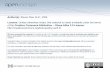
Bloodlines Above: From pluripotent cells arise the WBC and RBC and lymphoid s eries. Note that some cells will arise from the same mother cell. Anemias Reduction below normal limits of the total circulating red cell mass Reduced oxygen transport capacity of the blood Reduction below normal in the volume of packed red cell as measured by hematocrit or hemoglobin concentration IOW, the patient will appear pale and weak from lack of oxygen. Classification of Anemia According to Underlying Mechanism Blood loss Increased rate of destruction (hemolytic anemias) Impaired red cell production Anemia of Blood Loss Acute blood loss (microcytic, hypochromatic RBC’s may not be evident) Reflect loss of blood volumemay lead to shock, death Hemodilutionshift of water from interstitial fluid compartment into intravascular space Erythropoietin productionreticulocytosis (Immature RBC containing remnants of nuclei seen only in special stain. Bigger than usual RBC. Polychromatophilic bluish – red hue) reaching 10 – 15% Reticulocyte count normally 0.5 – 1.5% Chronic blood loss (microcytic, hypochromic RBC’s are more evident in chronic blood loss) GIT bleeding: gastric ulcer, hematemesis, hemorrhoids o Striking reticulocytosis may not be seen. Subject: Pathology Topic: RBC’s an d Ble eding Disorders Lecturer: Dr. Cagampan Date of Lecture: August 9, 2011 Transcriptionist: Mopster and Pinay Editor: Mopster and Pinay Pages: 16 S Y 2 0 1 1 2 0 1 2

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 1/15
1
Bloodlines
Above: From pluripotent cells arise the WBC and RBC and lymphoid series. Note that some cells will arise from
the same mother cell.
Anemias
Reduction below normal limits of the total
circulating red cell mass
Reduced oxygen transport capacity of the
blood
Reduction below normal in the volume of
packed red cell as measured by hematocrit
or hemoglobin concentration
IOW, the patient will appear pale and weak
from lack of oxygen.
Classification of Anemia According to Underlying
Mechanism
Blood loss
Increased rate of destruction (hemolytic
anemias)
Impaired red cell production
Anemia of Blood Loss
Acute blood loss (microcytic, hypochromatic RBC’s
may not be evident)
Reflect loss of blood volumemay lead to
shock, death
Hemodilutionshift of water from
interstitial fluid compartment into
intravascular space
Erythropoietin productionreticulocytosis
(Immature RBC containing remnants of
nuclei seen only in special stain. Bigger
than usual RBC. Polychromatophilicbluish
– red hue) reaching 10 – 15%
Reticulocyte count normally 0.5 – 1.5%
Chronic blood loss (microcytic, hypochromic RBC’s
are more evident in chronic blood loss)
GIT bleeding: gastric ulcer, hematemesis,
hemorrhoids
o Striking reticulocytosis may not be seen.
Gynecologic causes
Subject: PathologyTopic: RBC’s and Bleeding Disorders Lecturer: Dr. CagampanDate of Lecture: August 9, 2011Transcriptionist: Mopster and Pinay Editor: Mopster and Pinay Pages: 16
S Y
2 0 1 1 - 2 0 1 2

