13 C-detected protonless NMR spectroscopy of proteins in solution Wolfgang Bermel a , Ivano Bertini b, * , Isabella C. Felli b , Mario Piccioli b , Roberta Pierattelli b a Bruker BioSpin GmbH, Rheinstetten, Germany b Magnetic Resonance Center and Department of Chemistry, University of Florence, Via Luigi Sacconi 6, 50019 Sesto Fiorentino, Italy Received 2 August 2005 Available online 20 December 2005 Keywords: 13 C-detected NMR; Protonless NMR; Sequential assignment; Spin-state selection; Paramagnetic proteins Contents 1. Why the need of hetero detection ..................................................................... 25 2. Instrumental aspects .............................................................................. 27 3. The problem of the 13 C– 13 C coupling ................................................................. 28 4. A protocol for the assignment of backbone and side chains .................................................. 30 5. Detection of resonances in paramagnetic proteins ......................................................... 34 6. Conclusions .................................................................................... 36 Acknowledgements ............................................................................... 36 Appendix ...................................................................................... 37 References ..................................................................................... 44 1. Why the need of hetero detection The NMR determination of the structure of large biological macromolecules in solution is primarily limited by the fast transverse relaxation that broadens lines and reduces spectral resolution. Several steps ahead have been made recently to overcome this limitation. In particular, the constructive use of cross-correlated relaxation phenomena enables a reduction of the effective transverse relaxation rates of specific spins, such as backbone NH groups [1] and aromatic CH groups [2]. More recently, selected cross-correlation rates were exploited to obtain line narrowing for methyl [3] and methylene [4] groups. The other very efficient way to reduce transverse relaxation rates consists in 2 H isotopic enrichment [5–8]. The lower gyromagnetic ratio of 2 H compared to that of 1 H contributes to a drastic reduction of dipole–dipole interactions thus providing less efficient relaxation mechanisms. Selective isotope label- ling also simplifies crowded NMR spectra and allows the selection of specific resonances [9–11]. Direct-detection of heteronuclei, and of 13 C in particular, offers a valuable alternative to 1 H detection. The idea of 13 C direct-detection has its roots in the early days of NMR and has been used for decades to study small molecules such as organic and inorganic compounds [12] or metabolites [13]. However, 13 C direct-detection has not been widely applied to study biological macromolecules. Thanks its large 1 H gyromagnetic ratio, the 1 H sensitivity is indeed intrinsically much higher than that of 13 C, and proton-detected experiments are usually convenient. Moreover, most of the efforts to improve instrument technology have been devoted to increase the sensitivity of 1 H detection rather than that of the heteronuclei. The availability of increasingly high magnetic fields and the advent of cryogenically cooled probeheads [14] has now moved the sensitivity of NMR spectroscopy into regions that were unforeseeable less than a decade ago. As a consequence, 13 C sensitivity has been dramatically increased up to a level suitable to turn 13 C detected experiments on enriched samples into routine methods for biomolecular NMR applications. The features of 13 C NMR spectra can be realized from Fig. 1, which shows a 13 C– 15 N correlation experiment recorded on 13 C, 15 N labeled reduced monomeric superoxide dismutase (SOD, 15 kDa) at 14.1 T. The map shows all expected cross peaks with excellent resolution, including those of Pro residues as well as those of Asn and Gln side-chains. The spectrum Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 www.elsevier.com/locate/pnmrs 0079-6565/$ - see front matter q 2005 Elsevier B.V. All rights reserved. doi:10.1016/j.pnmrs.2005.09.002 * Corresponding author. Fax: C39 055 4574271. E-mail address: [email protected]fi.it (I. Bertini).

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

13C-detected protonless NMR spectroscopy of proteins in solution
Wolfgang Bermel a, Ivano Bertini b,*, Isabella C. Felli b, Mario Piccioli b, Roberta Pierattelli b
a Bruker BioSpin GmbH, Rheinstetten, Germanyb Magnetic Resonance Center and Department of Chemistry, University of Florence, Via Luigi Sacconi 6, 50019 Sesto Fiorentino, Italy
Received 2 August 2005
Available online 20 December 2005
Keywords: 13C-detected NMR; Protonless NMR; Sequential assignment; Spin-state selection; Paramagnetic proteins
Contents
1. Why the need of hetero detection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2. Instrumental aspects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
3. The problem of the 13C–13C coupling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
4. A protocol for the assignment of backbone and side chains . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
5. Detection of resonances in paramagnetic proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
6. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
Appendix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
1. Why the need of hetero detection
The NMR determination of the structure of large biological
macromolecules in solution is primarily limited by the fast
transverse relaxation that broadens lines and reduces spectral
resolution. Several steps ahead have been made recently to
overcome this limitation. In particular, the constructive use of
cross-correlated relaxation phenomena enables a reduction of
the effective transverse relaxation rates of specific spins, such
as backbone NH groups [1] and aromatic CH groups [2]. More
recently, selected cross-correlation rates were exploited to
obtain line narrowing for methyl [3] and methylene [4] groups.
The other very efficient way to reduce transverse relaxation
rates consists in 2H isotopic enrichment [5–8]. The lower
gyromagnetic ratio of 2H compared to that of 1H contributes to
a drastic reduction of dipole–dipole interactions thus providing
less efficient relaxation mechanisms. Selective isotope label-
ling also simplifies crowded NMR spectra and allows the
selection of specific resonances [9–11].
0079-6565/$ - see front matter q 2005 Elsevier B.V. All rights reserved.
doi:10.1016/j.pnmrs.2005.09.002
* Corresponding author. Fax: C39 055 4574271.
E-mail address: [email protected] (I. Bertini).
Direct-detection of heteronuclei, and of 13C in particular,
offers a valuable alternative to 1H detection. The idea of 13C
direct-detection has its roots in the early days of NMR and has
been used for decades to study small molecules such as organic
and inorganic compounds [12] or metabolites [13]. However,13C direct-detection has not been widely applied to study
biological macromolecules. Thanks its large 1H gyromagnetic
ratio, the 1H sensitivity is indeed intrinsically much higher than
that of 13C, and proton-detected experiments are usually
convenient. Moreover, most of the efforts to improve
instrument technology have been devoted to increase the
sensitivity of 1H detection rather than that of the heteronuclei.
The availability of increasingly high magnetic fields and the
advent of cryogenically cooled probeheads [14] has now
moved the sensitivity of NMR spectroscopy into regions that
were unforeseeable less than a decade ago. As a consequence,13C sensitivity has been dramatically increased up to a level
suitable to turn 13C detected experiments on enriched samples
into routine methods for biomolecular NMR applications.
The features of 13C NMR spectra can be realized from
Fig. 1, which shows a 13C–15N correlation experiment recorded
on 13C,15N labeled reduced monomeric superoxide dismutase
(SOD, 15 kDa) at 14.1 T. The map shows all expected cross
peaks with excellent resolution, including those of Pro residues
as well as those of Asn and Gln side-chains. The spectrum
Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45
www.elsevier.com/locate/pnmrs
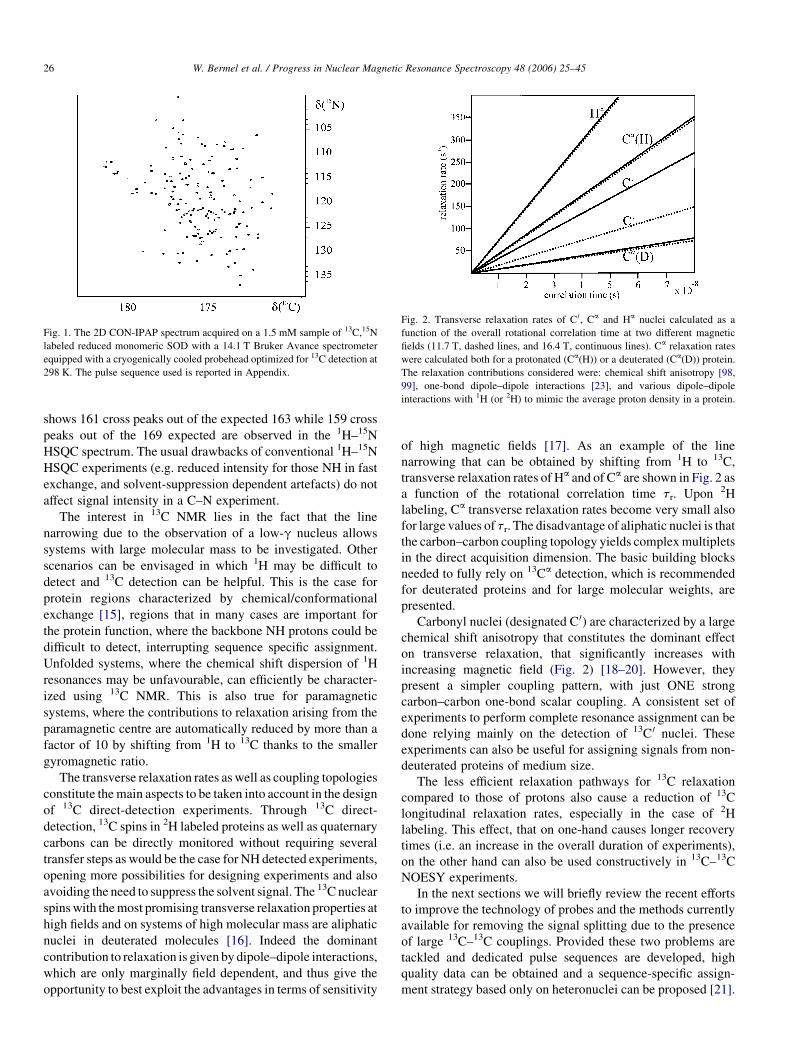
Fig. 1. The 2D CON-IPAP spectrum acquired on a 1.5 mM sample of 13C,15N
labeled reduced monomeric SOD with a 14.1 T Bruker Avance spectrometer
equipped with a cryogenically cooled probehead optimized for 13C detection at
298 K. The pulse sequence used is reported in Appendix.
Fig. 2. Transverse relaxation rates of C 0, Ca and Ha nuclei calculated as a
function of the overall rotational correlation time at two different magnetic
fields (11.7 T, dashed lines, and 16.4 T, continuous lines). Ca relaxation rates
were calculated both for a protonated (Ca(H)) or a deuterated (Ca(D)) protein.
The relaxation contributions considered were: chemical shift anisotropy [98,
99], one-bond dipole–dipole interactions [23], and various dipole–dipole
interactions with 1H (or 2H) to mimic the average proton density in a protein.
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4526
shows 161 cross peaks out of the expected 163 while 159 cross
peaks out of the 169 expected are observed in the 1H–15N
HSQC spectrum. The usual drawbacks of conventional 1H–15N
HSQC experiments (e.g. reduced intensity for those NH in fast
exchange, and solvent-suppression dependent artefacts) do not
affect signal intensity in a C–N experiment.
The interest in 13C NMR lies in the fact that the line
narrowing due to the observation of a low-g nucleus allows
systems with large molecular mass to be investigated. Other
scenarios can be envisaged in which 1H may be difficult to
detect and 13C detection can be helpful. This is the case for
protein regions characterized by chemical/conformational
exchange [15], regions that in many cases are important for
the protein function, where the backbone NH protons could be
difficult to detect, interrupting sequence specific assignment.
Unfolded systems, where the chemical shift dispersion of 1H
resonances may be unfavourable, can efficiently be character-
ized using 13C NMR. This is also true for paramagnetic
systems, where the contributions to relaxation arising from the
paramagnetic centre are automatically reduced by more than a
factor of 10 by shifting from 1H to 13C thanks to the smaller
gyromagnetic ratio.
The transverse relaxation rates as well as coupling topologies
constitute the main aspects to be taken into account in the design
of 13C direct-detection experiments. Through 13C direct-
detection, 13C spins in 2H labeled proteins as well as quaternary
carbons can be directly monitored without requiring several
transfer steps as would be the case for NH detected experiments,
opening more possibilities for designing experiments and also
avoiding the need to suppress the solvent signal. The 13C nuclear
spins with the most promising transverse relaxation properties at
high fields and on systems of high molecular mass are aliphatic
nuclei in deuterated molecules [16]. Indeed the dominant
contribution to relaxation is given by dipole–dipole interactions,
which are only marginally field dependent, and thus give the
opportunity to best exploit the advantages in terms of sensitivity
of high magnetic fields [17]. As an example of the line
narrowing that can be obtained by shifting from 1H to 13C,
transverse relaxation rates of Ha and of Ca are shown in Fig. 2 as
a function of the rotational correlation time tr. Upon 2H
labeling, Ca transverse relaxation rates become very small also
for large values of tr. The disadvantage of aliphatic nuclei is that
the carbon–carbon coupling topology yields complex multiplets
in the direct acquisition dimension. The basic building blocks
needed to fully rely on 13Ca detection, which is recommended
for deuterated proteins and for large molecular weights, are
presented.
Carbonyl nuclei (designated C 0) are characterized by a large
chemical shift anisotropy that constitutes the dominant effect
on transverse relaxation, that significantly increases with
increasing magnetic field (Fig. 2) [18–20]. However, they
present a simpler coupling pattern, with just ONE strong
carbon–carbon one-bond scalar coupling. A consistent set of
experiments to perform complete resonance assignment can be
done relying mainly on the detection of 13C 0 nuclei. These
experiments can also be useful for assigning signals from non-
deuterated proteins of medium size.
The less efficient relaxation pathways for 13C relaxation
compared to those of protons also cause a reduction of 13C
longitudinal relaxation rates, especially in the case of 2H
labeling. This effect, that on one-hand causes longer recovery
times (i.e. an increase in the overall duration of experiments),
on the other hand can also be used constructively in 13C–13C
NOESY experiments.
In the next sections we will briefly review the recent efforts
to improve the technology of probes and the methods currently
available for removing the signal splitting due to the presence
of large 13C–13C couplings. Provided these two problems are
tackled and dedicated pulse sequences are developed, high
quality data can be obtained and a sequence-specific assign-
ment strategy based only on heteronuclei can be proposed [21].

