Chemical interactions between additives in foodstuffs: a review M. J. Scotter* and L. Castle Central Science Laboratory, Sand Hutton, York YO41 1LZ, UK (Received 3 March 2003; revised 15 September 2003; accepted 30 September 2003) This paper critically reviews the key literature on food additive–additive chemical interactions published over the last 30 years together with appropriate relevant information on food additive–food component interac- tions. Five main classes of food additive are included, reflecting the research effort to date: the sulfur (IV) species of preservatives, synthetic food colouring mate- rials, nitrate and nitrite, ascorbic acid, and sorbic acid. Within each class, aspects of the chemistry (reac- tivity), functionality, stability, use and reactions with other specific food additives are reviewed. Where appropriate, the importance of interactions of food additives with other components of food (i.e. nutrients and non-nutrients) has been assessed and certain aspects of toxicology included. The practical outcome of this review is presented as a set of recommendations for future research in this area. The use of the data in this review is proposed as a training set to develop the framework into a diagnostic tool. This might be used ultimately for the development of a multilevel frame- work, operating systematically, to understand the important parameters that dictate the outcome of additive interactions. Keywords: food additives, contaminants, chemistry, stability, interactions, research, processing Introduction Food is a heterogeneous mixture of chemicals, both natural and man-made; hence, the science of food and food chemistry is complex. The subject area of food additives is similarly complex and includes both natural and synthetic substances, covering a wide range of chemical entities. The food additive industry is growing rapidly. Consumers have created a demand for processed foods requiring one or more additives as ingredients and low-calorie foods requiring sub- stitutes for (principally) fats and sugars. There are some 2500 chemicals that function as food additives that give rise to some 5000 trade name products on a world-wide basis (Ash and Ash 1995). For regula- tory compliance, not only must each additive have a demonstrated technological need, but also it must be subjected to extensive toxicity testing before approval. This is to establish a level of exposure, i.e. the acceptable daily intake (ADI), which is associated with acceptable risk (see below). The interaction (i.e. target function) of a food additive with food com- ponents, whilst being important for the appearance, nutritional value, taste, and safety of food, may have different implications in vivo. Food safety issues are of increasing concern to con- sumers. For food additives, concern has focussed mainly on adverse reactions to certain additives such as the artificial food colouring tartrazine or the sulfite group of preservatives. The consequences of chemical interactions between food additives permitted for use in the European Union (EU) are the main considera- tion in this review. The use of a threshold of no concern approach (i.e. threshold of regulation, linked to a threshold of no toxicological significance) might permit certain interaction products to be disregarded. For instance, for an additive that constitutes (say) 0.1 mg kg 1 in the diet and reacts to the extent of 1% to form unidentified products, these will be present at only 0.001 mg kg 1 in the diet and may be considered to be below the need for specific identification and toxicological assessment. Moreover, it may be that in a few cases, the nature of the interaction allows the conclusion that the products of an additive–food interaction (or an additive decomposition) was covered by the toxicological tests upon which the ADI for that additive was established. Food Additives and Contaminants, Vol. 21, No. 2 (February 2004), pp. 93–124 *To whom correspondence should be addressed. e-mail: [email protected] Food Additives and Contaminants ISSN 0265–203X print/ISSN 1464–5122 online # 2004 Taylor & Francis Ltd http://www.tandf.co.uk/journals DOI: 10.1080/02652030310001636912

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Chemical interactions between additives in foodstuffs:a review
M. J. Scotter* and L. CastleCentral Science Laboratory, Sand Hutton, York YO41 1LZ, UK
(Received 3 March 2003; revised 15 September 2003; accepted30 September 2003)
This paper critically reviews the key literature on foodadditive–additive chemical interactions published overthe last 30 years together with appropriate relevantinformation on food additive–food component interac-tions. Five main classes of food additive are included,reflecting the research effort to date: the sulfur (IV)species of preservatives, synthetic food colouring mate-rials, nitrate and nitrite, ascorbic acid, and sorbic acid.Within each class, aspects of the chemistry (reac-tivity), functionality, stability, use and reactions withother specific food additives are reviewed. Whereappropriate, the importance of interactions of foodadditives with other components of food (i.e. nutrientsand non-nutrients) has been assessed and certainaspects of toxicology included. The practical outcomeof this review is presented as a set of recommendationsfor future research in this area. The use of the data inthis review is proposed as a training set to develop theframework into a diagnostic tool. This might be usedultimately for the development of a multilevel frame-work, operating systematically, to understand theimportant parameters that dictate the outcome ofadditive interactions.
Keywords: food additives, contaminants, chemistry,stability, interactions, research, processing
Introduction
Food is a heterogeneous mixture of chemicals, bothnatural and man-made; hence, the science of food and
food chemistry is complex. The subject area of foodadditives is similarly complex and includes bothnatural and synthetic substances, covering a widerange of chemical entities. The food additive industryis growing rapidly. Consumers have created a demandfor processed foods requiring one or more additivesas ingredients and low-calorie foods requiring sub-stitutes for (principally) fats and sugars. There aresome 2500 chemicals that function as food additivesthat give rise to some 5000 trade name products ona world-wide basis (Ash and Ash 1995). For regula-tory compliance, not only must each additive havea demonstrated technological need, but also it mustbe subjected to extensive toxicity testing beforeapproval. This is to establish a level of exposure, i.e.the acceptable daily intake (ADI), which is associatedwith acceptable risk (see below). The interaction (i.e.target function) of a food additive with food com-ponents, whilst being important for the appearance,nutritional value, taste, and safety of food, may havedifferent implications in vivo.
Food safety issues are of increasing concern to con-sumers. For food additives, concern has focussedmainly on adverse reactions to certain additives suchas the artificial food colouring tartrazine or the sulfitegroup of preservatives. The consequences of chemicalinteractions between food additives permitted for usein the European Union (EU) are the main considera-tion in this review. The use of a threshold of noconcern approach (i.e. threshold of regulation, linkedto a threshold of no toxicological significance) mightpermit certain interaction products to be disregarded.For instance, for an additive that constitutes (say)0.1mg kg�1 in the diet and reacts to the extent of 1%to form unidentified products, these will be present atonly 0.001mgkg�1 in the diet and may be consideredto be below the need for specific identification andtoxicological assessment. Moreover, it may be that ina few cases, the nature of the interaction allows theconclusion that the products of an additive–foodinteraction (or an additive decomposition) wascovered by the toxicological tests upon which theADI for that additive was established.