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 2/15
2
Increased Rate of Destruction (Hemolytic Anemia)
Intrinsic (intracorpuscular) abnormalities of
red cells
o Hereditary
Red cell membrane disorders
Disorders of membrane
cytoskeleton: spherocytosis,
elliptocytosis Disorders of lipid synthesis:
selective increase in membrane
lecithin
Red cell enzme deficiencies
Glycolytic enzymes: pyruvate
kinase deficiency, hexokinase
deficiency
Enzymes of hexose
monophosphate shunt: G6PD,
glutathione synthetase
Disorders of hemoglobin synthesis
Deficient globin synthesis:
thalassemia syndromes
Structurally abnormal globin
synthesis
(hemoglobinopathies): sickle
cell anemia, unstable
hemoglobin
o Acquired
Membrane defect: paroxysmal
nocturnal hemoglobinuria
Extrinsic (extracorpuscular) abnormalities
o Antibody mediated Isohemagglutinins: transfusion
reactions, erythroblastosis fetalis
Autoantibodies: idiopathic
(primary), drug associated, SLE,
malignant neoplasm, mycoplasmal
infections
o Mechanical trauma to red cells
Microangiopathic hemolytic
anemia: thrombotic
thrombocytopenic purpura, DIC
Cardiac traumatic hemolytic anemia Infections: malaria
Chemical injury: lead poisoning
Sequestration in mononuclear
phagocyte system: hypersplenism
Impaired Red Cell Production
Disturbance of proliferation and
differentiation of stem cells: aplastic
anemia, pure red cell aplasia, anemia of
renal failure, anemia of endocrine disorders
Disturbance of proliferation and maturation
of erythroblasts:
o Defective DNA synthesis: deficiency or
impaired use of vitamin B12 and folic
acid (megaloblastic anemia)
o Defective hemoglobin synthesis
Deficient heme synthesis: iron
deficiency
Deficient globin synthesis:
thalassemias
Unknown or multiple mechanisms:
sideroblastic anemia, anemia of
chronic infections, myelophthisic
anemia due to marrow infiltration
Hemolytic Anemia
Premature destruction of red cells and a
shortened red cell life span below the
normal 120 days Elevated erythropoietin levels and a
compensatory increase in erythropoiesis
Markedly increased erythropoiesis with
associated reticulocytosis
Accumulation of hemoglobin degradation
products released by red cell breakdown
derived from hemoglobin (e.g., bilirubin)
Pigment stone formation as a result of
hemoglobin degradation.
Tend to produce extravascular hemolysis
although, on occasion, intravascularhemolysis may occur.
Tend to be autosomal dominant
Rare in the Philippines, except Thalassemia.
Intravascular hemolysis (causes):
o Mechanical injury: e.g., prosthetic
cardiac valves, thrombi
o Complement fixation to red cells: e.g.,
mismatched transfusion
o Toxic injury: e.g., malaria
Manifestations of intravascular hemolysis:
o Anemia
o Hemoglobinemia
o Hemoglobinuria
o Jaundice: a small percentage of gall
stones are of hemoglobin origin
o Hemosiderinuria
Extravascular hemolysis
o Occurs in mononuclear phagocytes of
spleen
o Predisposing factors:
Red blood cell membrane injury Reduced deformability
opsonization
o Sequestration of “deformed” or
“foreign” red blood cells followed by
opsonization phagocytosis as red
cells navigate sinusoids
o These sequestered RBC’s are rendered
“palatable” to macrophages due to
hypoxia and ATP depletion.
o Clinical features
Anemia Splenomegaly
Jaundice
Morphology of hemolytic anemias
o Normoblastic hyperplasia in marrow
o Reticulocytosis in peripheral blood
o Pigment gallstones
o Hemosiderosis
o Jaundice, anemia

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 3/15
3
Below: Defective RBC’s sequestered outside are
then phagocytized.
Hereditary Spherocytosis Intrinsic defect in RBC membraneankyrin
deficiency and other (usually spectrin)
skeletal membrane components
RBCspheroidal, less deformable,
vulnerable to splenic sequestration and
destruction
Ankyrin deficiencyassociated with
reduced stability and loss of membrane
fragments as cells traverse circulation
Inherited disorder, in Northern Europe
Autosomal dominant
Morphology:
o Spheroidal RBC (normal is biconcave
disc)
o No central pallor noted
o Moderate splenomegaly due to marked
congestion of the cords of Billroth
o Erythrophagocytes in the splenic cords
o Features of hemolytic anemia
Clinical course: treatment is splenomegaly
o Chronic hemolytic anemia mild to
moderate
o Aplastic crisis parvovirus infection o Hemolytic crisis
o Diagnosis: Osmotic Fragility Test
Below: Cell membrane defect leads to formation of
spherocytes, which are sequestered and rendered
palatable to macrophages.
Below: How primary membrane defect leads to
phagocytosis on a chemical basis. This
pathophysiology is common to most hemolytic
anemia and needs to be known by heart.
Below (next 2 photos): Note round shape of RBC’s
and absence of central pallor.
G6PD Deficiency
X – linked
One of the tests for newborn screening
Impaired or deficient enzyme function
which reduce ability of red cells to fight
against oxidative injuries
Abnormalities in Hexose Monophosphate
Shunt pathway or glutathione metabolism
Need reduced glutathione to protect RBC
against oxidants
Oxidant stress:
o Drugs: antimalarials, sulfonamides, etc
o Infection: viral hepatitis, TF,
pneumonia
o Fava bean ingestion