Fig. 3. The specified signal-to-noise ratio of 60% benzene-d6 in 40% p-dioxane
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 27
This alternative strategy, that does not actively exploit protons
and for this reason we called protonless NMR, offers an
alternative approach to the standard triple resonance assign-
ment strategy. Such an approach does not aim at replacing
methods relying on NH-detected TROSY experiments, which
combine favourable relaxation properties due to 2H labelling,
constructive use of relaxation interferences and the sensitivity
of 1H direct-detection [22]. However, the fact that protonless
sequential assignment of proteins is nowadays feasible
constitutes a breakthrough per se.
(the so-called ASTM sample) for 13C-observe probes plotted as a function ofthe year when a probe was first introduced at a particular magnetic field
(expressed as 1H operating frequency in MHz). Black dots denote the 13C
sensitivity of conventional 13C-observe probes. For the cryogenically-cooled
probes, the 13C sensitivity of 13C-detection probes are indicated with triangles,
the 13C sensitivity of the triple-resonance probes with both cryogenically
enhanced 1H and 13C capacities are denoted by diamonds and the four-nuclei
heteronuclear-observe probe is indicated with square (adapted with permission
from [14]).
2. Instrumental aspects
The sensitivity in NMR experiments is given by Eq. (1)
S=NzAgexcg3=2obsB
3=20 N1=2
scan (1)
were gexc and gobs are the gyromagnetic ratios of the excited
and of the observed nuclei, respectively, A is the number of
spins, Nscan is the number of scans and B0 is the strength of the
applied magnetic field [23]. Therefore, neglecting relaxation,1H observed experiments have a sensitivity gain of a factor of
32 (i.e. (gH/gC)5/2) compared to the corresponding experiments
starting and ending at 13C nuclei (i.e. HCCH COSY or HCACO
[24] vs 13C–13C COSY or CACO experiments [25]). Recent
developments in probe technology and the availability of NMR
spectrometers operating at high magnetic fields hold promise
for dramatic improvements in sensitivity, not only for 1H but
for 13C as well. 13C sensitivity, at least in NMR spectrometers
devoted to biomolecular NMR applications, has so far been
sacrificed in favour of 1H sensitivity. Indeed, as far as probe
design is concerned, it is generally necessary to achieve a
compromise between the performance of different nuclei in
terms of sensitivity and pulse lengths. Generally, optimal
sensitivity for a specific nucleus is achieved for a 2-coil probe
by using the inner coil for that nucleus, at the expense of the
performance of the other nuclei being on the outer coil.
Probeheads optimised for 13C are nowadays available, such as
dual probeheads, with the inner coil optimised for 13C and with
the possibility to pulse on 1H, and triple-resonance probeheads,
where the inner coil is used to pulse on 13C and 15N. The
evolution of 13C signal-to-noise (S/N) ratio for these probe-
heads over the years is shown in Fig. 3.
Changing the focus from 1H to 13C has prompted
manufacturers to concentrate their efforts in obtaining a new
generation of probeheads in which the main goal is optimized
sensitivity on 13C, coupled with the capability to excite and/or
decouple 1H, 2H and 15N. Indeed, a prototype room-
temperature 13C-observe triple-resonance probehead has been
built yielding a substantial increase in 13C sensitivity and has
greatly contributed to the expansion of the set of experiments
available [21,26,27].
A very efficient way to increase sensitivity consists in
reducing the thermal noise that contributes to the observed
signal, leading to the so-called cryogenically-cooled probe-
heads. This has recently been extensively exploited for 1H
allowing an increase in proton sensitivity by a factor of about
3–4 [14]. The improvement in the sensitivity is mainly due to
the reduced thermal noise achieved by decreasing the operating
temperature of the coils and of the 1H preamplifier. Inclusion of
a cryo-cooled preamplifier also for 13C without changing the
probe design of a standard probe for triple-resonance
experiments has given a significant increase in 13C sensitivity
without needing to make any compromise on 1H. The
behaviour of these probes in terms of S/N is shown in Fig. 3
(diamonds). Dual cryogenically-cooled probes optimised for13C sensitivity (Fig. 3, triangles) are also available but as yet
they do not allow pulsing on 15N and, therefore, are mainly
used to study small molecules. Obviously, a combination of the
cryo-technology with the design of triple-resonance probes
with improved 13C sensitivity is in principle possible and
would contribute to further increase the application of 13C
direct-detection experiments in biomolecular NMR. Further-
more, 13C detected experiments are less prone to sensitivity
losses due to high salt concentrations, particularly relevant in
cryogenically-cooled probeheads. It has been shown that 13C
detection significantly extends the range of sample conditions
under which 13C detection becomes competitive with proton
detection [28].
In order to perform a triple-resonance experiment using 13C
detection, we should also consider aspects other than
sensitivity. The minimal hardware requirements in terms of
transmitters and channels are the same as for standard triple-
resonance experiments (1H, 13C, 15N and 2H in the case of
deuterated proteins). In addition, filters may be required to
prevent cross talk between the 13C and 15N channel. This may
become an issue when 15N is decoupled during 13C detection.
These filters could be part of a band-selective preamplifier or
external devices. Additional hardware may also be needed to
perform band-selective 13C homodecoupling.
Band-selective homodecoupling (BH) of 13C 0 from 13Ca and
vice versa may be desirable in some experiments, but requires
some more consideration. During acquisition, data points are
sampled at a speed given by the amount of oversampling.

Fig. 4. Dwell cycle showing the acquisition status, the pulse and the dwell
window on the time axis. The durations of the decoupling pulse tp, of the
hdduty and of the dwell time are indicated. Time periods D are switching
delays, which are predetermined fractions of the dwell time.
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4528
In any case the receiver is unblanked during the whole period.
In order to achieve homodecoupling, radiofrequency has to be
irradiated during a fraction of the nominal dwell time, which in
turn is defined as the reciprocal of the spectral width of the
spectrum. This fraction is called hdduty, the homodecoupling
on/off ratio given as a percentage of the nominal dwell time.
During hdduty as well as some short delays for switching from
observe to transmit mode and vice versa, the receiver system is
blanked (Fig. 4). This results in a reduction in S/N with respect
to the theoretical factor of 2 according to [29]:
S=NðBHÞZ 2S=N
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi1K
hdduty
100
r(2)
A short value of hdduty would thus provide a higher S/N.
However, a smaller hdduty value implies higher RF power,
which may cause unwanted probe ringdown and sidebands. All
recent reports have used a 20% hdduty, which corresponds to a
maximum theoretical S/N of 1.79 [29–31]. Experimentally
measured S/N values rarely achieve such an ideal value and
show a significant frequency dependent variability. This
prevents a quantitative comparison of peak intensities within
the same spectrum, but does not prevent the use of
homodecoupling for R1 or R2 experiments, or any other
quantitative experiment. Following the convolution theorem in
Fourier transformation, an irradiation period spaced by a time
interval much longer than irradiation produces sidebands
spaced by n/r, where n is an integer and r is the dwell time,
(in the case where small hdduty values are used). Therefore, the
sweep width and the value of hdduty should be chosen to avoid
overlap of the signals of interest with sidebands arising from
homodecoupling. To perform homodecoupling, adiabatic chirp
pulses, with 10 ms pulse length, 25% smoothing and sweep
from low to high field have been used [32].
3. The problem of the 13C–13C coupling
In 13C direct-detection experiments we need to face the
problem of the large homonuclear one-bond carbon–carbon
couplings that evolve during the acquisition delay.
The presence of these couplings is, of course, beneficial for
coherence transfer efficiency but is detrimental to resolution in
the acquisition dimension. In addition to the large one-bond13C homonuclear scalar couplings (1JCCZ35–55 Hz), there are
also a variety of smaller homonuclear scalar couplings, such as2JCC, 3JCC, and 4JCC, that are generally within the resolution of
the experiments. For this reason, we will focus hereafter on the
one-bond scalar coupling constants, i.e. the 1JC 0Ca and the1JCaCb. In the case of experiments with detection of the 13C 0
nuclei, the splitting is quite uniform and leads to doublets
whose lines are separated by about 55 Hz. The same coupling
is responsible for the primary splitting of the Ca carbon signal,
to which the 35 Hz splitting of the Cb-coupling is added. In the
recent literature, several methods have been proposed to
collapse 1JCC splittings from the spectra [21,29,33,34].
Band-selective homodecoupling is the oldest method of
collapsing homonuclear couplings in the direct acquisition
dimension. In principle, the removal of the splitting due to two
coupled spins with BH allows an increase of the S/N of a factor
of about 1.8 (see Section 2), although this is seldom achieved.
Furthermore, even in the case of Ca carbon decoupling while
acquiring C 0, care should be taken in defining experimental
parameters such as hdduty and spectral width in order to avoid
irradiation of the region of interest by sidebands of the band
selective homodecoupling. Besides causing a Bloch-Siegert
shift [35] BH induces decoupling sidebands around the signals
of interest, since it uses composite pulse decoupling with cyclic
schemes [29,36]. The intensity of the induced sidebands
increases with the strength of the decoupling RF field and,
therefore, it is desirable to use the minimum necessary
decoupling power. Changing the phase of the sidebands in a
controlled manner by starting the decoupling sequence at
different times prior to acquisition permits one to cancel out to
a large extemt those that are due to the cyclic scheme [29,36].
An elegant solution for ‘virtual’ decoupling is the use of
spin-state selective schemes, such as in-phase anti-phase
(IPAP) [37–39] and spin-state selective excitation (S3E)
[39,40], widely used for the measurement of heteronuclear J
couplings [41,42], and recently applied for removal of 13C–13C
J-coupling in solid-state samples and in liquids [21,43].
In the IPAP approach (Fig. 5A,B), the removal of the
splitting is accomplished by recording two FIDs for each
increment, one for the anti-phase and one for the in-phase
components. The IPAP scheme relies on complete interconver-
sion between in-phase and anti-phase coherences and thus has
duration of 1/(2JCC), where JCC is the relevant coupling. For C 0
acquisition, the shortest possible duration of the IPAP block is
thus 9 ms. The in-phase and anti-phase components are stored
separately and are combined to yield the two multiplet
components. These are then shifted to the centre of the original
multiplet (by JC 0Ca/2 Hz) and summed to obtain a singlet
[21,44], as shown in Fig. 5C.
With the S3E scheme (Fig. 5D) two different experiments
are acquired and store separately in which one component is
absortive while the other is dispersive and which differ in
the sign of one of the two components [21,45]. The sum and
the difference of the two FIDs stored separately gives the