Food Additives and Contaminants, Vol. 21, No. 2 (February 2004), pp. 93–124
*To whom correspondence should be addressed.e-mail: [email protected]
Food Additives and Contaminants ISSN 0265–203X print/ISSN 1464–5122 online # 2004 Taylor & Francis Ltdhttp://www.tandf.co.uk/journals
DOI: 10.1080/02652030310001636912

The term ‘interaction’ may be interpreted differently,depending upon the particular scientific point of view.In toxicological terms, for instance, interaction maybe used as a general term to describe an alteredoutcome arising from the presence of two compoundsand could be antagonistic, additive or synergistic. Thework done by Groten et al. (2000), from which suchdescriptions arise, is a comprehensive and detailedanalysis of the possible health implications of suchjoint actions and interactions of food additives inrelation to their potential to increase or decrease therisk of a toxic effect. Risk assessment was based onthe potential for additives to share common sitesand mechanisms of action or common pathways ofelimination. All food additives approved for use inthe EU allocated with numerical ADIs were studied,initially based on reports by Food and AgriculturalOrganization (FAO)–World Health Organization(WHO) Joint Expert Committee for Food Additives(JECFA). In all but a very few cases, the possibility ofjoint actions or interactions could be excluded onscientific grounds. It was concluded that those addi-tives that could not be excluded did not represent asignificant health concern. Toxicokinetic interactionswere considered unlikely because of the low dosagesinvolved, the diverse nature of routes of metabolismand elimination, and the fact that enzyme inductionor inhibition would have influenced selection of theno observable adverse effect level (NOAEL). Manyof those additives that could not be excluded fromshowing joint actions or interactions would have lowintakes; in some cases, they were alternatives forthe same application, thereby lowering the combinedintake.
Over the last 30 years, research in the area of foodadditive–additive interactions has been wide-ranging.Several workers have reviewed specific areas of foodadditive interactions (Wedzicha 1984, 1995, Davıdeket al. 1990, Massey and Lees 1992, Adams andLangley 1995, Adams 1997), whilst others havefocussed on the interactions between additives andother food components, especially nutrients (Cort1987, Vanderveen 1988, Culik and Kellner 1995).
The first clue to an additive’s reactivity is its function.Many additives are intentionally reactive and exerttheir function by an interaction with biologicalmaterial. Examples are the preservatives sulfurdioxide and nitrite, and the phenolic antioxidantsbutylated hydroxyanisole (BHA) and butylatedhydroxytoluene. Their reactivity is well recognizedand evidenced, for example, by disappearance data.
Other additives are incidentally reactive. Examplesare food colours that confer their effect cosmetically,but may incidentally be photosensitive giving reactivespecies. Lastly, other additives can be consideredchemically relatively inert (e.g. gums and bulk sweet-eners), but even these may have important non-specific interactions, for example, by reducing thebioavailability of essential food components. Theboundary between undesirable interactions anddesirable interactions (such as those associated withfunctional foods for example) in many instances isnot clear and therefore merits further work.
Additive–food interactions are more problematicthan additive–additive interactions. This is becauseknowledge of the detailed chemical composition offood is still surprisingly limited, beyond the majorfood constituents and trace nutritional components.A ‘whole food’ approach, whereby not only the toxicfactors present in a food are taken into account, butalso the effects of other beneficial chemicals, suchas nutrients, protective factors and of substancesthat modify toxicity. While this approach does notattempt to provide a complete chemical descriptionof foods, it could be used to assess the balancebetween beneficial and detrimental effects of naturalfood constituents, individually and in combination.Such an approach may prove useful in assessingthe (arithmetical) additive and synergistic combina-tion of effects of all food additives that may bepresent in any given foodstuff. The identificationand quantification of degradation products may alsobe important in discussions of potential toxicity offood additives, as these products may react furtherwith other food components including other addi-tives, to form undesirable compounds.
So, how does one define the term ‘interaction’ withinthe context of food additives? In the simplest case, fortwo additives to interact, it can be presumed to a firstapproximation that encounter at the molecular level— the pre-exponential term A (the frequency factor)in an Arrhenius rate equation is second-order as theproduct of the concentration of the two species underconsideration (Moore 1972). Therefore, two specieseach at low concentration have a low rate of encoun-ter, and vice versa. Within the context of foodadditives and foodstuffs, the most important of thethermodynamic and kinetic factors to consider are(arguably) time, temperature, pH, water activity,oxygen, light and catalysis (including enzymes).
Wedzicha et al. (1993) have discussed the importanceof a correct approach to the design of model systems
94 M. J. Scotter and L. Castle

for additive–food component interactions. They notedthat many factors affect the rates of reaction betweenfood additives and food components and presenteda diverse set of examples to show the need for acritical approach to model system design. The effectsof concentration, ionic strength, non-electrolytes,pH, surfactants and solute transport were addressed.It was concluded that whilst identification of thekinetically significant food components was the mainproblem, they need not necessarily feature in reactionstoichiometry. Furthermore, the solution lies in theprinciples concerned with solute–solute and solute–solvent interactions that could be appropriatelyextended to solute–solid interactions.
This review focuses on five classes of food additive(and their reaction products) that reflect publishedliterature to date:
. Nitrates and nitrites.
. Synthetic food dyes and other food colouringmaterials.
. Sulfur (IV) species.
. Sorbic acid and sorbates.
. Ascorbic acid.
Caramel food colours are chemically heterogeneoushence their chemistry is complex and constitutes alarge subject area, which is covered elsewhere (Finotet al. 1990, O’Brien et al. 1998). For this reason, thisimportant group of food additives and Maillard-typereactions are not described here, except where otheradditives are involved too. Similarly, natural andsynthetic flavourings are also complex substances(and in some cases mixtures) used in foods, but theydo not appear on the EU list of permitted foodadditives and have separate regulations governingtheir use. Where appropriate, the importance of inter-actions of food additives with other components offood (i.e. nutrients and non-nutrients) is discussed, aswell as certain aspects of toxicology.
Nitrates and nitrites
The nitrate and nitrite salts of sodium and potassiumare permitted preservatives that may be added tofoods, such as cured and dried meats, canned meatproducts, certain cheeses, and pickled herring andsprat. Nitrate can be used up to a maximum levelof between 50 and 250mgkg�1, and nitrite between
100 and 175mgkg�1 (EC 1995a). The antibacterialeffect of nitrite and the formation of the so-calledPerigo-type factor (PTF) in certain heat-treated meatproducts has been discussed elsewhere (Christiansenet al. 1973, Skovgaard 1992). Walters (1992) hasreviewed the reactions of nitrate and nitrite infoods, with special reference to the determination ofN-nitroso compounds.