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 4/15
4
Pathogenesis: Oxidative stress results in
the oxidation of globin chains which causes
the globin chains to denature and
precipitate to form Heinz Bodies. Heinz
bodies render the RBC palatable to
phagocytes. Sometimes, the Heinz bodies
are so abundant, that intravascular
hemolysis can occur. Extravascular and
intravascular hemolysis can occur.
Below: Not the Bite Cell in the center of the larger
picture. In the smaller picture in the upper left
corner, note the presence of Heinz Bodies under
special stain.
Sickle Cell Anemia
Structurally abnormal hemoglobin
Substitution of valine for glutamic acid at
the 6th position of β globin chain
American blacks:o Heterozygote: 40% HgbS
o Homozygote: 100% HgbS
Under deoxygenation, the RBC will sickle.
At first the sickling is reversible until such
time that the RBC can no longer change its
shape and is sequestered and phagocytized.
Infarction: sickle cells have the unique
feature of having increased adhesion to
each other. This what causes them to
aggregate and form thrombi which can lead
to infarct. Morphology:
o Hyperplastic marrowlead to
resorption of bone
o Extramedullary hematopoiesis
o Sickle red cells
o Initial splenomegaly erythrocytosis
thrombosis and infarction scarring
autosplenectomy
o Infarction in bone, brain, kidney, liver,
and retina
o Leg ulcer, cor pulmonale
o Pigment gallstones
Clinical course:
o Severe anemia: reticulocytosis
o Vasoocclusive complications: acute
chest syndrome
o Chronic hyperbilirubinemia: gallstoneso Increased susceptibility to
infectionsepticemia and meningitis
o CNS hypoxia: seizures, stroke
o Aplastic crisis: triggered by parvovirus
infection
o Sequestration crisis
o Priapism: thrombi lead congestion of
blood vessels which can lead to
persistent, painful erection.
Diagnosis
o PBS, metabisulfite-induced sickling
o Hemoglobin electrophoresis
o Fetal DNA by chorionic biopsy of
amniocentesis
Below: Single point mutation leads to sickle cell
formation which leads to hemolysis in the spleen
and infract in the tissues.
Below: Sickle cell admixed with anisocytosis,
hypochromia, poikylocytosis
Below: Spleen shrunken down to 3 cm.

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 5/15
5
Below: Severe congestion because of trapping of
the RBC’s
Thalassemia
α type seen in the Philippines
o There are 2 genetic loci for the α chain,
thus there are 4 alleles. There are 4
types of α thalassemia, each type
coinciding to a loss a allele. Type 1: Loss or mutation of single
allele. Minimal symptomatology
because the other 3 chains are
present.
Type 2: 2 alleles affected. Mild
anemia.
Type 3: 3 alleles affected. Leads to
Hemoglobin H.
Type 4: 4 alleles affected. Hydrops
fetalis. No chance of survival.
Patients with mild forms of the disease are
usually asymptomatic and are noticed to
have anemia when a CBC is ordered. They
are then given iron, which does not improve
the anemia. The astute doctor may suspect
another diagnosis, order an electrophoresis
and this is when thalassemia is diagnosed.
Mendelian disorder characterized by a lack
of or decreased synthesis of either α or β
globin chain of HbA
β Thalassemialack of β globin chains with
excess α chain
o Thalassemia major: both alleles of theβ chain are affected.
o Thalassemia minor: only one allele of
the β chain is affected. Aka, Cooley’s
anemia.
α Thalassemialack of α globin chains,
with excess β, γ, δ
Excess chains will precipitate and result in
phagocytosis
Facie: prominent cheekbones because of
increased blood production
Target cells: typical cells seen inThalassemia but not exclusive to it.
Morphology: same as in all HA
o “Crew-cut” appearance of bone on X-
ray due to marrow expansion with
thinning of cortical bone with few bone
formation on the external aspect
o Hepatosplenomegaly
o Hemosiderosis
Clinical course:
o Growth retardation
o Death at early age of homozygous
patient
o Manifestation depends on severity
o Prone to infection
Below: Typical facie of patient with thalassemia.
Prominent cheekbones are a result of increasedblood production by the facial bones in order to
compensate for RBC loss.
Below: Pathophysiology of β thalassemia. There is
reduced β hemoglobin with a relative excess α
hemoglobin as a compensatory mechanism. The
excess α globin precipitates in RBC. Most die inbone marrow, but some will make it into circulation
where they will be sequestered in spleen and
destroyed. The resulting anemia causes increased
erythropoiesis and increased iron absorption as the
body attempts to correct the anemia. As a result,
bone marrow expansion occurs as does systemic
iron overload. The bone deformities that result in
the facie are a result of bone marrow expansion.
Note that iron overload also comes from
destruction of the erythroblasts within the bone
marrow and from regular blood transfusions that
are required by these patients.