Fig. 5. The IPAP and S3E approaches for C 0 direct-detection to remove the large Ca–C 0 splitting in the direct acquisition dimension. They are illustrated for the
simple case of 1D experiments, but can be implemented in any experiment based on C 0 direct-detection, as discussed in the text. Band-selective 13C pulses are
denoted by shapes (narrow and wide ones represent 90 and 1808 pulses, respectively). Panels A and B report the two variants of the pulse scheme for the IPAP
approach to acquire C 0 (A for the in-phase and B for the anti-phase components, respectively). The results of the two experiments reported in panels A and B on a set-
up sample of 13C, 15N labeled alanine in D2O are shown in panel C and indicated with IP and AP, respectively. For 13C 90 and 1808 pulses, Q5 (or time reversed Q5)
and Q3 shapes were used [100]. Decoupling of 1H and 15N was applied with waltz-16 [101] and garp-4 [102], respectively. The 13C 0, 13Ca, 1H and 15N pulses were
applied at 175, 55, 4.7 and 118 ppm, respectively. Panel C also shows schematically the approach employed to treat the data. Panel D shows the pulse scheme for the
S3E approach to acquire C 0. The two FIDs necessary to separate the in-phase and anti-phase components can be obtained by changing the phases of the pulses as
indicated below. The data are treated as described in the text. The delay D is 1/(2JCaC 0) (9 ms). The phases are: (A) fIPAPZx,Kx and frecZx,Kx; (B) fIPAPZKy, y
and frecZx,Kx; D) fS3E(1)Z458, 458; f1Zx, y; f2(1)Zx, y; frecZx,Kx and fS3E(2)Z458, 2258; f1Zx, y; f2(2)ZKx,Ky; frecZx,Kx where (1) and (2) are the
two experiments required to separate the in-phase and anti-phase components. It is worth noting that the S3E requires half the time with respect to the IPAP approach.
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 29
absortive and dispersive component, respectively. The
dispersive line in then phase-shifted by 908 and both
components are frequency shifted to the center of the
original multiplet [21,44]. The important difference with the
IPAP approach is that the overall duration of the building
block amounts to 1/(4JCC), where JCC is the relevant
coupling to suppress, and thus is half of that of the IPAP
approach. For C 0 acquisition, the block is 4.5 ms long.
These building blocks (IPAP and S3E) can be implemented in
any experiment based on C 0 direct-detection. In many of them
they can be embedded within the last coherence transfer element
already present in the experiment at no cost in terms of extra
relaxation (i.e. it is not necessary to extend the overall duration
of the experiment). In cases where the coupling to a 15N nucleus
needs to be refocussed (in order to decouple 15N during
acquisition) both building blocks can in principle be used. But
here the use of an S3E element would not shorten the refocussing
delay. So the IPAP approach is the preferred choice, since it is
more robust than S3E with respect to small variations in the
values of 1JCC [46]. Both versions can be implemented in those
experiments that end with the refocusing of the C 0–Ca anti-
phase signals (CACO, CBCACO, CCCO), or those in which the
blocks necessary for spin-state are added at the end of the
experiment prior to acquisition (NOESY, TOCSY). The S3E
approach, due to its shorter duration, may have some advantages
in terms of minimizing relaxation losses at high magnetic fields
and with high molecular mass systems [45].
Along the same lines, Pervushin and coworkers proposed
the acquisition of only the C 0 anti-phase component [47]. The
absolute value of the anti-phase component then looks like an
in-phase component and can be treated in an analogous way to
that described for the IPAP approach (see Fig. 5). A possible
drawback of this method is that the positive and negative
components of different signals in overlap will cancel out
causing loss of information for systems characterized by
extensive resonance overlap. On the other hand, as pointed out
by Dotsch et al., acquiring the anti-phase component allows the
removal of the last delays necessary to refocus it to in-phase
that inevitably causes some relaxation losses [28].
Along the same lines as TROSY, an additional approach to
select one of the two multiplet components in the direct
acquisition dimension of a C 0–Ca correlation experiment
(named COCAINE), has recently been proposed, and provides
an alternative way for ‘virtual’ decoupling of C 0 from Ca [48].
Other methods rely on signal processing algorithms to
deconvolute the C 0–Ca splitting. As an example, deconvolution
using maximum entropy reconstruction has been proposed for
this purpose [33,49]. These processing algorithms can be
applied to spectra where either the in-phase or the anti-phase C 0
signal component [28,33] is acquired.
Direct-detection of Ca is more complicated due to coupling to
C0 and Cb with large one-bond scalar-coupling constants (1JC0Ca,1JCaCb). Therefore, if compared to C0 direct-detection, two large
couplings should be quenched in order to simplify the Ca line and
obtain high-resolution. This can be achieved in principle with the
same approaches discussed for C0, i.e. band-selective homo-
decoupling, spin-state selection and deconvolution.
Pervushin and coworkers proposed triple band-selective
homodecoupling where the three regions irradiated during Ca
acquisition are the C 0 and the two Cb regions [31]. The
performance of this approach is satisfactory in terms of
collapsing the multiplet to a singlet, but the gain in S/N

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4530
achieved is much less than predicted. The loss of signal is
attributed to heteronuclear interference coming from simul-
taneous 15N and 2H decoupling [31].
Spin-state selective methods for Ca, such as the described
IPAP and S3E approaches, are sufficient only for those nuclei that
are singly split (e.g. Ca of Gly), but for the removal of the
additional large one bond coupling to Cb we must rely on a
scheme where the spin-state selection is applied repeatedly [21].
Two IPAP blocks, one specific for the C0–Ca coupling and one for
the Ca–Cb coupling can be combined, as suggested for solid state
applications [39]. Actually, the two IPAP blocks can be
concatenated into a shorter block in which the total duration is
determined by the value of the smaller coupling to consider. Fig. 6
shows the double IPAP implementation (DIPAP hereafter) that
has been proposed for Ca [21]. For each increment, four
experiments are acquired that differ in the selectivity of the Ca
pulse at the centre of the delay that determines the effective
evolution of the Ca–Cb coupling during this block and the
position of the C 0 pulses that determine the effective evolution of
the C0–Ca coupling during this delay. These four experiments,
which yield the four possible Ca multiplets, are stored separately,
combined through linear combinations and then shifted to
the centre of the original multiplet in order to remove the primary
splittings. The total length of the block is determined by 1/
(2JCaCb) and, despite compacting the two IPAP blocks into one,
the duration is rather long (14 ms). However, relaxation rates of
Ca nuclei are drastically reduced upon 2H labelling so that this
approach becomes convenient for large proteins at high magnetic
fields. Implementation of a ‘double’ S3E approach to Ca direct-
detection is also feasible.
The methods discussed for C 0 and Ca acquisition can in
principle be applied also to other carbon nuclei. However, the
diversity of the side-chains of aminoacids is responsible for the
large chemical shift dispersion of Cb nuclei, the partial overlap
IP-IP
IP-AP
A
C
B
D
13C´ 4
4
4
4
4
4
4
4
decoupling.1H/ N15
13C´
decoupling.1H/ N15
13C´
.1H/ N15
13C´
d.1H/ N15
FID
FID
4 4
4
4
φrec
13Cα
13CαφDIPAP
φDIPAPφDIPAP
φDIPAP
∆ ∆ ∆ ∆
∆ ∆ ∆ ∆
13Cα/β
13Cα/β13Cα/β
φrec13Cα
13Cα/β
13Cα
∆+ζ ∆
∆+ζ ∆
Fig. 6. The DIPAP method for Ca direct-detection to remove the two large Ca–C 0 a
simple case of 1D experiments and it can be implemented in any experiment based o
and wide ones represent 90 and 1808 pulses, respectively). The two lines labeled wi
unselective with respect to Cb, or both Ca and Cb spins. For 13C 908 and 1808 pulses,
applied with waltz-16 [101] and garp-4 [102], respectively. The 13C 0, 13Ca, 13Ca/b,
Panels A–D report the four variants of the pulse scheme to acquire and store sepa
respectively. The results of the four experiments performed on the set-up sample of 1
(14 ms) and z is 1/(2JCaC 0) (9 ms). The phases are: fDIPAP(B)Zx,Kx; fDIPAP(A)Z
with other carbon nuclei of the side-chain and the different
coupling topologies. Therefore, a general method of detecting
side-chain nuclei by removing large one-bond carbon–carbon
couplings in a single spectrum is not feasible. Partial solutions can
be employed for specific sets of spins. Multiple band-selective
homodecoupling has been proposed [31]. Also the DIPAP
approach has been implemented in the experiment to correlate Cb
and Cg in aromatic systems [50]. Similar approaches can be
designed for methyl groups, or adapted to specific aminoacid
types, as for example was done with the MUSIC approach [51].
Spin-state selective methods retain the information on the
effective C 0–Ca splitting. The latter contains a contribution
arising from partial orientation of the molecule that provides
precious structural information. These experiments are,
therefore, suitable to accurately measure 13C–13C residual
dipolar couplings, also when protons are not detectable or
absent due to perdeuteration. The possibility of determining13C–13C residual dipolar couplings from fully coupled13C–13C TOCSY spectra has recently been described by
Vogeli et al. [52].
4. A protocol for the assignment of backbone
and side chains
The methods described above to detect selected 13C nuclear
spins whilst collapsing the large one-bond homonuclear
carbon–carbon scalar couplings, as well as the availability of
probeheads with improved sensitivity for 13C [14,53], have
opened the way for the use of 13C direct-detection experiments
in biomolecular NMR. We summarize here a set of
experiments based exclusively on heteronuclei that allows
one to perform a complete sequence specific assignment of a13C,15N labeled protein without using 1H excitation and
detection. These experiments are based on the most efficient
AP-AP
AP-IPE
decoupling
ecoupling
FID
FIDIP-IP
AP-IP
IP-AP
AP-AP
596061 D ( C)13
4 4
4
4
φrec
φrec
δ
∆+ζ–ζ ∆–ζ
∆+ζ–ζ ∆–ζ
nd Ca–Cb splittings in the direct acquisition dimension. It is illustrated for the
n Ca direct-detection. Band selective 13C pulses are denoted by shapes (narrow
th Ca and Ca/b actually indicate band-selective pulses that only affect Ca spins,
Q5 and Q3 shapes were used, respectively [100]. Decoupling of 1H and 15N was1H and 15N pulses were applied at 175, 55, 39, 4.7 and 118 ppm, respectively.
rately the four components indicated with IP–IP, AP–IP, IP–AP and AP-AP,3C, 15N labeled alanine in D2O are shown in panel E. The delay D is 1/(2JCaCb’)
y,Ky; fDIPAP(C)ZKy, y; fDIPAP(D)Zx,Kx; frecZx,Kx.