Reactions of nitrate and nitrite withspecific additives
Sorbic acid. The reaction between nitrite ions andsorbic acid analogues is discussed separately and inmore detail below.
Sulfur (IV) oxo-anions (see also below). Nitratereacts with bisulfite to form sulfonates of ammoniaor hydroxylamine (Wedzicha 1984). Such reactionsare reported to destroy the preservative activity of theindividual additives and so they are rarely used incombination (Adams 1997). The destruction of excessnitrite using bisulfite has been suggested in a patentedprocess for curing meats, where the reaction wasreported not to affect the nitrosyl pigments (Colemanet al. 1974).
Lecithin. Lecithin preparations are used commer-cially as emulsifying agents, as dietary supplementsand as release agents. Lecithin is a source of choline,a quaternary ammonium compound that can decom-pose on heating to yield trimethylamine. Demethyla-tion can then produce dimethylamine, which in turncan react with nitrite to form dimethylnitrosamine,a known carcinogen. The formation of dimethyl-nitrosamine occurs on heating sodium nitrite andlecithin at pH 5.6 in a model system (Pensabeneet al. 1975).
Nitroso compounds
The occurrence of nitrosamines in the diet haswarranted much concern over the last 20 years orso, hence most of the data generated on the reactionsof nitrate and nitrite with other food componentshave focused on this area. Within the context of foodtoxicity, the reactions of nitroso compounds arisingfrom the interactions of food constituents with
95Chemical interactions between additives in foodstuffs

food additives have been reviewed (Culik and Kellner1995).
Inhibition of the formation of N-nitrosocompounds. Nitrite scavengers react over a widerange of pHs to produce predominantly nitric oxide,a poor nitrosating reagent (Walters and Taylor 1964).Nitric oxide is oxidized in air to nitrogen dioxide(NO2, in equilibrium with dinitrogen tetroxide,N2O4), which reacts readily with secondary aminesto form N-nitrosamines. However, the oxidation ofnitric oxide in air is trimolecular and is very slowat low concentrations. Thus, the inhibitory effect ofascorbic acid on the conversion of amines, amides,etc. into their N-nitroso derivatives is markedlydependent on the products of its reaction with nitriteunder the prevailing conditions. Where quantities oflipid are present, ascorbyl palmitate is particularlyuseful in inhibiting N-nitrosation.
Ascorbic acid and tocopherols inhibit nitrosationeffectively at concentrations of 500–1000mgkg�1
(ascorbic acid) and 100–500mgkg�1 (tocopherols).Mixtures of ascorbic acid and tocopherols have asynergistic effect. The inhibiting influence of ascorbicacid has been discussed in detail (Douglass et al. 1978)and the inhibiting effect of tocopherols on nitrosationhas been reviewed (Lathia and Blum 1989). Otherinhibitors include sulfur dioxide, sulfamate, cysteineand glutathione (Gray and Dugan 1975). The ammo-nium ion, hydroxylamine and retinol have beenreported as nitrosation inhibitors (Douglass et al.1978). The nitrosation of N-alkylureas can also beinhibited strongly by lowering the dielectric constantof the reaction medium (Garcia-Prieto et al. 1998).
Wedzicha and Wei (1989) postulated that 3-deoxy-hexosulose, and compounds derived from it, areimportant intermediates in the formation of colourin the non-enzymic Maillard browning reaction. Thereaction kinetics were consistent with C-nitrosation(equation 1):
Hþ þNO�2 þDHþ CH3COOH
ÐDH�NOþþ CH3COO�
�H2O: ð1Þ
Nitroso compound formation from phenoliccompounds. The formation of C-nitroso productsfrom phenolic compounds has been reported byKnowles et al. (1974, 1975) and Douglass et al.(1978), as has the role of phenols in the catalysisof nitrosamine formation from secondary amines(figure 1) (Walker et al. 1982).
Davies et al. (1978) found that the smoking of meatsprepared using nitrite can deposit up to 300mgkg�1
phenols in the matrix. This type of contaminant can beof great importance in relation to nitrite breakdown.The initial product of the nitrosation of phenol isa C-nitrosophenol, but oxidation of this to C-nitro-phenol can take place readily. Phenols thus playan interesting role, since they can inhibit or enhancethe formation of nitrosamines, depending uponthe relative concentrations of nitrite and phenol.Alcohols in high concentration inhibit the formationof N-nitrosodimethylamine and N-nitrosodiethyla-mine from sodium nitrite at pH 3.0, but enhance thereaction at pH 5.0 (Kurechi et al. 1980).
The reaction between nitrite and ferulic acid (anabundant plant phenolic cinnamic acid) under acidicconditions, gives complex mixtures of products(Rousseau and Rosazza 1998), including vanillin,2-methoxy-4,6-dinitrophenol and a new benzoxazin-4-one. It was suggested that the nature of theseproducts might give a basis to the role of ferulic acidas an effective antioxidant or chemoprotective agentin the quenching of nitrosating species.
Kalus et al. (1990) isolated and characterized someproducts of the reaction between BHA and nitrite andexamined their mutagenicity. Among the productsseparable by thin-layer chromatography (TLC) from
96 M. J. Scotter and L. Castle
O
NOH
+ HONO
O
NONO
+ H2O
O
NOH
+
OH
+ HNO2
O
N
H
+ H2O
O
O
O
O
O
H
H
+ 2 HNO2 + 2 NO + 2 H2O
(C2H5 NH2)
(C2H5) NNO2
Figure 1. Examples of the role of phenols in the forma-tion of nitrosamines (Walker et al. 1982).

the reaction in aerobic aqueous solution, 1-hydroxyl-2-tert-butyl-4-methoxy-6-nitrobenzene (BHA-NO2),tert-butyl substituted p-quinone and 3-tert-butyl-5-methoxy-1,2-benzoquinone (t-Bu-O-Q) were domi-nant (figure 2). One further compound with nitrosocharacter was also isolated along with traces oftert-butylhydroquinone. No evidence of mutagenicityin the Ames test was found and some of the productswere unstable. The authors concluded that onlya one-electron reduction and/or a reaction withamino acids may be involved in the cytotoxicity ofthe quinones, and that BHA appeared to exert aprotective effect against nitrosating agents in themedia containing it.