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 6/15
6
Below (next 3 pics): Target cells. Note: these are
not exclusive to thalassemias.
Below: “Crew cut” appearance of skull resulting
from increased erythropoiesis.
Below: Hepatic hemosiderosis in Beta Thalassemia
Assignment: Another name for target cell, why
does it happen?
Answer: Codocyte, leptocyte, or Mexican hat cell.
Seen in thalassemia, liver disease, post –
splenectomy patient. What causes targeting is
uneven distribution of hemoglobin.
Paroxysmal Nocturnal Hemoglobinuria
Acquired defect in cell membrane
Somatic mutation of the PIGA gene which is
essential for synthesis of the GPI(glycophosphatidylinositol) anchor
GPI – linked proteinsinactivate
complement
Other cells affected because GPI is found in
all blood cells, so patient can have
pancytopenia.
If patient is subject to an immune reaction
involving complement it can lead to lysis.
Ham’s Test: how it’s diagnosed.
Immunohemolytic Anemia Demonstration of the anti – RBC Ab
Coomb’s Test
Classification of Immune Hemolytic Anemias
Warm Antibody Type
o The antibody is of the IgG type, does
not usually fix complement and is active
at 37° C.
o Primary (Idiopathic)
o Secondary
Lymphomas and leukemias
Other neoplastic diseases
Autoimmune disorders (particularly
SLE)
Drugs
Cold Agglutinin Type
o The antibodies are IgM and are most
active in vitro at 0° C to 4° C.
Antibodies dissociate at 30° or above;
agglutination of cells by IgM and
complement fixation only in peripheral
cool parts of the body (eg, ears, toes,
and fingers)o Acute:
Mycoplasmal infection
Infectious Mononucleosis
o Chronic:
Idiopathic
Associated with lymphoma
Cold Hemolysin type (Paroxysmal
Hemoglobinuria)
o IgG antibodies bind red cells at cold
temperature, fix complement, and
cause hemolysis when the temperatureis raised above 30° C.
Hemolytic Anemia Resulting from Trauma to rBC
1. Prosthetic cardiac valves (mechanical)
2. Microangiopathic Hemolytic Anemia:
abnormally narrowed vessels

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 7/15
7
a. In DIC, malignant HTN, SLE, TTP,
Hemolytic-uremic syndrome,
disseminated cancer
b. See schistocytes, burr, helmet cells,
and triangle cells (couldn’t find a
pic, but some explanations basically
say these are RBC remnants that
resemble triangles). Note:
schistocytes are usually a sign of trauma, intravascular or
extravascular.
Below: Schistocyte
Below: Burr cell
Below: Target cell
Below: Helmet cell. Resembles Bite Cell, but Bite
Cell will have 1 bite, Helmet Cell will have >1 bite.
Anemias of Diminished Erythropoiesis
Megaloblastic Anemia
Impaired DNA synthesis leading to defective
nuclear maturation. DNA synthesis is
affected but RNA synthesis does not.
Asynchronism between nuclear and
cytoplasmic maturation. Immature nucleus
with very mature and often huge cytoplasm Due to folate or vitamin B12 deficiency.
o Vitamin B12 must be obtained through
the diet. It is absorbed in the ileum and
requires intrinsic factor.
o These are necessary for DNA synthesis
o RNA synthesis continues
o There is a lag so cell becomes
megaloblastic
Morphology:
o Macro – ovalocytes
o Hypersegmented neutrophils (>6 lobes)
o Bone marrow hypercellular (1:1)
o Megakaryocytes large with bizarre,
multilobate nuclei
o Ineffective
erythropoiesisintramedullary
destruction of megaloblast
o Increased hemolytic destruction of RBC
o Leukopenia and thrombocytopenia
o In short, pancytopenia