Table 1
Selection of the protonless NMR experiments available, with the correlations observed in each experiment and an estimate of the number of scans necessary to
acquire them compared to a CACO acquired with N number of scans
Experiment Correlations observed Scans Reference
2D
CACO–IPAP/S3E Cai KC0
i N [58,83]
CBCACO–IPAP/S3E Cbi KC0
i; Cai KC0
iN [21,58]
CCCO–IPAP/S3E Cb;g;d;3i KC0
i; Cai KC0
i2N [58]
CON–IPAP NiKC0iK1 2N [58,80]
CANCO–IPAP Cai KC0
iK1 CaiK1KC0
iK1 16N [26,27]
CBCANCO–IPAP Cai KC0
iK1; CaiK1KC0
iK1; Cbi KC0
iK1; CbiK1KC0
iK116N [58]
3D
CBCACO–IPAP/S3E Cbi KCa
i KC0i; Ca
i KCai KC0
iN [21]
CCCO–IPAP/S3E Cb;g;d;3i KCa
i KC0i; Ca
i KCai KC0
i2N [58]
CANCO–IPAP Cai KN0
iKC0iK1; Ca
iK1KNiKC0iK1 16N [26,27]
CBCACON–IPAP CaiK1KNiKC0
iK1; CbiK1KNiKC0
iK12N [58]
CCCON–IPAP CaiK1KNiKC0
iK1; Cb;g;d;3iK1 KNiKC0
iK14N [58]
CBCANCO–IPAP Cai KNiKC0
iK1 CaiK1KNiKC0
iK1; Cbi KNiKC0
iK1; CbiK1KNiKC0
iK116N [58]
Fig. 7. A schematic representation of sequence specific assignment through
protonless NMR spectroscopy. In each panel, the key correlations necessary to
perform spin system identification and sequence specific assignment are shown.
The inset reports a schematic representation of the backbone of a protein and
the magnetization transfer pathways responsible for the observed correlations.
The experiments based on C 0 acquisition are reported on the left side. The
CBCACO and the CCCO can also be acquired in the 3D mode evolving Ca in
the third dimension in order to remove ambiguities deriving from C 0
degeneracy. The CANCO has been designed as a 3D with N evolution. The
CON experiment is reported in an oblique way to indicate that it can be
combined with the other C 0-based experiments by including N evolution and
obtain the corresponding 3D experiment. The TOCSY experiment with Ca
acquisition is reported on the right.
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 31
building blocks of the conventional triple-resonance experi-
ments [54–57] and are described in detail in the Appendix. In
Table 1 the list of the experiments, including a summary of the
correlations observed and the relative sensitivity, is reported.
This heteronuclear assignment strategy is then critically
discussed in terms of advantages and drawbacks with respect
to the conventional sequence specific assignment strategy and
is compared to other proposed approaches that partly rely on
heteronuclear direct-detection. The exclusively heteronuclear
assignment strategy starts with acquisition and analysis of the
most sensitive experiments based on C 0 acquisition and on the
exploitation of the large one-bond coupling constants for
coherence transfer (JC 0Ca, JCaCb, JCC and JNC 0, Fig. 7).
The CACO experiment [29] yields the intraresidue C0i–Ca
i
correlation (n correlations for a protein ofn aminoacids) as well as
the correlations of carbonyl/carboxylate spins and the attached
carbon of Asp/Asn ðCbi –C
gi Þ, Glu/Gln ðC
gi –Cd
i Þ. The C0i–Ca
i
correlation can be detected through a variety of different schemes,
by acquiring the in-phase or the anti-phase C 0 components [28]
and eventually using signal processing algorithms to deconvolute
the splitting [33,47], by using one of the techniques described in
Section 2 to eliminate the JC0Ca coupling in the direct acquisition
dimension (BH [29], IPAP [21], S3E [21], COCAINE [48]), by
evolving single-quantum or double-quantum coherences (or
both). Some of the different experimental schemes used to detect
the C0i–Ca
i correlation are outlined in Fig. 8.
The next experiment to consider is the CBCACO sequence
[21] (Appendix, panel 1 for the IPAP version and panel 2 for
the S3E version). This exploits the Ca–Cb coupling (JCaCb)
through in COSY-type approach and yields for each residue, in
addition to the C0i–Ca
i , also the C0i–Cb
i correlation. The Cb
chemical shift dispersion, which is much larger than that of Ca,
allows one to reduce the overlap and to identify the aminoacid
on the basis of the Cb chemical shift. The remaining carbons of
the side chains of non-aromatic residues can be identified
through the CCCO experiment [58] (Appendix, panels 3 and 4
for the IPAP and S3E versions, respectively) where C 0 is
correlated, through Ca and by means of an additional TOCSY
step, to the rest of the side chain. By changing the spin-lock
time, the relative intensities of the correlations observed can be
modulated to detect all expected correlations. It is worth noting
that the analysis of these experiments also yields in a
straightforward way the assignment of acidic and amidic
side-chain resonances as each aliphatic carbon is correlated to
the backbone carbonyl and to the side chain carbonyl/
carboxylate nuclei, making the assignment procedure very
simple. When overlap is severe, the CBCACO and the CCCO
experiments can be extended in a third dimension by evolving
the chemical shift of Ca to resolve ambiguities due to C 0
resonance overlap [58]. For aromatic residues, the correlation
with the aromatic ring carbons ðCbi –C
gi Þ can be detected

y
PFG
13C´
y 13C
2 2
decoupling.D1H/15N
2
2
decoupling.
BH
13C
13C´
2-t
2-t
1H15N
A
B
C
D
E
Dec..
BH
FID
FID
PFG
13C´
y
13C 2 2
decoupling.D1H/15N
IP
2
2
AP
13C´
13C 4 44 4
decoupling.
FID
PFG
13C´
13C 2 2
decoupling.D1
1 1
H/15N
4
4
decoupling.FID
PFG
13C´
y
13C 2 2
decoupling.D decoupling.D1H/15N
FID
3
φ1 φ2
φ43φS3E φ φrec
φ3
φ1 φ21α/β
α/β
∆− t1∆− t ∆ ∆
φ1 1 1
φ2
3φ
∆ ∆ φrec
φrec
φIPAP
φ11 1
φ2∆+ t ∆−t
φIPAP
α/β
α/β
∆
∆ ∆
∆∆ ∆
φrec
∆+t ∆− t ∆ ∆
α/β
α/β
φ11 1
φ2∆+t ∆− t
φrecφ
Fig. 8. Some of the different experiments to detect the C 0–Ca correlation: (A)
with multiple quantum evolution during the indirect dimension and
bandselective homodecoupling; (B–D) with single quantum evolution during
the indirect dimension and various approaches to remove the Ca–C 0 splitting
(BH, IPAP, and S3E, respectively); and (E) with detection of the anti-phase
component. Band selective 13C pulses are denoted by shapes (narrow and wide
ones represent 90 and 1808 pulses, respectively). Two different panels indicate
the variants necessary to separate the in-phase and anti-phase components for
the CACO–IPAP experiment. Experiments were tested on different spec-
trometers. Shaped pulses, 1H and 15N decouplings, position of carriers and data
treatment for IPAP and S3E experiments have been described in the caption of
Fig. 5. The delay D is 1/(2JC 0Ca) (9 ms). In (B) and (E) the delay D can be
shortened to reduce relaxation losses with paramagnetic or large systems at the
expense of transfer efficiency. The phases are: (A) f1Zx,Kx; f2Zx; f3Z2(x),
2(Kx); frecZx, 2(Kx), x; (B) f1Zx,Kx; f2Z4(x), 4(y); f3Z2(x), 2(Kx);
frecZx, 2(Kx), x,Kx, 2(i),Kx; (C) f1Zx,Kx; f2Z4(x), 4(y); fIPAP(IP)Z2(x), 2(Kx); fIPAP(AP)Z2(Ky), 2(y); frecZx, 2(Kx), x,Kx, 2(x),Kx; (D)
f1Zx,Kx; f2Z4(x), 4(y); fS3E (1)Z4(458); fS3E (2)Z2(458), 2(2258); f3Z2(x), 2(y); f4Z2(x), 2(y); frecZx, 2(Kx), x,Kx, 2(x),Kx; (E) f1Zx,Kx; f2Z4(x), 4(y); f3Z2(x), 2(Kx); frecZx, 2(Kx), x,Kx, 2(x),Kx.
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4532
through the CGCB–DIPAP experiment that employs the
DIPAP approach for Cb spins to eliminate the splitting due to
coupling with Cg and Ca spins [50] (Appendix, panel 5).
The identification of 13C nuclei within each aminoacid is
thus complete. Correlation with backbone nitrogen nuclei is
performed using the CON-IPAP experiment [21,58] through
the one bond C 0–N coupling (Appendix, panel 6). The
experiment yields the sequential C0i–NiC1 correlation (nK1
correlations for a protein of n aminoacids) and the correlations
for side chains of Asn ðCbi –N
gi Þ and Gln ðC
gi –Nd
i Þ residues. This
experiment is quite sensitive (the most sensitive one involving
N nuclei) and characterized by a very good chemical shift
dispersion for both N and C 0 (Fig. 1). Therefore, it can be
combined with the other most sensitive blocks (CBCACO,
CCCO) to yield 3D experiments (CBCACON-IPAP, CCCON-
IPAP) [58] with C 0 in the direct acquisition dimension, Ca (or
Ca/b, or Ca/b/g.) in the second dimension and N in the third
(Appendix, panels 7 and 8).
With the above mentioned set of experiments, complete
identification of spin-systems can be achieved and each residue
can be correlated with the following backbone amide nitrogen.
However, in order to perform complete sequence-specific
assignment an additional correlation through the backbone is
necessary. This correlation can be detected by using the
CANCO-IPAP experiment [26,58]. In this experiment (Appen-
dix, panel 9) magnetization is transferred from Cai and Ca
iC1 to
NiC1 and then to C0i giving the two correlations (Ca
i NiC1–C0i and
CaiC1–NiC1–C0
i) necessary for sequence specific assignment
(Fig. 7). Actually the experiment is optimised to detect the
CaiC1–NiC1–C0
i peak, which is the ‘sequential one’, with most
sensitivity [26]. Two available variants of the experiment allow
one to discriminate between the two correlations (Appendix,
panel 10, for the most sensitive) [27]. A further variant of the
CANCO-IPAP experiment includes the transfer to the Cb,
allowing the exploitation of Cb chemical shift information as
an aid for spin-system identification (Appendix, panel 11).
Therefore, complete sequence specific assignment using
protonless NMR can be achieved through C 0 direct-detection
[58]. An example of the sequence-specific assignment strategy
employing these experiments is shown in Fig. 9. The 3D
experiments are necessary when dealing with large systems in
order to increase the resolution.
As mentioned in the previous paragraphs, methods used to
collapse the large one-bond couplings in the direct acquisition
dimension for Ca spins allow the design of experiments based
on Ca direct-detection [21]. Indeed the C0i–Ca
i correlation can
also be detected in the COCA experiment employing the
DIPAP approach in the acquisition dimension (Appendix,
panel 12). The Ca-TOCSY [21], a TOCSY experiment where
DIPAP has been implemented prior to acquisition (Appendix,
panel 13), yields the correlations of Cai with all the aliphatic
13C nuclei in the side chain (Ca=b=g.i ), providing an additional
way to assign all 13C spins in non-aromatic aminoacid side-
chains. Sequential correlations can be detected through the
CAN-DIPAP experiment in which each Cai is correlated to the
one and two bonds distant backbone nitrogen nucleus (Cai –Ni,
CaiC1–Ni) (Appendix, panel 14).

