1.4
Allosteric activators AMP KA (mM) 0.66 0.52 0.40nH 1.2 1.2 1.2Activation ratio (V : Vo) 72.6 14.1 3.6
Fru 2,6-P2 KA (µM) 40.0 1.7 47.0nH 1.3 1.5 1.5Activation ratio (V/Vo) 40.6 21.5 3.8
Without ATPinhibition
Lo 4.30 4.76 0.81
KATPS (mM) 0.012 0.03 0.07
KATPT (mm) 0.024 0.54 0.094
KATPR (mM) 17.4 0.26 0.145
AMP Lo 0.03 0.52 0.63KATP
S (mM) 0.02 0.03 0.07KATP
T (mM) 0.029 0.51 0.084KATP
R (mM) 38.1 0.27 0.139Fru 2,6-P2 Lo 0.057 0.02 0.62
KATPS (mM) 0.012 0.03 0.07
KATPT (mM) 0.055 0.57 0.084
KATPR (mM) 33.0 0.27 0.139
1 Nissler et al. (1983).2 Bar et al. (1997).
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

6-phosphofructokinase from Pichia pastoris 945
same range as obtained with AMP. Under theseconditions, S.c.-Pfk possesses the highest affinityto the substrate but the reduction of the Hillcoefficient is less pronounced, as found for the twoother enzymes. For all the enzymes, no differencebetween the effect of AMP- and Fru 2,6-P2 on themaximum activity was observed (data not shown).
Effects of allosteric activators
The concentration of AMP, necessary to generatethe half maximum activation (KA) is nearly thesame for all Pfks investigated. No cooperativeeffects (nH) towards AMP were detected in allcases. However, a significantly higher activationratio (V : Vo) was found for the P.p.-Pfk (five-fold increased in comparison to the enzyme fromS. cerevisiae and 20-fold increased in comparisonto the enzyme fom K. lactis). The KA-value forFru 2,6-P2 of P.p.-Pfk was similar to that ofK.l.-Pfk, but significantly higher than that of theenzyme from S. cerevisiae. P.p.-Pfk showed thehighest activation ratio (V : Vo) with Fru 2,6-P2in comparison to the other enzymes but reachedonly 56% of that value obtained with AMP. Nosignificant cooperative effects towards Fru 2,6-P2were detected for any of the enzymes.
ATP inhibition
The inhibiting effect of ATP for P.p.-Pfk is morepronounced than that of the other two enzymes(Nissler et al., 1983; Bar et al., 1997). Both, AMPand Fru 2,6-P2 were shown to diminish the extentof ATP inhibition. However, while the ATP inhibi-tion of the enzyme from S. cerevisiae can be com-pletely abolished by Fru 2,6-P2, a significant ATP-inhibition remains for the enzymes from P. pastorisand K. lactis. In the case of P.p.-Pfk, at physiolog-ical levels of ATP a more than 60-fold increaseof the enzyme activity is achieved by additionof AMP.
The apparent affinity of ATP to the substratebinding site (K ATP
S ) of P.p.-Pfk is found to besignificantly higher than the respective values ofthe two other enzymes.
While the effect of AMP on the ATP velocitycurves of the enzymes from S. cerevisiae andK. lactis does not indicate an interaction of AMPtowards the substrate binding site of ATP, thedata of the enzyme from P. pastoris point to a
competitive interaction of ATP and AMP to thesubstrate binding site. However, Fru 2,6-P2 has noinfluence on the K ATP
S -value of any of the threeenzymes.
The description of the ATP velocity curves forP.p.-Pfk by equation 2 (Monod–Wyman–Chang-eux model) allows a rather good fit of the data.However, a variation of both, the allosteric con-stant (L0) and the affinity constants for ATP in theactive R – (K ATP
R ) and the inactive T-state (K ATPT )
caused by AMP or Fru 2,6-P2 has to be taken intoaccount. This indicates a rather complicated acti-vation mechanism of the enzymes. In the absenceof both activators, P.p.-Pfk possesses in compari-son to the enzymes from S. cerevisiae and K. lactisan ATP effector binding site of high affinity in theinactive state (K ATP
T ).The addition of AMP has no influence on the
affinity of ATP towards all three enzymes in theinactive state (K ATP
T ). Fru 2,6-P2 does not changethe values of K ATP
T for S.c.-Pfk and K.l.-Pfk,while the affinity of ATP towards the enzymefrom P. pastoris in the inactive state decreasessignificantly.
AMP and Fru 2,6-P2 do not influence the affinityof ATP towards S.c.-Pfk and K.l.-Pfk in the activestate (K ATP
R ).In summary, P.p.-Pfk is more efficiently acti-
vated by AMP than by Fru 2,6-P2. The extentof ATP inhibition is more pronounced than thatfor the enzymes from S. cerevisiae and K. lactis.The apparent affinity of Fru 6-P to the P. pastorisenzyme in the absence of activators is significantlylower than for the both other enzymes.