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 8/15
8
Below: Normal vitamin B12 metabolism. R –
binder is produced by the salivary gland. Vitamin
B12 is digested in the stomach via gastric acids and
binds with R – binder. It is carried through the small
intestine, where the B12 – R-binder complex is
digested by proteases. Vitamin B12 is now bound
to Intrinsic Factor and carried to the ileum, where it
is absorbed into the portal circulation. Intrinsic
factor is the switch by which vitamin B12 isabsorbed. Disruption of the GI tract, loss of R –
binder or Intrinsic Factor can all lead to
malabsorpton of vitamin B12.
Below: Macroovalyctes. Bone marrow would be
hypercellular, composed of RBC series, all
megaloblasts. Because megaloblastic anemia
affects all cell lines, PMN’s will also be affected.
They will appear, as below, as hypersegmented (>5
lobes) because of the asynchrony between DNA and
RNA synthesis. Sometimes, the deficiency is so bad,
RBC’s can be destroyed in the bone
marrowineffective erythropoiesis just likeThalassemia. Again, megaloblastic anemia may
manifest as pancytopenia in the bone marrow.
Below: Bone marrow looks busy, hypercellular.
The cells are very large nucle, hyperchromatic, and
granular. There appears to be maturation of the
cytoplasm with a lag in the nucleus:cytoplasm ratio.
Below (next 3 pictures): Macro – ovalocytes in the
PBS with schistocytes.

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 9/15
9
Pernicious Anemia
Autoimmune destruction of gastric mucosa
Chronic atrophic gastritis lack of intrinsic
factor
Presence of autoantibodies against parietal
cellsblocking Ab, Type II Ab and parietal
canalicular Ab
Morphology:
o GIT Atrophic glossitis
Diffuse chronic gastritis. This is
specific to pernicious anemia. If it is
not due to pernicious anemia, you
won’t find this.
Intestinalization of gastric glands
o CNS lesion
Myelin degeneration of the dorsal
and lateral tractssensory motor
deficits
Diagnostic features:o Moderate to severe megaloblastic
anemia
o Leucopenia with hypersegmented
granulocytes
o Mild to moderate thrombocytopenia
o Neurologic changes: “subacute
combined degeneration”
o Achlorydia even after histamine
stimulation. Remember, histamine is
supposed to release gastric acids. The
patient with pernicious anemia will not
do this.
o Inability to absorb oral dose of
cobalamine – “Schilling Test”
o Low serum B12
o Excretion of methylmalonic acid in urine
o Improvement after parenteral B12
o Demonstration of antibody to instrinsic
factor
Below: Atrophic glossitis
Below: Atrophic gastritis
Below: myelin degeneration of the dorsal tracts