Fig. 9. The assignment strategy based only on 13C direct-detection 3D experiments is shown for a fragment Gly 73-Pro 74-Lys 75-Asp 76 in 13C,15N labeled
monomeric SOD. The portions of Ca/ali–C 0 planes of 3D spectra reported in the Fig. are taken from the following experiments recorded at 14.1 T: (A) 3D CANCO–
IPAP, (B) 3D CBCACO–IPAP, (C) 3D CCCO–IPAP, (D) 3D CBCACON–IPAP, (E) 3D CCCON–IPAP. For each residue, the figure shows in panel A the region of
the Ca–C 0 plane of 3D CANCO at the Ni chemical shift (129.2 ppm for Pro 74, 115.8 ppm for Lys 75), in panels B and D a portion of the Cali–C 0 plane of the
CBCACO and CCCO at the Cai chemical shift (63.7 ppm for Pro 74, 54.9 ppm for Lys 75), and in panels C and E the portions of the Cali–C 0 plane of the CBCACON
and CCCON at the NiC1 chemical shift (115.8 ppm for Lys 75, 121.5 ppm for Asp 76). All experiments were acquired using the IPAP approach that allows one to
remove the effect of the large C 0–Ca scalar coupling in the direct dimension.
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 33
The relative performance of experiments based on C 0 and Ca
direct-detection, once the problem of the homonuclear coupling
in the direct acquisition dimension is solved, strongly depends on
the relaxation properties of the system, both in terms of
longitudinal and transverse relaxation and on the chemical shift
dispersion. Indeed, for medium sized proteins at intermediate
fields and for non-2H labelled samples, the C0-based acquisition
experiments are the preferred choice as transverse relaxation rates
of C 0 are still tolerable. Actually these can be the preferred
choice for unfolded systems to exploit the residual chemical
shift dispersion of 13C 0, especially in combination with that of 15N
[59]. At high fields, with 2H labelled proteins, the relaxation
properties of Ca and of aliphatic nuclei in general are favourable
and thus experiments based on Ca detection offer valuable
alternatives. Indeed, by focusing mainly on Ca or on other
aliphatic 13C nuclei, experiments can be designed to minimize
relaxation losses due to C 0 magnetization in the plane.
A combination of the above-mentioned experiments, namely
those based on C 0 and on Ca direct-detection, can also be used to
provide redundant information useful in studying systems
characterized by crowded spectra.
A comment is due about alternative possibilities to the
CANCO-IPAP [58] and CAN-DIPAP experiments used to obtain
the sequential correlations in the protonless NMR sequence
specific assignment strategy. Indeed in both cases described, the
key correlations used to link aminoacid spin systems in a
sequence specific manner are provided by the Ca–N scalar
couplings. However, even if much smaller, there are also
homonuclear C 0–C0 (about 3 Hz) and Ca–Ca (about 2 Hz)
coupling constants that can be exploited for sequence specific
assignment by correlating a C 0 or a Ca of a specific residue with
the previous and with the following one through a spin-lock on a
very narrow bandwidth (C0 or Ca) that thus requires a fairly low
RF power [60].
The exploitation of 13C–13C cross-relaxation rates has been
proposed as an alternative approach to detect correlations
between directly bound 13C nuclei for large molecules using
inverse-detected experiments [61]. With the increase in
molecular mass and with the increase in percentage of 2H
isotopic enrichment, it is actually more convenient to opt for
experiments that start and end on 13C [62]. Indeed, the cross
relaxation rate sCC increases, while the longitudinal relaxation
rates decrease with increasing molecular mass. This makes
dipolar-coupling based transfer competitive with scalar-
coupling based transfer [61].
IPAP and DIPAP versions of the NOESY experiment
have been implemented to increase resolution by removing
complex multiplet structures in C 0 [43] and in Ca regions,
respectively [63] (Appendix, panels 15 and 16). The one-
bond correlations are indeed observed with good sensitivity.
With long NOESY mixing times, two and three bonds
correlations have also been identified and explained by spin-
diffusion [43]. This effect can be exploited as an alternative
approach used to extend the assignment to side-chains in
large macromolecules. The identification of 13C–13C corre-
lations in NOESY spectra between nuclei not directly bound
and not mediated by spin diffusion would mean a break-
through in solution structure determination of large
macromolecules by providing distance constraints. An
extra leap in sensitivity for 13C is required to be able to
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4534
detect truly long-range correlations to obtain 13C–13C
distance constraints.
Summarizing, the protonless NMR strategy in biomole-
cules, that was pioneered by Markley in 1988 [25,64,65], has
been expanded to a large set of multidimentional experiments
characterized by high sensitivity and simplified 13C patterns in
the acquisition dimension thanks to spin-state selective
methods and to homodecoupling.
A hybrid approach, that combines 13C direct-detection with
starting on 1H has been proposed by Dotsch and co-workers
[53]. On the grounds that 13C has several advantages they
proposed to ‘cut’ out-and-back versions of existing triple
resonance experiments into ‘out-and-stay’ versions where the
nuclei used to acquire are either C 0 (as in the example of
HACACO [49]) or aliphatic nuclei (as in the case of HCC
[49]). Improved versions of these experiments were later
proposed by Pervushin and co-workers [66,67]. These
additional experiments can also be added to the conventional
triple-resonance assignment strategy. However, the drawback
is the re-introduction of 1H transverse relaxation in one of the
building blocks of the sequences.
5. Detection of resonances in paramagnetic proteins
Paramagnetic systems are characterized by additional
contributions to chemical shifts and nuclear relaxation
arising from the so-called hyperfine interaction, i.e. the
interaction between the nuclear spin I and the electron spin
S [68]. This interaction occurs via two different mechanisms,
a through-bond interaction, which depends on the amount of
unpaired electron spin-density delocalized onto the investi-
gated nuclear spin (contact interaction) [69,70] and a
through-space interaction [71,72], which depends on the
dipole–dipole interaction between nuclear and electron spins.
Both effects contribute to chemical shifts and nuclear
relaxation. The interplay between the two different contri-
butions and their absolute and relative magnitudes depends
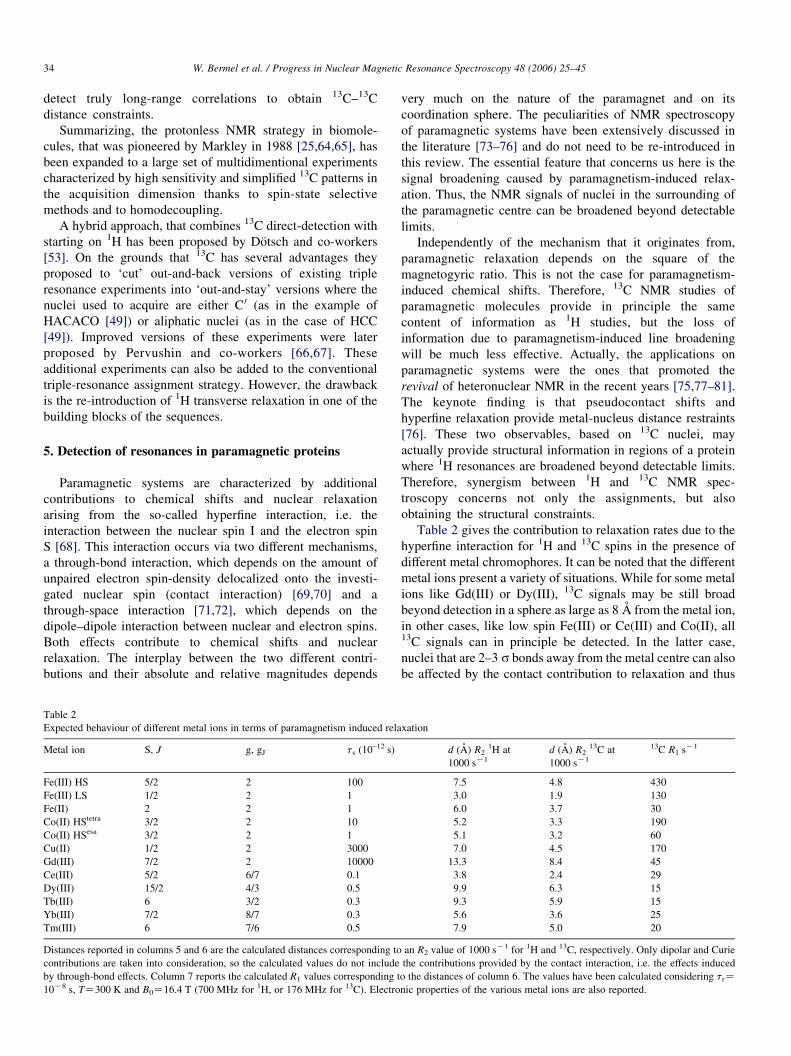
Table 2
Expected behaviour of different metal ions in terms of paramagnetism induced rela
Metal ion S, J g, gJ ts (10–12 s)
Fe(III) HS 5/2 2 100
Fe(III) LS 1/2 2 1
Fe(II) 2 2 1
Co(II) HStetra 3/2 2 10
Co(II) HSesa 3/2 2 1
Cu(II) 1/2 2 3000
Gd(III) 7/2 2 10000
Ce(III) 5/2 6/7 0.1
Dy(III) 15/2 4/3 0.5
Tb(III) 6 3/2 0.3
Yb(III) 7/2 8/7 0.3
Tm(III) 6 7/6 0.5
Distances reported in columns 5 and 6 are the calculated distances corresponding to
contributions are taken into consideration, so the calculated values do not include
by through-bond effects. Column 7 reports the calculated R1 values corresponding t
10K8 s, TZ300 K and B0Z16.4 T (700 MHz for 1H, or 176 MHz for 13C). Electro
very much on the nature of the paramagnet and on its
coordination sphere. The peculiarities of NMR spectroscopy
of paramagnetic systems have been extensively discussed in
the literature [73–76] and do not need to be re-introduced in
this review. The essential feature that concerns us here is the
signal broadening caused by paramagnetism-induced relax-
ation. Thus, the NMR signals of nuclei in the surrounding of
the paramagnetic centre can be broadened beyond detectable
limits.
Independently of the mechanism that it originates from,
paramagnetic relaxation depends on the square of the
magnetogyric ratio. This is not the case for paramagnetism-
induced chemical shifts. Therefore, 13C NMR studies of
paramagnetic molecules provide in principle the same
content of information as 1H studies, but the loss of
information due to paramagnetism-induced line broadening
will be much less effective. Actually, the applications on
paramagnetic systems were the ones that promoted the
revival of heteronuclear NMR in the recent years [75,77–81].
The keynote finding is that pseudocontact shifts and
hyperfine relaxation provide metal-nucleus distance restraints
[76]. These two observables, based on 13C nuclei, may
actually provide structural information in regions of a protein
where 1H resonances are broadened beyond detectable limits.
Therefore, synergism between 1H and 13C NMR spec-
troscopy concerns not only the assignments, but also
obtaining the structural constraints.
Table 2 gives the contribution to relaxation rates due to the
hyperfine interaction for 1H and 13C spins in the presence of
different metal chromophores. It can be noted that the different
metal ions present a variety of situations. While for some metal
ions like Gd(III) or Dy(III), 13C signals may be still broad
beyond detection in a sphere as large as 8 A from the metal ion,
in other cases, like low spin Fe(III) or Ce(III) and Co(II), all13C signals can in principle be detected. In the latter case,
nuclei that are 2–3 s bonds away from the metal centre can also
be affected by the contact contribution to relaxation and thus
xation
d (A) R21H at
1000 sK1
d (A) R213C at
1000 sK1
13C R1 sK1
7.5 4.8 430
3.0 1.9 130
6.0 3.7 30
5.2 3.3 190
5.1 3.2 60
7.0 4.5 170
13.3 8.4 45
3.8 2.4 29
9.9 6.3 15
9.3 5.9 15
5.6 3.6 25
7.9 5.0 20
an R2 value of 1000 sK1 for 1H and 13C, respectively. Only dipolar and Curie
the contributions provided by the contact interaction, i.e. the effects induced
o the distances of column 6. The values have been calculated considering trZnic properties of the various metal ions are also reported.

Fig. 10. (A) CACO–IPAP and (B) CACO–AP spectra of 13C,15N labeled
CaTmCb recorded at 16.4 T. The additional peaks observed in (B) are indicated
by arrows. The CACO–IPAP experiment has been recorded using standard
experimental conditions for the detection of diamagnetic resonances using the
pulse sequence shown in Fig. 8C. The CACO–AP spectrum was acquired using
the pulse sequence reported in Fig. 8E, with Ca–C 0 transfer delays shortened
from 9.0 to 5.5 ms and a recycle period shortened from 1.0 to 0.3 s. Both
spectra were acquired using a 13C-optimized triple-resonance probehead at
16.4 T and 300 K.
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 35
may become undetectable (this contribution is not included in
the calculations as it depends on the molecular structure and on
the electron spin delocalisation and thus is difficult to account
for in a general way [68,75]).
Direct-detection of 13C intrinsically offers a way to detect
resonances close to the metal ion where 1H resonances are too
broad to be detected. However, the 13C-based approach can be
further optimised for paramagnetic systems by selecting the
most efficient coherence transfer pathways and identifying
those that are least affected by fast relaxation. In principle, the
most sensitive experiments are those based on coherence
transfer mechanisms mediated by large scalar couplings;
however, when transverse relaxation is much faster than
longitudinal relaxation, dipolar-based transfers can be usefully
exploited, especially at higher fields.
The 13C–13C COSY-based experiments were the first to be
used for paramagnetic systems [77–79,81,82]. As an alterna-
tive, several other experiments based on coherence transfer via
the C 0–Ca scalar couplings, and also via the smaller N–C 0
scalar coupling, exploiting either the single quantum or
multiple quantum coherence transfer can be used [29,80,81].
Signal losses due to fast relaxation can be reduced by
shortening the coherence transfer delays [29] and even further
by completely removing the building block in which the anti-
phase C 0–Ca coherence is refocused and detecting directly the
anti-phase component [28,83]. Several CACO schemes have
been compared and the most suitable experiment for
paramagnetic systems turned out to be the single quantum
CACO experiment used without refocusing prior to C 0
detection (CACO–AP) [83], outlined in Fig. 8E. Fig. 10
shows the comparison of a standard CACO–IPAP experiment
vs the single-quantum CACO–AP experiment for the Tm(III)
substituted calcium binding protein Calbindin D9k [84].
Several additional peaks were observed in the experiment
which was tailored for paramagnetic signals. This result is due
both to the different pulse sequence used and to the choice of
experimental parameters, such as optimized coherence-transfer
and recycle delays [84].
As pointed out for large molecules, the 13C–13C NOESY
experiment can be useful when the limiting factor consists in
fast transverse relaxation [61]. Indeed in many paramagnetic
systems the longitudinal relaxation rates are influenced to a
smaller extent than the transverse relaxation rates. Table 2,
column 6, reports the longitudinal relaxation rate for a given13C resonance with R2Z1000 sK1. The large variability of the
values in column 6 provides an estimate of the potential interest
in the use of homonuclear 13C–13C NOE-based transfer. For
example, all lanthanides, including Gd(III) but especially the
more far-shifting ones like Dy(III) or Tb(III), have a relatively
small contribution to R1 arising from paramagnetic relaxation.
For the above cases, the use of 13C–13C NOE can be a useful
alternative to overcome the quench of scalar coupling based
transfer. Of course, this will be particularly true on increasing
the size of the molecule, as the magnitude of the NOE effect
depends on tr.
An interesting application of using 13C direct-detection to
get closer to a paramagnetic centre is for those metalloproteins
in which the metal ion is coordinated via carboxylate ligands.
Indeed, for bound Asp or Glu residues, the carboxyl carbon
atom is only two bonds away from the metal centre, while the
closest 1H spin is four bonds away. Therefore, 13C–13C COSY
has been successfully used in lanthanide substituted calcium
binding proteins [78]. Furthermore, the intrinsic asymmetry of
an homonuclear 13C–13C COSY experiment provides a clever
way of identifying the coordinating residues of a paramagnetic
metal ion and to discriminate between monodentate and
bidentate carboxylate ligands [85]. It is the only NMR method
available to distinguish between monodentate and bidentate
coordinating side-chain CO groups. Finally, lanthanide
substituted calcium binding proteins can also be used as
examples to monitor the different performances of various type
of experiments. This is graphically summarized in Fig. 11 for
the Tm(III) substituted derivative of calbindin D9k [84]. The
use of low g nuclei direct-detection provided the identification
of all residues while only about 50% of aminoacids were