Reaction with thiols. Thiols such as glutathioneare readily converted by nitrite into their S-nitrosoderivatives (see below), although most such com-pounds are relatively unstable. In the presence ofsources of Fe and nitrite, thiols can give rise to ironthionitrosyls, such as the salts of Roussin, which exerta bactericidal effect.
Reaction with haems. The primary action of nitritein relation to the haem proteins, myoglobin andhaemoglobin in preserved meats is to oxidize thenatural oxygenated ferrous to the ferric met- forms,whilst itself being oxidized to nitrate. Subsequently,reduction occurs both of nitrite to nitric oxide and offerric met-haem to the ferrous forms of haem protein,nitrosylmyoglobin and nitrosohaemoglobin. Thesecompounds give cured meat its characteristic pinkcolour. These reductions can be effected by residualrespiratory enzymes in uncooked meat products andby sulfhydryl systems in heat-processed products
(Walters and Taylor 1964). Lee and Cassens (1976)have demonstrated that the nitrosylhaemochromeobtained on heating contains two nitric oxide moie-ties coordinated to the iron atom.
Reaction with amines. Primary amines, such ascertain amino acids, react with nitrous acid, theprincipal products being the corresponding hydroxycompounds. Secondary amines are converted intotheir N-nitroso derivatives, along with those fromsubstituted amides and guanidines, etc. (Walters1992). Walters also reported that despite manyreports to the contrary, nitrous acid reacts withtertiary amines and even with quaternary ammoniumcompounds. The principal products formed are acarbonyl compound, resulting from oxidation of asubstituent group, and the N-nitroso derivative of thesecondary amine arising from its cleavage. An exam-ple of this is the reaction between the alkaloidhordenine and oxides of nitrogen during the directfired drying of malt, which is considered to beresponsible for the formation of N-nitrosodimethyl-amine. Nitrite may undergo a number of reactionswith various components of food matrices, includingC-, S-, O- and N-nitrosation reactions. Isotope (15N)labelling studies have indicated that the majority ofnitrite nitrogen is still present in cured meat, but nolonger as nitrite (Massey and Lees 1992).
Reaction with thiocyanates. Thiocyanates are widelydistributed in vegetables of the Brassica type andare known to catalyse the formation of N-nitrosocompounds. Catalytic activity is greatest at low pH,such as that found in the human stomach (Boylandand Walker 1974).
Synthetic food dyes
The most common artificial food colours are azo dyesthat include mono-, dis- and tris-azo types. Triphe-nylmethyl (triarylmethyl) compounds, as well asa small number of dyes based on quinoline, xantheneand indigoid structures, constitute the remainder.Table 1 shows the EU maximum permitted levels ofusage for those synthetic dyes with numerical ADIs.
All of the synthetic colouring materials permitted forfood use in the EU and USA (except lithol rubine BKand citrus red, which have restricted use, and the lakecolours) are soluble in water to a greater or lesser
97Chemical interactions between additives in foodstuffs
O
O
N
CH3
H
O2
O
O
+
O
O
OCH3
NO+
N O2 3or
BHA t-BuQ
BHA-NO2
O
OCH3
H
NO2
t-Bu-O-Q
Figure 2. Dominant products from the reaction betweenBHA and nitrite (Kalus et al. 1990).

extent and insoluble in oils and fats. The chemistryand stability of synthetic food dyes has been coveredin other texts (Scotter 2003), but many factorscan and do contribute to colorant stability, such asheat, light, pH, redox systems, other food ingredients(especially preservatives) and trace metals. Rosenthalet al. (1988) screened 43 certified US Food, Drug andCosmetic (FD&C) colours for photosensitizing acti-vity using photogeneration of singlet oxygen (1(O2))as an index. Only xanthine-type dyes substituted withBr or I (e.g. erythrosine) (figure 3 (I)) generated 1(O2).This was monitored by oxidation of 2,2,6,6-tetra-methyl-4-piperidone (figure 3 (II)) to the corres-ponding N-oxide radical (figure 3 (III)) measured byelectron spin resonance (ESR) spectrometry.The formation of reactive species of oxygen suchas 1(O2) implied potential for dye phototoxicity asa result of photodynamic activity, but this wasconsidered a low risk due to the deactivation of suchspecies in food matrices, especially those whichcontain antioxidants.
Indigo carmine showed mutagenic activity andgrowth inhibition of Bacillus subtilis following ultra-violet light (UV) irradiation (Ozaki et al. 1998).Mutagenic activity reached a maximum after 6 daysof irradiation and DNA-damaging activity after14 days. No toxicity or mutagenicity was observedafter 30 days, indicating that the photodegradation
products became inactive on further degradation.Six intermediate products of photodegradation wereobserved after 6 days using high-performance liquidchromatography (HPLC) analysis, which was redu-ced to 5 after 14 days. No diagnostic peaks couldbe characterized. Superoxide radicals and hydrogenperoxide were postulated as intermediates in thephoto-induced generation of hydroxy radicals undercertain conditions (Ishimitsu et al. 1995, Ozaki et al.1998). Heat can cause loss of colour during foodprocessing and cooking. For example, during thethermal degradation of indigo carmine (Taru andTakoka 1982), the heterocyclic ring was decomposedat 340–470�C, leading to loss of colour.
The majority of colour additives are unstable incombination with oxidizing and reducing agents.Since colour depends on the existence of a conjugatedunsaturated system within the dye molecule, anysubstance which modifies this system (e.g. oxidizingor reducing agents, sugars, acids, and salts) will affectthe colour. All dyes, those with an azo group inparticular, will exhibit accelerated fading under bothacid and alkaline conditions in the presence of metals,including zinc, tin, aluminium, iron and copper,especially at higher temperatures. This is mostly dueto the reducing effect of liberated hydrogen. Dyes willoften react with the metal in food cans.
98 M. J. Scotter and L. Castle
II
II
H
21[O ]
.
(I)
(II) (III)
OO O
N
O
N
O
O
COO
Figure 3. Food colour erythrosine (I), a xanthine-typedye, and the scheme for oxidation of 2,2,6,6-tetramethyl-4-piperidone (II) to the corresponding N-oxide radical(III) (Rosenthal et al. 1988).
Table 1. EU-permitted synthetic foodcolouringmaterialswith numerical ADIs.