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 10/15
10
Below: Myelin degeneration of the lateral tracts
Folate Deficiency
Same as B12 deficiency but without
neurologic changes
Iron Deficiency Anemia
Most common nutritional deficiency
Iron is absorbed in the duodenum and can
be recycled. Storage pool of Fe: hemosiderin and
ferritin
Ferritin:
o Protein – iron complex; stored in
parenchymal cells or within RES
o Level is a good indicator of adequacy of
body iron stores
o Iron deficiency
anemia↓iron↓Ferritin
Transferrin:
o Iron – binding glycoprotein whichtransports iron in plasma; deliver iron
to cells including erythroid precursors
(TIBC)
o When iron is deficient↑TIBC
(because the body is scavenging for
iron)
Causes of iron deficiency:
o Dietary lack: in elderly, poor, infants,
and children
o Impaired absorption: in malabsorption
o Increased requirement: growing infants
and children, adolescents,
premenopausal, pregnancy
o Chronic blood loss: hemorrhoids, GIT
Ca, parasitism, menstrual
abnormalities, urinary tract bleeding
Morphology:
o Normoblastic hyperplasia in marrow
o Microcytic, hypochromic RBC
Diagnosis:
o PBS findings, decreased hemoglobin
and hematocrit, low serum Fe and
serum Ferritin TIBC (transferrin
concentration) is high
Below: Microcytic, hypochromic RBC’ssmall and
paler central pallor. The PMN is used as a point of
reference to determine the relative size of RBC.
PMN’s are around 12μm. The RBC’s in the PBS
below are ¼ the size, so around 4 – 5 μm. Normal
RBC’s are usually 6 – 7 μm, or 1/3 the size of a PMN.
Anemia of Chronic Disease
Reduced erythroid proliferation and
impaired Fe utilization
Chronic infection: osteomyelitis, bacterial
endocarditis, lung abscess
Chronic immune disorder: RA, Crohn’s
Neoplasms: Hodgkin’s CA of lung and
breast
Pt. peripheral blood smear may appear like
iron deficiency anemia, but stores are
normal
The failure is in the utilization of iron, not
in the amount.
Diagnosis:
o Low serum Fe, decreased TIBC but
abundant stored iron in marrow
macrophage
o Low erythropoietin levels marrow
hypoproliferation
Aplastic Anemia
Pancytopenia characterized by anemia,
neutropenia, and thrombocytopenia Bone marrow is almost converted to fat.
Cells present are usually lymphocytes.
Normal BM is 50:50.
Morphology:
o Markedly hypocellular marrow – “fatty
marrow”
o Fibrous tissue with scattered
lymphocytes

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 11/15
11
Below: Most of the time cause is unknown, but
when it is known, it is usually drug induced.
Below: Pancytopeniaall cells are decreased.
Markedly hypocellular marrow. Sometimes fibrotic
with scattered lymphocytes.
Below: Note the abundance of fat in the marrow.
Other Forms of Marrow Failure
Myelophthisic anemia
o Due to space – occupying lesions in
marrow; metastatic carcinoma; multiple
myeloma, leukemia, Tb
Diffuse liver disease
Chronic renal failure
Below: myelophthisic anemia secondary to
leukemia. Leukemias typically fill up the marrow
with abnormal cells.
Bleeding Disorders
Can be caused by :
o Increased blood vessel fragility/ Vessel
wall abnormality
o Platelet disorders/ abnormality (both in
function and in number)
o Coagulation defects
Evaluation requires laboratory testing:o Bleeding time: tests platelet function
o Platelet counts
o Prothrombin time: tests extrinsic
pathway (Mnemonic: PeT, prothrombin
extrinsic time)
o Partial Thromboplastin time: tests
intrinsic pathway (Remember: PiTT,
partial intrinsic thromboplastin time)
o Specialized tests (e.g., clotting factor
levels)
I. Vessel Wall Abnormality
relatively common but usually do not
cause serious bleeding
typically induce only petechial and
purpuric hemorrhages
Can be caused by infections, drug
reactions, autoimmune diseases, vitamin
deficiency, immune complex deposits, or
hereditary disorders
normal platelet count, BT, PT, and PTT
Conditions which causes increased vascular fragility:1. Infections
a. Meningococcemia (Waterhouse –
Friedrichsen syndrome), gram (-)
septicemia, infective endocarditis,
rickettsiosis
b. Microbiologic damage to vessels
(vasculitis) or DIC (Disseminated
intravascular coagulation) underlying
mechanism
2. Drug reactions – often secondary to
immune complex deposition in vessel wallswith resulting hypersensitivity vasculitis
3. Poor vascular support
a. Abnormal collagen synthesis (Scurvy,
Ehlers- Danlos Syndrome: impaired
collagenous support
b. Loss of perivascular supporting tissue
(Cushing syndrome)
c. Vascular wall amyloid deposition
4. Henoch – Schonlein Purpura: systemic
hypersensitivity reaction of unknown cause
characterized by purpuric rash, abdominal
pain, polyarthralgia, and acute
glomerulonephritis. Associated with
vascular and glomerular mesangial
deposition of immune complexes.
5. Hereditary hemorrhagic telangiectasia
II. Reduced platelet number:
Thrombocytopenia= characterized principally by
petechial bleeding, most often from small vessels of