Fig. 11. Radii of the spheres within which signals cannot be observed in the case of
CaTmCb. Different spheres are reported for each different set of NMR
experiments. Radii are as follows: 17.5 A for standard 1H detected NMR
experiments, 15 A for 1H experiments optimized to fast relaxing signals, 14 A for
standard 13C detected experiments, 9 A for 13C detected experiments optimized to
fast relaxing signals, 5 A from 13C 1D spectra, 4.2 A for 1D 15N spectra.
W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4536
assigned with standard 1H detected experiments. In this case,15N 1D spectra were also used to identify previously
unobserved resonances. Indeed, the development of 15N
optimized probes, characterized by a 15N selective inner coil,
would certainly contribute to increase the S/N achievable for
this nucleus, thus providing better quality 15N direct-detected
experiments.
6. Conclusions
We have shown that the combination of upgraded
instrumental performances and the design of novel pulse
sequences allows 13C direct-detected protonless NMR spec-
troscopy to be used to conveniently perform the complete13C,15N assignment for proteins. Development of the exclu-
sively heteronuclear NMR experiments summarized in this
review has been stimulated by the need for a different approach
to study paramagnetic systems, for which the main problems
are fast transverse relaxation and difficulties with 1H detection.
These two features, however, are common to large proteins, as
well as to exchanging and partially unfolded systems.
Heteronuclear protonless NMR can thus be useful for making
assignments in a wide range of applications.
When one aims at extending this strategy to protein
structural determination, 13C-based restraints are needed.
The present state of the art allows one to fully exploit chemical
shift values [86] as Ca, Cb, and CO chemical shifts are
available from the present approach. Of course, the wealth of13C data available can be further exploited as a source of 13C-
chemical shift structural restraints.13C–13C residual dipolar couplings can in principle be
accurately determined through IPAP-type experiments and
therefore residual dipolar coupling restraints may be
available. Cross-correlation rates and J-values can also be
determined using 13C direct-detection experiments, providing
some dihedral angle restraints. Selected protonated amino
acids can provide 1H–1H NOEs, which are most valuable
within this strategy [11]. Of course, the measurement of
long-range 13C–13C NOEs would be a significant
breakthrough.
In the case of paramagnetic systems, nuclear relaxation
can be related to metal-nucleus distances [71]. This
approach has already been exploited for 13C nuclei [81].
However, reports are available in the literature, which point
to a break-down in the relaxation–distance relationship for
heteronuclei far from the metal ion [87,88], presumably due
to the occurrence of other phenomena not yet fully
understood. Paramagnetic probes and spin-labels were
recently used to exploit the paramagnetism-derived structural
information in natively diamagnetic proteins [89–97]. The
availability of further data via protonless NMR may provide
additional information at shorter distances than those
obtainable through 1H NMR.
Pseudocontact shifts are also very valuable restraints [30].
Here a problem can be the determination of the diamagnetic
reference to be subtracted from the paramagnetic chemical
shift. One should check that the structural features of the
two systems are absolutely the same, as the diamagnetic
chemical shift of carbon nuclei is highly sensitive to
dihedral angle variations. Another paramagnetic-based
restraint is the cross-correlation between Curie-relaxation
and 13C–13C dipolar relaxation. Further studies are needed to
implement their use. The combined use of paramagnetic and
diamagnetic-based restraints is expected to provide a good
method for determination of the structure of proteins in
solution.
Technological advancements in 13C solid-state spec-
troscopy, where 13C is the nucleus of detection, have been
beneficial to the development of direct-detection of hetero-
nuclei in solution and it is likely that the present development
will have a fall-out in solid-state NMR. Optimization of the
hardware and the development of dedicated cryogenically-
cooled probeheads may further establish heteronuclear pro-
tonless NMR as a routine spectroscopic approach for studying
biomolecules in solution.
Acknowledgements
This work has been supported in part by the EC (Contract
QLG2-CT-2002-00988) and by the Italian Ministero per la
Universita e la Ricerca (COFIN 2003).

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 37
Appendix
The pulse sequences used to acquire the experiments discussed in the review are described in detail here. In particular we
report the experiments based on C 0 direct-detection with IPAP and/or S3E spin state selective methods to remove the effects
of the C 0–Ca coupling in the direct acquisition dimension (CBCACO–IPAP, CBCACO–S3E, CCCO–IPAP, CCCO–S3E,
CBCACON–IPAP, CCCON–IPAP, CON–IPAP, CANCO–IPAP, CANCO–IPAP selective) and the experiments based on Ca
direct-detection with the DIPAP approach to remove the C 0–Ca and Ca–Cb couplings in the direct acquisition dimension
(COCA–DIPAP, Ca–TOCSY–DIPAP, CAN–DIPAP). We also report the NOESY experiment with implementation of spin-
state selective approaches to remove the one bond splittings in the direct acquisition dimension (CACO–NOESY–IPAP and
Ca–NOESY–DIPAP) and the CGCB–DIPAP experiment for connecting the aromatic side-chains to the backbone. These
experiments were tested using different instruments and on several proteins. We report here the parameters used with a 14.1 T
Bruker Avance instrument, equipped with a cryogenically-cooled probehead optimized for 13C sensitivity, on 13C,15N labelled
reduced monomeric superoxide dismutase. When acquiring experiments on a deuterated protein, 2H decoupling should be
applied as indicated for 1H. In all the figures (unless otherwise specified), band selective 13C pulses are denoted by shapes.
For 13C bandselective 908 and 1808 pulses Q5 (or time reversed Q5) and Q3 shapes [100] were used with durations of 320
and 256 ms, respectively except for the 1808 pulse indicated in grey (Q3, 1.0 ms) and for the 1808 pulse indicated in crossed
stripes (adiabatic inversion pulse over the C 0 and Ca regions, smoothed chirp 500 ms, sweep width 60 KHz, 20% smoothing
[103]). The rectangular wide and narrow pulses correspond to 180 and 908 hard pulses. Pulse field gradients (PFG line) are
also indicated by shapes. All the gradients, used for purging and not for coherence selection, have a duration of 1.0 ms and a
sine-shape. The 1H and 15N carriers were placed at 4.7 and 118 ppm, respectively. The change in the position of the 13C
carrier (39 ppm for Cali, 55 ppm for Ca and 173 ppm for C 0) is indicated by vertical arrows. The RF power used for the 13C
FLOPSY16 spin-lock was 10 kHz (applied for durations ranging from 10 to 22 ms in the 2D versions and 22 ms in the 3D
version). Decoupling of 1H and 15N was applied with 2.9 kHz (waltz-16) [101] and 1.0 kHz (garp-4)[102] respectively. For
experiments that employ the IPAP approach to suppress the C 0–Ca coupling, the in-phase (IP) and anti-phase (AP)
components are acquired and stored separately using the pulse schemes illustrated that differ only for the two panels indicated
with IP and AP respectively. For experiments that employ the S3E approach to suppress the effect of the C 0–Ca coupling, the
two components that need to be acquired and stored separately differ by the phase fS3E and by a p increment of phase f4 in
the CCCO-S3E experiment and f5 in the CBCACO-S3E experiment. For experiments that employ the DIPAP approach to
suppress the C 0–Ca and the Ca–Cb couplings, the four variants of the experiment that should be acquired and stored
separately are shown in the four panels indicated with IP-IP, AP-IP, IP-AP, AP-AP respectively. The phase cycle, the method
used for quadrature detection and the durations of the delays shown in the pulse sequences are reported case-by-case.
Panel 1
CBCACO–IPAP: The delays are:DZ9 ms,D1Z8 ms. The phase cycle is:f1Zx,Kx;f2Z8x,8(Kx);f3Z2y,2(Ky);fIPAP(IP)Z4(x),
4(Kx); fIPAP(AP)Z4(Ky),4(y); frecZx,(Kx),(Kx),x,(Kx),x,x,(Kx). Quadrature detection in the F1 and F2 dimensions is obtained by
incrementing f1 and f3 in a States-TPPI manner.
Transfer pathway: F1(Ca/b, t1)/F1(Ca, t2)/F1(C 0,t3)
Correlations observed : Cbi –Ca
i –C 0i ; Ca
i –Cai –C 0
i
Panel 2
CBCACO–S3E; The delays are: DZ9 ms, D1Z8 ms and 3Z4 ms. The phase cycle is: f1Zx,Kx; f2Z8x, 8(Kx); f3Z2y, 2(Ky); f4Z4x, 4(y); f5Z4x, 4(y); fS3E(1)Z4(458), 4(2258); fS3E(2)Z8(458); frecZx, (Kx), (Kx), x, (Kx), x, x, (Kx). Quadrature
detection in the F1 and F2 dimensions is obtained by incrementing f1 and f3 in a States-TPPI manner.

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4538
Transfer pathway: F1(Ca/b, t1)/F1(Ca, t2)/F1(C 0,t3)
Correlations observed : Cbi –Ca
i –C 0i ; Ca
i –Cai –C 0
i
Panel 3
CCCO–IPAP; The delays are: DZ9 ms, 3Zt1(0). The phase cycle is: f1Zx,Kx; f2Z2x, 2(Kx); fIPAP(IP)Z4x, 4(Kx);
fIPAP(AP)Z4(Ky),4y; frecZx, (Kx), (Kx), x, (Kx), x, x, (Kx). Quadrature detection in the F1 and F2 dimensions is obtained by
incrementing f1 and f2, respectively, in a States-TPPI manner.
Transfer pathway: F1(Cali, t1)/F1(Ca, t2)/F1(C 0,t3)
Correlations observed : Calii –Ca
i –C 0i ; Ca
i –Cai –C 0
i
Panel 4
CCCO–S3E: The delays are: DZ9 ms, 31Zt1(0) and 3Z4 ms. The phase cycle is: f1Zx,Kx; f2Z4x, 4(Kx); f3Z2x, 2(y); f4Z2x, 2(y); fS3E(1)Z4(458); fS3E(2)Z2(458), 2(2258); frecZx, (Kx), (Kx), x, (Kx), x, x, (Kx). Quadrature detection in the F1 and F2
dimensions is obtained by incrementing f1 and f2, respectively, in a States-TPPI manner.
Transfer pathway: F1(Cali, t1)/F1(Ca, t2)/F1(C 0, t3)
Correlations observed : Calii –Ca
i –C 0i ; Ca
i –Cai –C 0
i