ColourADI
(mgkg�1 bw day�1)Maximum permitted
level (mg kg�1)
Allura red AC 7 500a
Amaranth 0.8 100Black PN 5 500a
Brilliant blue FCF 10 500a
Brown FK 0.15 20b
Brown HT 3 500a
Carmoisine 4 500a
Green S 5 500a
Indigo carmine 5 500a
Patent blue V 15 500a
Ponceau 4R 4 500a
Quinoline yellow 10 500a
Sunset yellow FCF 2.5 500a
Tartrazine 7.5 500a
aUse at this level in sauces, seasonings, salmon substitutes, surimi,decorations and coatings; main range of application 50–200mgkg�1
and Quantum satis permitted in, for example, edible casings;buse restricted to kippers.

Interaction of synthetic food dyes with otherfood additives
Preservatives. The added colour in canned productsmay degrade in the presence of tartaric and citricacids, because the acids can react with the metal ofthe container to liberate hydrogen. A study of ninered colours in comminuted meat products foundthat most of the dyes were destroyed to some extent,but, with nitrite added, more of the colour survived(Knowles et al. 1974). Subsidiary dye componentsand colourless fluorescent products were formed asa result of heat processing and in certain casesadditional products were observed in the presence ofnitrite. Nitrite can also cause rapid de-tinning toproduce Sn2þ, a strong reducing agent. Sulfur dioxideis known to cause rapid decolourization of dyesolutions (see below).
Model food systems and foods. McCormick (1971)studied the degradation of allura red AC in 10%solutions of acetic acid, citric acid, malic acid andtartaric acid, as well as solutions of sucrose, glucose,SO2, sodium benzoate and buffers, ranging from pH 3to 8. No colour fading was detected, but increasesin the blue hue were observed in 10% solutions ofvarious alkalis after 1 week. The fading of colour attemperatures above 135�C has been reported (StangeCo. 1970, Lancaster and Lawrence 1986). Severalfactors have been listed as possible causes, includingexcessive heat and reaction with protein, reducingions and reducing sugars (Krukar 1980). When heatedin aqueous solution in the presence of fructose,amaranth decomposes slowly (Ross 1975, Bibeauand Clydesdale 1978a), whereas sunset yellow FCFis reported to be much more resistant to fructose-mediated decomposition (Bibeau and Clydesdale1978b).
Synthetic azo colour additives are often used in foodsprocessed at 100–170�C or even higher, especiallysugar-based confectionery commodities, whichaccount for a significant proportion of the consump-tion of azo food colours (MAFF 1987). Within thistemperature range, reducing sugars are degraded tohighly reactive species (Fleming et al. 1968, Ross1975), which are capable of reducing azo dyes,demonstrated by heating aqueous solutions underreflux for several hours. Amino R-salt (1-amino-2-hydroxynaphthalene-2,7-disulfonic acid) is a reactivedegradation product of amaranth and has been thesubject of several studies. Singh (1970) reported
that amino R-salt completely decomposed whenheated with an aqueous mixture of sucrose or dex-trose. Baking soda was also reported to accelerate thedecomposition. In a separate study, the compoundwas degraded by 60% when heated at 103�C for15min in a 10% sucrose solution. In a candy mixture,it was reported to degrade faster than naphthionicacid (4-aminonaphthalene-1-sulfonic acid).
Simulated candy making at 120–162�C showed azodye degradation by 10–25%. Amaranth degraded tonaphthionic acid and amino R-salt, whereas tartra-zine and sunset yellow FCF showed no increasein intermediates (Lancaster and Lawrence 1986).No significant increases in the levels of intermediates(above regulatory limits) were found in commercialcandies. The same authors also reported that reactiveintermediate compounds arising from the manu-facture of food colouring materials may be presentat low levels (<1%) in food colouring preparationsand thus may also be affected by heat during product-ion of confectionery commodities. Candies containingthe amaranth intermediates naphthionic acid andR-salt at levels of 4–19mgkg�1 in the absence of foodcolour were prepared at 162�C. Naphthionic acid wasreported to decompose by an average of 40% whilstR-salt degraded by about 20%. Similar studies withsunset yellow FCF at 120–162�C using Schaeffers saltshowed no significant decomposition whereas sulfa-nilic acid (4-aminobenzene-1-sulfonic acid) showed46% decomposition at 126�C. The tartrazine inter-mediate pyrazolone-T (PyT or 1-(4-sulfophenyl)-3-carboxy-5-hydroxypyrazolone) was shown to degradesubstantially (80–90%) over the temperature range120–162�C.
The stability of azo dyes in food systems dependsupon a number of parameters, the most importantof which is related to the presence of reducingspecies. These result in the cleavage of the azo doublebond, ultimately to form primary amines such asaniline, sulfanilic acid and naphthionic acid. Theseproducts can degrade further to ammonia (Nurstenand Williams 1969, Fogg and Summan 1983, 1984)(see below). The current EU regulations limit theamounts of unsulfonated aromatic amines allowed to0.01% (EC 1995b), so the degradation of azo dyescould produce a significant contribution to this risk.
Nursten and Williams (1969) reported that carmoi-sine decolourized more rapidly at pH 7 than at pH 3at 121�C in the presence of ascorbic acid (AA).Ponceau 4R, however, was stable at this pH. Theresults suggested that the sulfonate group peri to
99Chemical interactions between additives in foodstuffs

the azo bond was the source of the increased stabi-lity of ponceau 4R. The disazo dye black PN wasalso found to be particularly unstable in thepresence of AA. This was attributed to the reductivecleavage of both azo groups to yield 1,4-diamino-naphthalene-6-sulfonic acid, a compound that hadpreviously been shown to be capable of catalysingthe breakdown of the original dye (after Eisenbrandtand Lang 1966). AA-mediated degradation ofazo dyes was reduced by the presence of ethylene-diaminetetracetic acid (EDTA), but enhanced bytungsten light at pH 5.5 (Fogg and Summan 1983,1984).
A major source of synthetic dyes in the diet is fromsoft drinks, to which AA is added as an antioxidant/vitamin supplement. This provides a chemically redu-cing environment in which azo dyes have the potentialto be degraded. Marovatsanga and Macrae (1987)studied the stability of amaranth under a range ofconditions similar to those found in soft drinks. Thedye was reduced under reference conditions toprovide authentic degradation products as referencesubstances. Soft drink model solutions containingamaranth were made in citrate buffer preserved withsodium benzoate and analysed for dye degradationin the presence of AA during storage. The effects ofother components such as sugar were investigatedand also the effect of removing oxygen by flushingwith nitrogen. These findings were rationalizedin terms of the amount of AA available. Sugars havebeen reported as having a protective effect on AAin solution by virtue of their ability to chelate metalions, which would otherwise catalyse AA oxidation.AA is also oxidized by atmospheric oxygen and,in this instance, particularly by dissolved oxygen(Birch and Parker 1974, Birch and Pepper 1983).In a study on the fate of black PN used to stainmeat (Scotter 1983), TLC was used to identify degra-dation products both with and without autoclavingof the dyed meat samples. The expected productsof azo-bond cleavage, sulfanilic acid, 1,7-cleve’s acid,and acetyl-K acid (4-acetamido-5-hydroxynaphtha-lene-1,7-disulfonic acid), were identified along withcoloured monoazo dyes, corresponding to the resi-dual coupled products sulfanilic acid ! 1,7-cleve’sacid and 1,7-cleve’s acid ! acetyl-K-acid (figure 4).The deacetylated analogues were also identified in theautoclaved samples.