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 12/15
12
skin and mucous membranes. Count is
<100,000/mm3
Normal: 150,000 – 450,000/mm3
Thrombocytopenia: <100,000/mm3
Spontaneous bleeding: <20,000/mm3
***Sometimes patient has <10,000/mm3 and
patients don’t bleed, or has <50,000/ mm3 and
already experienced spontaneous bleeding therefore clinically it really depends on when to
start your management; as physicians we should
know when to act
Causes:
a. Decreased production: due to ineffective
megakaryopoiesis (e.g., megaloblastic
states) or to generalized marrow disease
that also compromises megakaryocyte
number (e.g., aplastic anemia, disseminated
cancer).
b. Decreased survival: due to immune-
mediated platelet destruction, usually with
a compensatory megakaryocytic marrow
hyperplasia
-it can follow drug exposure or
infections
-platelet deficiencies due to
consumption often occur in systemic
coagulopathies (DIC, hemolytic uremic
syndrome, thrombotic
thrombocytopenia purpura).
c. Sequestration: platelets are retained in thered pulp of enlarged spleens
d. Dilution: massive whole blood transfusions
can cause a relative reduction in the
number of circulating platelets because
storage for longer than 24 hours at 4°C
results in rapid hepatic platelet
sequestration upon infusion.
e. HIV: results from immune complex injury,
antiplatelet antibodies, and HIV- induced
suppression of megakaryocytes.
Idiopathic Thrombocytopenic Purpura (ITP)/
Immune Thrombocytopenic Purpura
: antibody- mediated platelet destruction
Acute (children)
o Self-limiting
o Seen most often in children after a viral
infection (e.g., rubella, cytomegalovirus
infection, viral hepatitis, infectiousmononucleosis)
o Platelet destruction is due to transient
antiplatelet autoantibodies.
Chronic (adult<40 y/o), mostly female of
childbearing age
o Long history of easy bruising or
nosebleeds
o Platelet autoantibodies (synthesized in
the spleen) are usually directed toward
one of two platelet antigens (platelet
membrane glycoprotein complexes IIb/
IIIa or Ib/IX).
o Destruction of antibody-coated
platelets occurs in the spleen.
o Splenectomy benefits 75- 80% of
patients
Antiplatelet antibodies
Pathogenesis: opsonized platelet
susceptible to phagocytosis by RES cells (in
spleen)
Morphology: spleennormal in size
o Congestion of sinusoids and enlarged
follicles
o Prominent germinal centers
o Megakaryocytes within sinusoids
o Bone marrowincrease number of
megakaryocytes
Below: Bone marrow in ITPincreased number of
megakaryocytes, because it is a compensatory
mechanism. If ITP count is not increased in the
bone marrow, you can pretty much rule out ITP.