Pseudoaffinity ligand–enzyme interaction
Cibacron Blue F3G-A is well known as a pseudo-biospecific ligand interacting with the ATP-bindingsite(s) of Pfk from S. cerevisiae and other yeastenzymes Kopperschlager et al., 1982; Kopper-schlager, 1994). Since the kinetic data displayeddifferences in the ATP and AMP effect betweenthe enzymes from P. pastoris and S. cerevisiae,dye-ligand affinity partitioning was applied to anal-yse the nucleotide binding site(s) of both enzymes.No significant differences were found in the rel-ative affinities of the two enzymes for the dye-ligand. The competitive effect of ATP, especiallyat high dye-ligand concentration, is stronger pro-nounced for the enzyme from P. pastoris. This can
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

946 J. Kirchberger et al.
be explained by the higher affinity of P.p.-Pfk forATP as found in the kinetic studies (Table 4) andby the lower affinity for the dye-ligand under theconditions used. However, no competitive effectof AMP on the interaction between Pfk from bothstrains and the dye-ligand were found, althoughAMP seems to compete with ATP (at low ATP con-centration) as shown by the kinetic data. The sig-nificant higher extraction power (�log Kmax) indi-cates a more complex type of interaction for theenzyme from P. pastoris. In summary, remarkabledifferences in the structure of the nucleotide bind-ing site(s) of Pfk from P. pastoris and S. cerevisiaewere not detected by affinity partitioning.
Summary
Pfk from P. pastoris was found to be hetero-oligomeric and behaves as an allosteric 6-phosphofructokinase. However, the regulatoryproperties of the enzyme differ from those observedfor Pfk from S. cerevisiae and K. lactis. Also,the structural organisation seems to be distinct incomparison with hetero-octameric Pfks from otheryeasts, by virtue of the presence of a third proteincomponent which does not resemble to any knownprotein. The physiological function of this is alsonot clear so far, but it seems to be necessary forthe catalytic activity of the enzyme.
This particular structural behaviour of Pfk incombination with the finding that the α-subunitcontributes to the process of glucose-inducedmicro-autophagy in Pichia pastoris make it desir-able to continue the investigation of the functionof the third protein component in more detail.
Acknowledgements
The authors thank Dr W. Babel (UmweltforschungszentrumLeipzig-Halle, Germany) for providing the strain of Pichiapastoris (Collection No. 70382). The skilful technicalassistance of Ms M. Rockstroh is gratefully acknowledged.
References
Arvanitidis A, Heinisch JJ. 1994. Studies on the function of yeastphosphofructokinase subunits by in vitro mutagenesis. J BiolChem 269: 8911–8918.
Asara JM, Allison J. 1999. Enhanced detection of phosphopeptidesin matrix-assisted laser desorption/ionization mass spectrometryusing ammonium salts. J Am Soc Mass Spectrom 10: 35–44.
Bar J, Schellenberger W, Kopperschlager G. 1997. Purificationand characterization of phosphofructokinase from the yeastKluyveromyces lactis . Yeast 13: 1309–1317.
Bar J, Golbik R, Hubner G, Kopperschlager G. 2000. Denatura-tion of phosphofructokinase-1 from Saccharomyces cerevisiaeby guanidium chloride and reconstitution of the unfoldedsubunits to their catalytically active form. Biochemistry 39:6960–6968.
Blangy D. 1968. Phosphofructokinase from E. coli : evidence fora tetrameric structure of the enzyme. FEBS Lett 2: 109–111.
Bradford MM. 1976. A rapid and sensitive method for thequantification of microgram quantities of proteins utilizing theprinciple of protein–dye binding. Anal Biochem 72: 248–254.
Cohn EJ, Edsall JT (eds). 1943. Proteins, Amino Acids andPeptides as Ions and Dipolar Ions. Reinhold: New York.
Cregg JM, Barringer KJ, Hessler AY, Madden KR. 1985. Pichiapastoris as a host system for transformation. Mol Cell Biol 5:3376–3385.
Dai Y, Whittal RM, Li L. 1999. Two-layer sample preparation:a method for MALDI–MS analysis of complex peptide andprotein mixtures. Anal Chem 71: 1087–1091.
Dean PDG, Watson DH. 1979. Protein purification usingimmobilized triazine dyes. J Chromatogr 165: 301–316.
Dihazi H, Kessler R, Eschrich K. 2001. One-step purificationof recombinant yeast 6-phosphofructo-2-kinase after theidentification of contaminants by MALDI–TOF MS. ProteinExpr Purif 21: 201–209.
Gevaert K, Demol H, Martens L, et al. 2001. Protein identificationbased on matrix assisted laser desorption/ionization–post sourcedecay–mass spectrometry. Electrophoresis 22: 1645–1651.
Goldhammer AR, Paradies HH. 1979. Phosphofructokinase: struc-ture and function. Curr Top Cell Regul 15: 109–141.
Heinisch JJ. 1986. Isolation and characterization of the twostructural genes coding for phosphofructokinase in yeast. MolGen Genet 202: 75–82.
Heinisch JJ, Ritzel RG, von Borstel RC, Aguilera A, Rodicio R,Zimmermann FK. 1989. The phosphofructokinase genes ofyeast evolved from two duplication events. Gene 78: 309–321.