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 39
Panel 5
CGCB–DIPAP: The experiment to correlate the Cb of aromatic aminoacids with the quaternary Cg carbon that employs
the DIPAP approach to obtain singlets in the direct acquisition dimension. Pulses on Cg and on Cb and Ca/b were centered
at 130, and 33 ppm, respectively. The phase cycle is: f1Zx,Kx; f2Z8(x), 8(y), 8(Kx), 8(Ky); f3Z4(y), 4(Ky); frecZx,
(Kx),(Kx), x,(Kx), x, x,(Kx), (Kx), x, x,(Kx), x,K(x),(Kx), x; fDIPAP(IP-IP)Z2(x), 2(Kx); fDIPAP(AP-IP, IP-AP)Z2(Ky), 2(y); fDIPAP(AP-AP)Z2(Kx), 2(x). Quadrature detection is achieved by incrementing f1 in a States-TPPI manner.1H and 15N decoupling is applied during all the experiment, except during the relaxation delay at 7 ppm in the first part
and at 3.5 ppm in the second part (1H) and at 197 ppm (15N). The duration of the delays is: DZ4 ms, D1Z14.4 ms, dZ9 ms.
Transfer pathway: F1(Cg, t1)/F1(Cb,t2)
Correlations observed : Cgi –Cb
i
Panel 6
CON–IPAP: The delays are: DZ9 ms, D1Z25 ms, 3Zt1(0). The phase cycle is: f1Zx,Kx; f2Z2x, 2(Kx); f3Z4x, 4(Kx);
fIPAP(IP)Zx; fIPAP(AP)ZKy; frecZx, (Kx), x, (Kx), (Kx), x, (Kx), x. Quadrature detection in the F1 dimension is obtained by
incrementing f1 in a States-TPPI manner.
Transfer pathway: F1(C 0)/F3(N, t1)/F1(C 0, t2)
Correlations observed : Ni–C0iK1
Panel 7
CBCACON–IPAP: The delays are: DZ9 ms, D1Z25 ms, D2Z8 ms and 3Zt2(0). The phase cycle is: f1Zx,Kx; f2ZKy;
f3Z2x, 2(Kx); f4Z4x, 4(Kx); f5Z8x, 8(Kx); fIPAP(IP)Zx; fIPAP(AP)ZKy; frecZ2(x, (Kx), (Kx), x), 2((Kx), x, x, (Kx))
. Quadrature detection in the F1 and F2 dimensions is obtained by incrementing f1 and f3, respectively, in a States-TPPI
manner.
Transfer pathway: F1(Ca/b, t1)/F1(Ca)/F1(C 0)/F3(N, t2)/F1(C 0, t3)

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4540
Correlations observed : Cbi –C 0
i–NiC1; Cai –C 0
i–NiC1
Panel 8
CCCON–IPAP: The delays are: DZ9 ms, D1Z25 ms, 31Zt1(0), 3Zt(0). The phase cycle is: f1Zx,Kx; f2Z2x, 2(Kx); f3Z4x, 4(Kx); f4Z8x, 8(Kx); fIPAP(IP)Zx; fIPAP(AP)ZKy; frecZ2(x, (Kx), (Kx), x), 2((Kx), x, x, (Kx)). Quadrature detection in
the F1 and F2 dimensions is obtained by incrementing f1 and f2, respectively, in a States-TPPI manner.
Transfer pathway: F1(Cali, t1)/F1(Ca)/F1(C 0)/F3(N, t2)/F1(C 0, t3)
Correlations observed : Calii –C 0
i–NiC1; Cai –C 0
i–NiC1
Panel 9
CANCO–IPAP: The delays are: DZ9 ms, D1Z25 ms, D2Z32 ms. The phase cycle is: f1Zx,Kx; f2Z8x, 8(Kx); f3Z2x,
2(Kx); fIPAP(IP)Z4x, 4(Kx); fIPAP(AP)Z4(Ky), 4y; frecZx, (Kx), (Kx), x, (Kx), x, x, (Kx). Quadrature detection in the F1
and F2 dimensions is obtained by incrementing f1 and f3, respectively, in a States-TPPI manner.
Transfer pathway: F1(Ca, t1)/F3(N, t2)/F1(C 0, t3)
Correlations observed : Cai –Ni–C
0iK1; Ca
iK1–NaiK1–C 0
iK1

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 41
Panel 10
CANCO–IPAP ‘selective’: The delays are: DZ9 ms, D1Z25 ms, D2Z32 ms and 3Zt2(0). The phase cycle is: f1Zx,Kx; f2Z8x, 8(Kx); f3Z2x, 2(Kx); fIPAP(IP)Z4x, 4(Kx); fIPAP(AP)Z4(y), 4(Ky); frecZx, (Kx), (Kx), x, (Kx), x, x, (Kx). Quadrature detection in the F1 and F2 dimensions is obtained by incrementing f1 and f3, respectively, in a States-TPPI
manner.
Transfer pathway: F1(Ca, t1)/F3(N, t2)/F1(C 0, t3)
Correlations observed : CaiK1–Ni–C
0iK1
Panel 11
CBCANCO–IPAP: The delays are: DZ9 ms, D0Z8 ms, D1Z25 ms, D2Z32 ms. The phase cycle is: f1Zx,Kx; f2Z8x,
8(Kx); f3Z2x,2(Kx); fIPAP(IP)Z4x,4(Kx); fIPAP(AP)Z4(Ky),4y; frecZx,(Kx),(Kx)x,(Kx),x,x,(Kx). Quadrature detection
in the F1 and F2 dimensions is obtained by incrementing f1 and f3, respectively, in a States-TPPI manner.
Transfer pathway: F1(Ca/b, t1)/F3(N, t2)/F1(C 0, t3)
Correlations observed : Cai –Ni–C
0iK1; Ca
iK1–Ni–C0iK1; Cb
i –Ni–C0iK1; Cb
iK1–Ni–C0iK1
Panel 12
COCA-DIPAP: The delays are: DZ9 ms, D1Z14.4 ms. The phase cycle is: f1Zx,Kx; f2Z4x, 4y; fDIPAP(IP-IP)Z2x, 2(Kx);
fDIPAP(AP-IP)ZfDIPAP(IP-AP)Z2(Ky), 2y; fDIPAP(AP-AP)Z2(Kx), 2x; frecZx, (Kx), (Kx), x, (Kx), x, x, (Kx). Quadrature
detection in the F1 dimension is obtained by incrementing f1 in a States-TPPI manner.
Transfer pathway: F1(C0
, t1)/F1(Ca, t3)
Correlations observed : C 0i–C
ai :

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4542
Panel 13
Ca–TOCSY–DIPAP: The delays are: DZ9 ms, D1Z14.4 ms, 3Zt1(0). The phase cycle is: f1Zx,Kx; f2Z4x, 4y; f3Z8x, 8(Kx);
fDIPAP(IP-IP)Z2x, 2(Kx); fDIPAP(IP-AP)Z2(Ky), 2y; fDIPAP(AP-IP)Z2y, 2(Ky); fDIPAP(AP-AP)Z2x, 2(Kx); frecZx, (Kx),
(Kx),x, (Kx),x, x, (Kx), (Kx),x, x, (Kx),x, (Kx), (Kx),x. Quadrature detection in the F1 dimension is obtained by incrementingf1 in a
States-TPPI manner.
The experiment is generally performed with all the pulses before the spin-lock, as well as the spin-lock itself, to cover the
aliphatic region. In this case, the experiment yields all the correlations of Ca with the remaining aliphatic carbon atoms within
the side chain of the aminoacid. However, the experiment can also be performed with all the pulses before the spin-lock, as well as
the spin-lock itself, that are selective over the Ca region and with a much longer duration of the spin-lock. In this case the
experiment yields Ca–Ca sequential correlations.
Transfer pathway: F1(Cali, t1)/F1(Ca, t3)
Correlations observed : Calii –Ca
i
Panel 14
CAN–DIPAP: The delays are: DZ13.6 ms, dZ3.6 ms, xZ2.25 ms, 3Zt1(0). The phase cycle is: f1Zx,Kx; f2Z2x, 2(Kx);
fDIPAP(IP-IP)Z4x, 4(Kx); fDIPAP(AP-IP)Z4(Ky), 4y; fDIPAP(IP-AP)Z4y, 4(Ky); fDIPAP(AP-AP)Z4x, 4(Kx); frecZx,
(Kx), x, (Kx), (Kx), x, (Kx), x. Quadrature detection in the F1 dimension is obtained by incrementing f1 in a States-TPPI
manner.

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 43
Transfer pathway: F1(Ca)/F3(N, t1)/F1(Ca, t2)
Correlations observed : Cai –Ni; Ca
iK1–Ni
Panel 15
CACO–NOESY–IPAP: The delays are: DZ9 ms, 3Zt1(0), tMZmixing time. The phase cycle is: f1Zx,Kx; f2Z4x, 4(Kx);
fIPAP(IP)Z2x, 2(Kx); fIPAP(AP)Z2(Ky), 2y; frecZx, (Kx), (Kx), x, (Kx), x, x, (Kx). Quadrature detection in the F1 dimension
is obtained by incrementing f1 in a States-TPPI manner.
Transfer pathway: F1(Ca, t1)/F1(C0
, t2)
Correlations observed : Cai –C 0
i ; and other correlations arising from spin � diffusion
Panel 16
Ca–NOESY–DIPAP: The delays are: DZ9 ms, D1Z14.4 ms, 3Zt1(0), tMZmixing time. The phase cycle is: f1Zx,Kx; f2Z4x, 4(Kx); fDIPAP(IP-IP)Z2x, 2(Kx); fDIPAP(IP-AP)Z2(Ky), 2y; fDIPAP(AP-IP)Z2y, 2(Ky); fDIPAP(AP-AP)Z2x, 2(Kx);
frecZx, (Kx), (Kx), x, (Kx), x, x, (Kx). Quadrature detection in the F1 dimension is obtained by incrementing f1 in a States-TPPI
manner.
Transfer pathway: F1(Cb/ali, t1)/F1(Ca, t2).
Correlations observed : Cbi –Ca
i and other correlations arising from spin � diffusion