Indigo carmine is known to be unstable in solution toheat, acids and alkalis (Nursten and Williams 1969).Investigations into the losses incurred during indigo
carmine analysis of boiled sweets revealed thatoxygen was essential for photochemical degradationof the dye to sodium 2,3-dioxo-5-indolinesulfonate(Boley et al. 1981). Whilst this is not an additive–additive interaction per se (figure 5), it could bepostulated that mediation by other oxidizing agentscould occur and any products such as indolinederivatives formed as a result may be reactive towardsother food additives.
Sulfur (IV ) species. The stability of mono-azo dyesin the presence of S(IV) oxo-species is reported to bevariable (Wedzicha 1984). Reduction to hydrazoproducts would be expected to be reversible, butthat to the amino compound would not. Irreversibleformation of the amine is proposed if one of the
100 M. J. Scotter and L. Castle
O2hv
HN
O
O
(II)
(I)
N
O
N
ONaO3
S S 3
HH
NaO3S
O Na
Figure 5. Degradation of indigo carmine (I) to sodium2,3-dioxo-5-indolinesulfonate (II) (Boley et al. 1981).
NN
O N
NN
NaO3S
SO3Na
SO3Na
H HCOCH 3
ab
c
SO3Na
Figure 4. Structure of black PN showing the points of azocleavage (a, b) and deacetylation (c) (Scotter 1983).

OH- groups present in the aromatic ring is in the2-position. Where the OH- group occurs in the4-position, relatively stable hydrazo compoundsmay be formed. Carmoisine is reduced by S(IV)oxo-anions with the transfer of two electrons thusfacilitating the conversion of S(IV) to sulfate.
The stabilities of the triarylmethane dyes permitted infoods (green S, brilliant blue FCF and patent blue V)have not been studied in any depth. However, thetriarymethyl cation may be decolourized by combina-tion with sulfite ion, e.g. in the use of pararosanilineas a Schiff reagent for the histochemical determina-tion of S(IV) oxoanions. Malachite green and crystalviolet are triarylmethane dyes that are not permittedfor use in foodstuffs, but their reactivities towardsS(IV) oxoanions have been studied. At the pH ofacetic acid/acetate buffer (3.7–5.7), both dyes reactwith bisulfite to give colourless sulfonates (Wedzicha1984):
ðArÞ3Cþ þHSO�3 Ð ðArÞ3C�SO�
3 þHþ
The stability of permitted artificial food colouringagents with respect to S(IV) oxospecies is shown intable 2.
Wedzicha and Rumbelow (1981) investigated thekinetics and mechanisms of the interaction betweensulfite species and carmoisine. The kinetic data
suggested a 1:1 reaction of sulfite with the azocompound to form a complex and it was suggestedthat a hydrazo compound could then be formed viahydrolysis. Saxby and Stephen (1981) reported thatsunset yellow in the presence of SO2 forms a second-ary dye. Further work by Damant et al. (1989) andDamant (1990) using nuclear magnetic resonance(spectroscopy) (NMR) and fast atom bombardment-mass spectrometry (FAB-MS) revealed the structureof this secondary dye to be 1-(40-sulfo-1-phenylhy-drazo)-2-keto-3,3,4-trihydronaphthalene-4,6-disulfo-nic acid and elucidated the mechanism of formation.Because these dyes exist predominantly as hydrazonetautomers, the addition of bisulfite to the paraposition is facilitated, the observed secondary rateconstants being proportional to pH and hence bisul-fite availability.
Studies of interactions between amaranth and SO2
were also undertaken, where amaranth was found todegrade to an orange-yellow substance which couldnot be identified due to its instability. Adams andLangley (1995) repeated aspects of this work in theirstudies on the reactivity of six food colours selectedon the basis of their frequent use in combination withsulfite, ascorbate and nitrite, and reported similarresults, but were unable to identify the structureof the secondary dyes. Kinetic studies of the foodcolours, amaranth, black PN, ponceau 4R, sunsetyellow FCF and tartrazine, were carried out todetermine the conditions necessary to obtain degra-dation products that could be identified by massspectrometry.
The rate of degradation of sunset yellow FCF at pH 3in the presence of AA and bisulfite to a lemon yellowcompound was found to be proportional to bisulfiteconcentration and temperature (Damant et al. 1989,Damant 1990). In the absence of bisulfite, AA causedoxidative degradation of sunset yellow FCF.Conversely, black PN, tartrazine and carmoisinedegraded without an observed shift in their corre-sponding lmax values. Disazo and other morecomplex azo dyes are reported to have fair to goodstability to sulfur dioxide, but the degradation prod-ucts do not appear to have been studied in depth.The most likely products of degradation in thiscase are thought to be sulfonate derivatives. Indigocarmine has poor stability to SO2; even so, theproducts are unknown. Quinoline yellow, the onlypermitted quinoline food colouring material, isreported to have excellent stability to SO2 (Adamsand Langley 1995).
101Chemical interactions between additives in foodstuffs
Table 2. Reactivity of artificial food colour additivestowards S(IV) oxospecies (Wedzicha 1984).
Common nameChemical
classStability towards
S(IV)
Tartrazine monoazo excellentQuinoline yellow quinoline excellentSunset yellow FCF monoazo fairCarmoisine monoazo fairAmaranth monoazo fairPonceau 4R monoazo fairErythrosine xanthene goodRed 2G monoazo goodAllura red AC monoazo not listedPatent blue V triarylmethane not listedIndigo carmine indigoid poorBrilliant blue FCF triarylmethane not listedGreen S triarylmethane goodBlack PN disazo fairBrown FK azoa goodBrown HT disazo fair
aMixture of six main components; only used for the colouring ofkippers.