Heinisch JJ, Hollenberg CP. 1993. Yeasts. In Biotechnology,Rehm HJ, Reed G (eds). VCH Verlagsgesellschaft: Weinheim;469–514.
Heinisch JJ, Kirchrath L, Liesen T, Vogelsang K, Hollenberg CP.1993. Molecular genetics of phosphofructokinase in the yeastKluyveromyces lactis . Mol Microbiol 8: 559–570.
Heinrich R, Rapoport SM, Rapoport TA. 1977. Metabolic regula-tion and mathematical models. Prog Biophys Mol Biol 32: 1–82.
Herrmann K, Diezel W, Kopperschlager G, Hofmann E. 1973.Immunological evidence for non-identical subunits in yeastphosphofructokinase. FEBS Lett 36: 190–192.
Hofmann E. 1976. The significance of phosphofructokinase to theregulation of carbohydrate metabolism. Rev Physiol BiochemPharm 75: 1–69.
Hudson PJ, Hengartner H, Kolb E, Harris JI. 1979. The pri-mary structure of phosphofructokinase from Bacillus stearother-mophilus . In Proceedings of the 12th FEBS Symposium, vol 52,Hofmann E, Pfeil W, Aurich H (eds). Pergamon: Oxford, NewYork; 341–348.
Johansson G. 1984. Affinity partitioning. Methods Enzymol 104:356–364.
Kemp RG, Foe LG. 1983. Allosteric regulatory properties ofmuscle phosphofructokinase. Mol Cell Biochem 57: 147–154.
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.

6-phosphofructokinase from Pichia pastoris 947
Klinder A, Kirchberger J, Edelmann A. Kopperschlager G. 1998.Assembly of phosphofructokinase-1 from Saccharomycescerevisiae in extracts of single-deletion mutants. Yeast 14:323–334.
Kopperschlager G, Bar J, Nissler K, Hofmann E. 1977. Physico-chemical parameters and subunit composition of yeastphosphofructokinase. Eur J Biochem 81: 317–325.
Kopperschlager G, Bohme HJ, Hofmann E. 1982. Cibacron BlueF3G-A and related dyes as ligands in affinity chromatography.Adv Biochem Eng Chromatogr 25: 101–138.
Kopperschlager G, Bar J, Stellwagen E. 1993. Limited proteolysisof yeast phosphofructokinase. Sequence locations of cleavagesites created by the actions of different proteinases. Eur JBiochem 217: 527–533.
Kopperschlager G. 1994. Affinity extraction with dye ligands.Methods Enzymol 228: 121–143.
Kopperschlager G. 2000. Effects of specific binding reactions onthe partitioning behaviour of biomaterials. Int Rev Cytol 192:61–97.
Kotlarz D, Buc H. 1982. Phosphofructokinases from Escherichiacoli . Methods Enzymol 90: 60–70.
Larsson C, Pahlman II, Gustafsson L. 2000. The importance ofATP as a regulator of the glycolytic flux in Saccharomycescerevisiae. Yeast 16: 797–809.
Lorberg A, Kirchrath L, Ernst JF, Heinisch JJ. 1999. Genetic andbiochemical characterization of phosphofructokinase from theopportunistic pathogenic yeast Candida albicans . Eur J Biochem260: 217–226.
McRorie DK, Voelker PJ. 1993. Self-associating Systems in theAnalytical Ultracentrifuge. Beckman Instruments Inc: Palo Alto,CA.
Monod J, Wyman J, Changeux JP. 1965. On the nature ofallosteric transitions: a plausible model. J Mol Biol 12: 88–118.
Nissler K, Otto A, Schellenberger W, Hofmann E. 1983. Similar-ity of activation of yeast phosphofructokinase by AMP andfructose 2,6-bisphosphate. Biochem Biophys Res Commun 111:294–300.
Poorman RA, Randolph A, Kemp RG, Heinrikson RL. 1984.Evolution of phosphofructokinase — gene duplication andcreation of new effector sites. Nature 309: 467–469.
Reuter R, Naumann M, Bar J, Haferburg D, Kopperschlager G.2000. Purification, molecular and kinetic characterization ofphosphofructokinase-1 from the yeast Schizosaccharomycespombe: evidence for an unusual subunit composition. Yeast 16:1273–1285.
Rodicio R, Strauss A, Heinisch JJ. 2000. Single point mutations ineither gene encoding the subunits of the hetero-octameric yeastphosphofructokinase abolish inhibition by ATP. J Biol Chem275: 40 952–40 960.
Sols A. 1981. Multimodulation of enzyme activity. Curr Top CellRegul 19: 77–101.
Yuan W, Tuttle DL, Shi YJ, Ralph GS, Dunn WA Jr.1997. Glucose-induced microautophagy in Pichia pastorisrequires the α-subunit of phosphofructokinase. J Cell Sci 110:1935–1945.
Copyright 2002 John Wiley & Sons, Ltd. Yeast 2002; 19: 933–947.
Related Documents