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 13/15
13
Below: normal spleen. But there may be
megakaryocytes in the spleen.
Thrombotic Microangiopathies:
-characterized byo Thrombocytopenia
o Microangiopathic hemolytic anemia
o Fever
o Transient neurologic deficits (in TTP)
o Renal failure (HUS)
-most of the clinical manifestations are due to
widespread hyaline microthrombi in arterioles and
capillaries (microcirculation) composed of dense
aggregates of platelets and fibrin
-platelets adhere more to the thrombi
Thrombotic Thrombocytopenic Purpura(TTP)
o Associated with inherited or acquired
deficiencies in ADAMTS13 (a
matalloprotease that limits the size of
von Willebrand factor multimers in the
plasma.
o In adult female
o Pentad of: fever, thrombocytopenia,
microangiopathic hemolytic anemia,
neurologic defects, renal failure
o Probably viral - induced
Hemolytic – uremic syndrome (HUS)
o Commonly follows gastrointestinal
infections with verotoxin-producing
E.coli.
Verotoxin injures endothelial cells
promotes dysregulated platelet
activationaggregation
o Microangiopathic hemolytic anemia,
thrombocytopenia and acute renal
failure o Onset in childhood
o Follow infection with verotoxin –
producing E. coli
Both show widespread formation of hyaline
thrombi in microcirculation
Bleeding Related to Defective Platelet Function
A. Congenital Disorders
1. Defective adhesion
a. “Bernard – Soulier Syndrome” :
caused by deficient platelet
membrane glycoprotein complex
GpIb/ IX- platelet receptor for vWF
and necessary for platelet-collagen
adhesion
b. Inherited deficiency of platelet
membrane glycoprotein
2. Defective aggregation
a. Thrombasthenia: caused by
deficient platelet membraneglycoprotein GpIIb/ IIIa- involved in
binding fibrinogen
3. Defective secretion- platelet will secrete
an enzyme to stabilize the plug
B. Acquired disorders
1. Aspirin ingestion- potent inhibitor of
cyclooxygenase and can suppress the
synthesis of thromboxane A2 – for
platelet aggregation
2. Uremia
III. Bleeding due to Abnormalities in Clotting
Factors
von Willebrand’s Disease
Autosomal dominant
Characterized by spontaneous bleeding
from mucous membranes; excessive
bleeding from wounds, menorrhagia

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 14/15
14
Prolonged BT, PTT, normal platelet count,
reduced vWF and Factor VIII levels
May have quantitative or qualitative effect
in VWF
Compound defects involving platelet
function and coagulation pathway
Hemophilia A (Factor VIII Deficiency)
X – linked recessive trait; in male andhomozygous female
Reduction in amount or activity of Factor
VIII
Severity depends on Factor VIII activity;<1%
Factor VIII activity is severe disease
Easy bruising and massive hemorrhage after
trauma or operation
Spontaneous joint hemorrhages –
hemarthrosesrecurrent
bleedingdeformities
Normal BT, platelet count, and PT,prolonged PTT
Below: Easy bruising is common
Below: Bleeding gums is common
Below: Genogram showing the X – linked
inheritance of Hemophilia in the royal families of Europe.
Disseminated Intravascular Coagulation (DIC)
-an important complication= not a disease but a
complication of some other disease
Acute, subacute, and chronic
thrombohemorrhagic disorder occurring as
a secondary complication in a variety of
disease
Characterized by activation of the
coagulation sequence that leads toformation of microthrombi throughout the
microcirculation
Consumption of platelets, fibrin,
coagulation factors with secondary
activation of fibrinolytic
mechanisms”Consumptive
Coagulopathy”
2 major mechanisms which trigger DIC
o Release of tissue factors or
thromboplastic substances
o Injury to endothelial cells
releasingthromboplastic substances, which
causes:
Massive thrombosis
Bleeding to death
***massive thrombosis release of thromboplastic
substancestrigger the coagulation system
consume more factorsbleed spontaneously
death.
***Can be difficult to treat as there are 2 stages:
thrombotic and bleeding. If patient is in the
thrombotic stage, then giving fibrinogen can
potentially worsen the condition because itconsumes more factors to be used. If during the
bleeding stage, giving thrombotic agents can also
hasten the bleeding.
***Thrombi can lead to ischemia tissue damage
Morphology:
o Multiple thrombi in one or several
organs
o ARDS in lungs, microinfarcts in brain,
adrenal hemorrhages
Below: common causes of DIC. Most commoninfective agent is Gram negative sepsis. Whatever,
the cause, it triggers DIC through the release of
coagulation factors.

8/4/2019 2011-08-PATHO-Red Blood Cells and Bleeding Disorders (1)
http://slidepdf.com/reader/full/2011-08-patho-red-blood-cells-and-bleeding-disorders-1 15/15
Below: Pathophysiology of DIC
Below: DIC causes thrombi that can lead to occlusion of the blood vesselsischemia in various organs.
Note: With DIC the patient is usually admitted for another condition, but develops clotting disorders because
DIC is a complication and rarely a primary disorder itself.
--------------------------------------------------End of Transcription--------------------------------------------------------
“And God showed His love for us by sending His own Son into the world.” 1 John 4:9-10
Related Documents