Other food colours
Within the EU, only the natural colouring materialsannatto, carmines, carotenes and the Cu complexesof chlorophyll have been allocated numerical ADIs(0.065, 5, 5 and 15mgkg�1 bwday�1, respectively).Annatto is permitted for use in a restricted range offoodstuffs up to 50mgkg�1, whereas the carmines andcarotenes can be used up to a level of 500mgkg�1.The Cu complexes of chlorophylls may be usedquantum satis.
Anthocyanins
The chemistry of anthocycanins is complex andseveral detailed reviews have been published(Markakis 1982, Jackman et al. 1987, Harborne et al.1988, Francis 1989, Hendry and Haughton 1992,Mazza 1997). Anthocyanins are derivatives of 2-phe-nylbenzopyrylium salts with the substitution patternsuch as those shown in figure 6(a).
Where these substances occur in fruit juices and otherplant cell materials, and as purified food colouringmaterials in foodstuffs, they will interact readily withother naturally occurring materials, such as flavonols.Such interactions depend upon intermolecular asso-ciation through, for instance, hydrogen bonds, andmay therefore affect anthocyanin stability.
The flavylium nucleus is electron deficient and there-fore highly reactive. Its reactions usually involvedecolourization. The rate of anthocyanin degradationdepends on pH, temperature, O2 partial pressure,metal ion (i.e. Mnþ), ascorbate concentration, andother components (Davıdek et al. 1990). In most foodprocessing operations, the anthocyanins are relativelystable, especially when low pH conditions are main-tained. Naturally present ascorbic acid can be proble-matic. In the presence of iron or copper ions andoxygen, the oxidation of ascorbic acid to dehydro-ascorbic acid is accompanied by the formationof hydrogen peroxide, which in turn can oxidizeanthocyanins to colourless malvones, a reaction impli-cated in the loss of colour from canned strawberries(Coultate 2002). The mechanism, however, is notclear. Under anaerobic conditions, anthocyaninsand ascorbic acid may react (in the presence ofamino acids, phenols, sugar derivatives, etc.) viacondensation-type mechanisms, but the products
of these interactions, some of which are complexpolymers, have not been well characterized (Jurd1964, Harborne et al. 1975).
Mazza (1997) has reviewed the structure, stability andanalysis of anthocyanins and concludes that in addi-tion to pH, structure, concentration, co-pigmentationand metal complexing, the intensity and stability ofanthocyanin colours are influenced by other factors.These include temperature, light, oxygen, acetal-dehyde, ascorbic acid, sugars and their degradationproducts sulfur dioxide, amino acids and catechins.Oxygen is an important factor influencing the rate ofanthocyanin degradation, due to the partial open-ringstructure. Anthocyanins are oxidized by peroxides,depending upon the substructure at the C3 hydroxyl.The hydrogen peroxide oxidation of flavyliumsalts produces a number of different compounds(Chichester 1972, Havlikova 1985).
Anthocyanins form adducts with S2O5� as a result of
its common use as an antimicrobial preservative infruit juices and wines, as well as in other foodcommodities, such as dried fruits, comminuted meatproducts and compound foodstuffs (Hendry and
102 M. J. Scotter and L. Castle
O+
HO
OH
OH
R
R
1
2
OG35
7
3'4'
5'
(a)
N+
NH
COOH
RO
OHCOO-
H
H
COOH
NH
COOH
H
COOH
N+
RH
(b) (c)
Figure 6. (a)2-Phenylbenzopyrylium(flavylium)nucleus;G, glycoside; R1, R2¼H, OH or OCH3; (b) betalains(R, glucose¼ betacyanin) and (R¼H¼ betanidin); (c)vulgaxanthin-I (R, glutamine), vulgaxanthin-II (R, glu-tamic acid).

Houghton 1992). The bisulfite ion adds to position 2or 4 of the flavylium nucleus, discolouring the pig-ment and simultaneously conferring ‘high’ heat sta-bility on the glycosidic bond (Adams 1973, Adamsand Woodman 1973). According to Wedzicha (1984),the reaction involves the reversible formation of a1:1 adduct between S(IV) and the flavylium cation,which is pH dependent. Nucleophilic attack on thecation leads to the formation of Meisenheimer-typesigma-complex in which positions 2 and 4 are reactive(figure 7). (A Meisenheimer complex, or adduct, isa cyclohexadienyl derivative formed as a Lewisadduct from a nucleophile (Lewis base) and anaromatic or heteroaromatic compound.)
According to Timberlake and Bridle (1968), attack atthe 4-position is preferred. However, polymericanthocyanins are resistant to S(IV) reaction, sincethe 4-position is already occupied. Anthocyanin reac-tivity and the status of the flavylium 4-substituenthave been reported to play an important role in thecolour stability of anthocyanins (Garcia-Viguera andBridle 1999).
Chlorophylls
Chlorophyll degradation during food processing hasbeen reviewed by Davıdek et al. (1990), who cite thework carried out by White et al. (1963). Chlorophylldestruction can proceed as an acid-, base- or enzyme-catalysed reaction. Weak acids liberate the Mg atombound to the porphyrin ring to form pheophytins bysubstitution with hydrogen. This results in a colourchange from green to dull brown. Magnesium may bereplaced by copper (as in copper chlorophyll additives,i.e. chemically modified chlorophylls) and by tin andzinc. Alkaline salts may also be produced, but theseare very unstable. Chlorophylls may also undergophoto-oxidation accompanied by the loss of desirablecolour. The rate of oxidation has been shown to bedependent upon water activity and the temperatureand duration of blanching in dehydrated products.
Lipoprotein-bound chlorophylls are somewhat pro-tected against acids, but can be affected by cookingand processing. In alkaline media, the decompositionof chlorophylls is very rapid and they are not stableagainst the action of free-radicals, e.g. during lipo-xygenase-catalysed oxidation of lipids, probably dueto the effect of hydroperoxides. The allomerizationreaction of chlorophylls takes place spontaneously ina polar medium and is metal-ion catalysed; allomeric10-hydroxychlorophylls and 10-methoxylactones havebeen detected (Schaber et al. 1984). Chlorophyllsalso form Schiff bases, the colour maximum of whichis pH dependent (Maggiora et al. 1985). The majororganic acids involved in the degradation of chloro-phylls are acetic acid and 5-oxopyrrolidinecarboxylicacid (Lin et al. 1971).