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–4544
References
[1] K. Pervushin, R. Riek, G. Wider, K. Wuthrich, Proc. Natl Acad. Sci. USA
94 (1997) 12366.
[2] K. Pervushin, R. Riek, G. Wider, K. Wuthrich, J. Am. Chem. Soc. 120
(1998) 6394.
[3] V. Tugarinov, P.M. Hwang, J.E. Ollerenshaw, L.E. Kay, J. Am. Chem.
Soc. 125 (2003) 10420.
[4] E. Miclet, D.C. Williams Jr., G.M. Clore, D.L. Bryce, J. Boisbouvier,
A. Bax, J. Am. Chem. Soc. 126 (2004) 10560.
[5] D.M. LeMaster, F.M. Richards, Biochemistry 27 (1988) 142.
[6] M.A. Markus, K.T. Dayie, P. Matsudaira, G. Wagner, J. Magn. Reson.
Ser. B 105 (1994) 192.
[7] M. Sattler, S.W. Fesik, Structure 4 (1996) 1245.
[8] R.A. Venters, R. Thompson, J. Cavanagh, J. Mol. Struct. 602-603 (2002)
275.
[9] C.H. Arrowsmith, Y.S. Wu, Progr. NMR Spectrosc. 32 (1998) 277.
[10] L.Y. Lian, D.A. Middleton, Progr. NMR Spectrosc. 39 (2001) 171.
[11] V. Tugarinov, W.Y. Choy, V.Y. Orekhov, L.E. Kay, Proc. Natl Acad.
Sci. USA 102 (2005) 622.
[12] R.K. Harris, Nuclear Magnetic Resonance Spectroscopy, Longman,
London, 1986.
[13] J.C. Lindon, E. Holmes, J.K. Nicholson, Prog. NMR Spectrosc. 45 (2005)
109.
[14] H. Kovacs, D. Moskau, M. Spraul, Prog. NMR Spectrosc. 46 (2005)
131.
[15] E.R. Zuiderweg, Biochemistry 41 (2002) 1.
[16] S. Grzesiek, J. Anglister, H. Ren, A. Bax, J. Am. Chem. Soc. 115 (1993)
4369.
[17] J. Cavanagh, W.J. Fairbrother, A.G. Palmer III, N.J. Skelton, Protein
NMR Spectroscopy. Principles and Practice, Academic Press, San Diego,
1996.
[18] K.T. Dayie, G. Wagner, J. Am. Chem. Soc. 119 (1997) 7797.
[19] M. Pellecchia, Y. Pang, L. Wang, A.V. Kurochkin, A. Kumar,
E.R.P. Zuiderweg, J. Am. Chem. Soc. 121 (1999) 9165.
[20] L. Zeng, M.W.F. Fischer, E.R.P. Zuiderweg, J. Biomol. NMR 7 (1996)
157.
[21] W. Bermel, I. Bertini, L. Duma, L. Emsley, I.C. Felli, R. Pierattelli,
P. Vasos, Angew. Chem. Int. Ed. 44 (2005) 3089.
[22] M. Salzmann, K. Pervushin, G. Wider, H. Senn, K. Wuthrich, Proc. Natl
Acad. Sci. USA 95 (1998) 13585.
[23] R.R. Ernst, G. Bodenhausen, A. Wokaun, Principles of Nuclear Magnetic
Resonance in One and Two Dimensions, Oxford University Press,
London, 1987.
[24] A. Bax, G.M. Clore, P.C. Driscoll, A.M. Gronenborn, M. Ikura,
L.E. Kay, J. Magn. Reson. 87 (1990) 620.
[25] B.-H. Oh, W.M. Westler, P. Darba, J.L. Markley, Science 240 (1988)
908.
[26] I. Bertini, L. Duma, I.C. Felli, M. Fey, C. Luchinat, R. Pierattelli,
P.R. Vasos, Angew. Chem. Int. Ed. 43 (2004) 2257.
[27] W. Bermel, I. Bertini, I.C. Felli, R. Pierattelli, P.R. Vasos, J. Magn.
Reson. 172 (2005) 324.
[28] N. Shimba, H. Kovacs, A.S. Stern, A.M. Nomura, I. Shimada, J.C. Hoch,
C.S. Craik, V. Dotsch, J. Biomol. NMR 30 (2005) 175.
[29] W. Bermel, I. Bertini, I.C. Felli, R. Kummerle, R. Pierattelli, J. Am.
Chem. Soc. 125 (2003) 16423.
[30] E. Babini, I. Bertini, F. Capozzi, I.C. Felli, M. Lelli, C. Luchinat, J. Am.
Chem. Soc. 126 (2004) 10496.
[31] B. Vogeli, H. Kovacs, K. Pervushin, J. Biomol. NMR 31 (2005) 1.
[32] R. Tycko, A. Pines, J. Guckenheimer, J. Chem. Phys. 83 (1985)
2775.
[33] N. Shimba, A.S. Stern, C.S. Craik, J.C. Hoch, V. Dotsch, J. Am. Chem.
Soc. 125 (2003) 2382.
[34] K. Pervushin, A. Eletsky, J. Biomol. NMR 25 (2003) 147.
[35] F. Bloch, A. Siegert, Phys. Rev. 57 (1940) 522.
[36] J. Weigelt, A. Hammarstroem, W. Bermel, G. Otting, J. Magn. Reson.
Ser. B 110 (1996) 219.
[37] M. Ottiger, F. Delaglio, A. Bax, J. Magn. Reson. 131 (1998) 373.
[38] P. Andersson, J. Weigelt, G. Otting, J. Biomol. NMR 12 (1998) 435.
[39] L. Duma, S. Hediger, A. Lesage, L. Emsley, J. Magn. Reson. 164 (2003)
187.
[40] M.D. Sørensen, A. Meissner, O.W. Sørensen, J. Biomol. NMR 10 (1997)
181.
[41] M. Ottiger, F. Delaglio, J.L. Marquardt, N. Tjandra, A. Bax, J. Magn.
Reson. 134 (1998) 365.
[42] J.H. Prestegard, K.L. Mayer, H. Valafar, G.C. Benison, Methods
Enzymol. 394 (2005) 175.
[43] I. Bertini, I.C. Felli, R. Kummerle, C. Luchinat, R. Pierattelli, J. Biomol.
NMR 30 (2004) 245.
[44] N.C. Nielsen, H. Thøgersen, O.W. Sørensen, J. Am. Chem. Soc. 117
(1995) 11365.
[45] A. Meissner, J.O. Duus, O.W. Sørensen, J. Magn. Reson. 128 (1997)
92.
[46] L. Duma, S. Hediger, B. Brutscher, A. Bockmann, L. Emsley, J. Am.
Chem. Soc. 125 (2003) 11816.
[47] K. Hu, A. Eletsky, K. Pervushin, J. Biomol. NMR 26 (2003) 69.
[48] D. Lee, B. Vogeli, K. Pervushin, J. Biomol. NMR 31 (2005) 273.
[49] Z. Serber, C. Richter, V. Dotsch, ChemBioChem. 2 (2001) 247.
[50] E. Babini, I.C. Felli, M. Lelli, C. Luchinat, R. Pierattelli, J. Biomol.
NMR, 33 (2005) 137.
[51] M. Schubert, D. Labudde, D. Leitner, H. Oschkinat, P. Schmieder,
J. Biomol. NMR 31 (2005) 115.

W. Bermel et al. / Progress in Nuclear Magnetic Resonance Spectroscopy 48 (2006) 25–45 45
[52] B. Vogeli, H. Kovacs, K. Pervushin, J. Am. Chem. Soc. 126 (2004)
2414.
[53] Z. Serber, C. Richter, D. Moskau, J.-M. Boehlen, T. Gerfin, D. Marek,
M. Haeberli, L. Baselgia, F. Laukien, A.S. Stern, J.C. Hoch, V. Dotsch,
J. Am. Chem. Soc. 122 (2000) 3554.
[54] M. Sattler, J. Schleucher, C. Griesinger, Progr. NMR Spectrosc. 34
(1999) 93.
[55] L.E. Kay, M. Ikura, R. Tschudin, A. Bax, J. Magn. Reson. 89 (1990) 496.
[56] S. Grzesiek, A. Bax, J. Magn. Reson. 99 (1992) 201.
[57] S. Grzesiek, A. Bax, J. Am. Chem. Soc. 114 (1992) 6291.
[58] W. Bermel, I. Bertini, I.C. Felli, R. Kummerle, R. Pierattelli, J. Magn.
Reson., in press.
[59] J. Yao, H.J. Dyson, P.E. Wright, FEBS Lett. 419 (1997) 285.
[60] S. Balayssac, B. Jimenez, M. Piccioli, unpublished results.
[61] M.W.F. Fischer, L. Zeng, E.R.P. Zuiderweg, J. Am. Chem. Soc. 118
(1996) 12457.
[62] I. Bertini, I.C. Felli, R. Kummerle, D. Moskau, R. Pierattelli, J. Am.
Chem. Soc. 126 (2004) 464.
[63] W. Bermel, I. Bertini, I.C. Felli, R. Kummerle, R. Pierattelli,
unpublished results.
[64] W.M. Westler, M. Kainosho, H. Nagao, M. Tomonaga, J.L. Markley,
J. Am. Chem. Soc. 110 (1988) 4093.
[65] W.M. Westler, B.J. Stockman, J.L. Markley, J. Am. Chem. Soc. 110
(1988) 6256.
[66] K. Hu, B. Vogeli, K. Pervushin, J. Magn. Reson. 174 (2005) 200.
[67] A. Eletsky, O. Moreira, H. Kovacs, K. Pervushin, J. Biomol. NMR 26
(2003) 167.
[68] I. Bertini, C. Luchinat, G. Parigi, Solution NMR of Paramagnetic
Molecules, Elsevier, Amsterdam, 2001.
[69] E. Fermi, Z. Phys. 60 (1930) 320.
[70] H.M. McConnell, J. Chem. Phys. 25 (1956) 709.
[71] I. Solomon, Phys. Rev. 99 (1955) 559.
[72] N. Bloembergen, E.M. Purcell, P.V. Pound, Phys. Rev. 73 (1948)
679.
[73] D.R. Eaton, R.D. Fischer, C.J. Hawkins, W.D. Horrocks Jr., J.P. Jesson,
R.W. Kreilick, R.J. Kurland, G.N. La Mar, C.H. Langford,
W.D. Phillips, L.H. Pignolet, M.F. Rettig, T.J. Swift, NMR of
Paramagnetic Molecules, Academic Press, New York, 1973.
[74] I. Bertini, C. Luchinat, M. Piccioli, Methods Enzymol. 339 (2001) 314.
[75] T.E. Machonkin, W.M. Westler, J.L. Markley, Inorg. Chem. 44 (2005)
779.
[76] I. Bertini, C. Luchinat, G. Parigi, R. Pierattelli, ChemBioChem. 6 (2005)
1536.
[77] U. Kolczak, J. Salgado, G. Siegal, M. Saraste, G.W. Canters,
Biospectroscopy 5 (1999) S19–S32.
[78] I. Bertini, Y.-M. Lee, C. Luchinat, M. Piccioli, L. Poggi, ChemBio-
Chem. 2 (2001) 550.
[79] T.E. Machonkin, W.M. Westler, J.L. Markley, J. Am. Chem. Soc. 124
(2002) 3204.
[80] M. Kostic, S.S. Pochapsky, T.C. Pochapsky, J. Am. Chem. Soc. 124
(2002) 9054.
[81] F. Arnesano, L. Banci, I. Bertini, I.C. Felli, C. Luchinat,
A.R. Thompsett, J. Am. Chem. Soc. 125 (2003) 7200.
[82] T.E. Machonkin, W.M. Westler, J.L. Markley, J. Am. Chem. Soc. 126
(2004) 5413.
[83] I. Bertini, B. Jimenez, M. Piccioli, J. Magn. Reson. 174 (2005) 125.
[84] S. Balayssac, B. Jimenez, M. Piccioli, J. Biomol. NMR, in press.
[85] I. Bertini, B. Jimenez, M. Piccioli, L. Poggi, J. Am. Chem. Soc. 127
(2005) 12216.
[86] D.S. Wishart, B.D. Sykes, J. Biomol. NMR 4 (1994) 171.
[87] L. Ma, A.M.M. Jørgensen, G.O. Sorensen, J. Ulstrup, J.J. Led, J. Am.
Chem. Soc. 122 (2000) 9473.
[88] S.J. Wilkens, B. Xia, B.F. Volkman, F. Weinhold, J.L. Markley,
W.M. Westler, J. Phys. Chem. B 102 (1998) 8300.
[89] V. Gaponenko, A. Dvoretsky, C. Walsby, B.M. Hoffman, P.R. Rosevear,
Biochemistry 39 (2000) 15217.
[90] L.W. Donaldson, N.R. Skrynnikov, W.-Y. Choy, D.R. Muhandiram,
B. Sarkar, J.D. Forman-Kay, L.E. Kay, J. Am. Chem. Soc. 123 (2001)
9843.
[91] V. Gaponenko, A.S. Altieri, J. Li, R.A. Byrd, J. Biomol. NMR 24 (2002)
143.
[92] J. Wohnert, K.J. Franz, M. Nitz, B. Imperiali, H. Schwalbe, J. Am.
Chem. Soc. 125 (2003) 13338.
[93] V. Gaponenko, S.P. Sarma, A.S. Altieri, D.A. Horita, J. Li, R.A. Byrd,
J. Biomol. NMR 28 (2004) 205.
[94] T. Ikegami, L. Verdier, P. Sakhaii, S. Grimme, P. Pescatore, K. Saxena,
K.M. Fiebig, C. Griesinger, J. Biomol. NMR 29 (2004) 339.
[95] M. Prudencio, J. Rohovec, J.A. Peters, E. Tocheva, M.J. Boulanger,
M.E. Murphy, H.J. Hupkes, W. Koster, A. Impagliazzo, M. Ubbink,
Chem. Eur. J. 5 (2004) 3252.
[96] M.R. Jensen, C. Lauritzen, S.W. Dahl, J. Pedersen, J.J. Led, J. Biomol.
NMR 29 (2004) 175.
[97] G. Pintacuda, A. Moshref, A. Leonchiks, A. Sharipo, G. Otting,
J. Biomol. NMR 29 (2004) 351.
[98] Y. Hiyama, C.H. Niu, J.V. Silverston, A. Bavoso, D.A. Torchia, J. Am.
Chem. Soc. 110 (1988) 2378.
[99] M. Tessari, H. Vis, R. Boelens, R. Kaptein, G.W. Vuister, J. Am. Chem.
Soc. 119 (1997) 8985.
[100] L. Emsley, G. Bodenhausen, J. Magn. Reson. 97 (1992) 135.
[101] A.J. Shaka, J. Keeler, R. Freeman, J. Magn. Reson. 53 (1983) 313.
[102] A.J. Shaka, P.B. Barker, R. Freeman, J. Magn. Reson. 64 (1985) 547.
[103] J.M. Boeheen, G. Bodenhausen, J. Magn. Reson. Ser A 102 (1993)
293.
Related Documents


![13C NMR AND DENSITY FUNCTIONAL THEORY STUDY OF …lfd/Lfz/474/21/Ljp47421.pdf · tion of 13C NMR critical o w [12] is the only appli-cation of 13C NMR to study critical phenomena.](https://static.cupdf.com/doc/110x72/5fa0728b929a19569c69b999/13c-nmr-and-density-functional-theory-study-of-lfdlfz47421-tion-of-13c-nmr.jpg)