Betalains
Betalains are the red and yellow plant pigmentsobtained from members of the order Centrospermae,the most food-significant of these being the beetroot(Beta vulgaris L.). The betalains are divided into twogroups, the betacyanins which are purplish-red incolour and the less common yellow-coloured beta-xanthins (figure 6 (b, c)). The betacyanins compriseabout 90% of beetroot betalains. They are all eitherthe glycoside or the aglycon of betanidin or its C-15isomer. Iso forms account for only about 5% of thetotal betacyanins and some 95% of betanidin andisobetanidin carry a glucose residue at C-5, and a verysmall proportion of these glucose residues, esterifiedwith sulfate. In beetroot, the betaxanthins arecharacterized by the vulgaxanthins (types I and II inroughly equal proportions), which are characterizedby the lack of an aromatic ring system attached to N-1and by the absence of sugar residues (Coultate 2002).Betacyanins differ from other naturally occurringwater-soluble plant pigments, especially anthocyanins,in that their colour is hardly affected by pH changesin the range normally encountered in foodstuffs.Betacyanins are fairly stable under food-processingconditions, although heating in the presence of air atneutral pH causes breakdown to brown compounds.
According to Davıdek et al. (1990), the most import-ant betalain is betanin (or phytolaccanin), theb-D-glucopyranoside of betanidin, which may beenzymically hydrolysed to the corresponding aglycon.In the presence of acids, it is transformed into itsisomer, and further, to yellow betalamic acid prod-ucts, containing an open ring system, and finally to
103Chemical interactions between additives in foodstuffs
O+
HO
OH
OG
Ph
:N
O+
HO
OH
OG
Ph
:N
or
Figure 7. Sites for nucleophilic attack on the flavyliumcation (Wedzicha 1984).

brown products (Saguy et al. 1978). In alkalinemedium, the red-violet pigment is decomposed intocolourless products (Mabry 1980). Vulgaxanthin I,the most important betaxanthin, and its amidevulgaxanthin II, are hydrolysed in acid medium toamines or amino acids bound to the dihydropyridinemoiety. The stability of betalains during foodprocessing is influenced by many factors, most signi-ficantly by temperature, pH, aw, Mnþ, O2, and hn(Singer and Von Elbe 1979).
Carminic acid
Carminic acid (7-a-D-glucopyranosyl-9,10-dihydro-3,5,6,8-tetrahydroxy-1-methyl-9,10-dioxoanthracene-2-carboxylic acid) from cochineal is a pale yellowcompound which complexes readily with aluminium(as in cochineal carmines), tin and other metals togive brilliant red pigments. The cochineal complexesare relatively stable under food processing conditions,but are easily bleached by SO2 (Davıdek et al. 1990).Carminic acid has close structural and chromogenicsimilarities to the highly toxic anthracycline antitu-mour drug doxorubicin and is reported to redoxcycle to produce free radicals (Gutteridge andQuinlan 1986). These radicals, in the presence oftrace amounts of iron salts, readily damage mem-brane lipid and degrade the carbohydrate deoxyri-bose. Damage to membrane lipid appears to involvemainly organic oxygen radicals, such as alkoxyand peroxy radicals, whereas that to deoxyriboseimplicates the hydroxyl radical formed in a Fenton-type reaction. a-Tocopherol, the phenolic antioxi-dants BHA and butylated hydroxytoluene (BHT),the hydroxyl radical scavengers mannitol, benzoate,and thiourea, and iron chelators, such as EDTA, wereall reported to prevent such damage. The productof the reaction between carmine and hot ammoniasolution has been reported to be the acid-stable4-amino analogue, which is permitted for food usein the USA (Sugimoto et al. 2002).
Curcumin
Curcumin is the collective term given to the yellow-coloured compounds obtained from rhizomes ofCurcuma domestica Val. (syn. Curcuma Longa Koenignon L.), which themselves are used as turmericspice (Hendry and Haughton 1992). The colouring
principles comprise one major and two minor struc-turally related species, characterized by the presenceof a dione system with two substituted aromaticmoieties, each conjugated to a keto group. Theseare curcumin itself (major), demethoxycurcumin andbisdemethoxycurcumin. Curcumin is relatively stableto heat processing except in alkaline medium, whereit degrades to vanillin and diferuloylmethane.Curcumin is sensitive to light, which limits its appli-cations in food. Curcumin gives a lemon-yellow witha distinct green shade in solution. Cations will tend toinduce a more orange-brown shade and SO2 reducesthe colour intensity.
Majeed et al. (2000) have reviewed the chemistry andpharmacology of curcumin. Bisdemethoxycurcuminwas observed to be less susceptible to degradationat pH 10.2 than curcumin or demethoxycurcumin.In buffer solutions, curcumin was observed to decom-pose in a pH-dependent manner, with faster reactionsat neutral-basic conditions, trans-6-(40-hydroxy-30-methoxyphenyl)-2,4-dioxo-5-hexanal being predictedto be the major degradation product. Vanillin, ferulicacid and feruloylmethane were the minor degradationproducts, with vanillin increasing with the time ofincubation. The differential physical properties of thenaturally occurring curcuminoids and selected deri-vatives were explained in a study that investigatedintra- and intermolecular hydrogen-bond formationin these compounds using absorption and emissionproperties (Tonnesen et al. 1995).
Carotenoids (see also below). The carotenoidsinclude the carotenes, which are hydrocarbons,and the xanthophylls, which are oxygen-containinganalogues (Britton et al. 1995). Within the contextof food additives, their reactivities and general pro-perties may be considered to be similar. Carotenoidsmay exist in the free state, in solution in the lipidphase, or associated with proteins or polysaccharides.They can also exist as esters and glycosides, etc.In food, the main causes of carotenoid degradationare various oxidative reactions mainly involvingoxygen (principally 1(O2)), hydroperoxides and per-oxy-radicals. The presence of a polar (e.g. hydroxyl)group in a carotenoid molecule may effect the reac-tivity of neighbouring carbon atoms. Carotenoidsact as very effective 1(O2) scavengers in the photo-sensitized oxidation of unsaturated fatty acids, wherechlorophylls may act as sensitizers. The oxidativestability of carotenoids is improved by the additionof antioxidants, such as BHA, BHT, tocopherolsand ascorbyl palmitate. The addition of ascorbic
104 M. J. Scotter and L. Castle

acid is reported to improve the colour stability(Bunnell 1968). Flour bleaching agents used to deco-lourize carotenoids, include benzoyl peroxide, nitro-syl chloride and chlorine dioxide (Fortmann andJoiner 1971).
Degradation of carotenoids. Several studies havereported on the formation of volatile aromatic hydro-carbons as anaerobic thermal degradation productsof (principally) b-carotene (Kuhn and Winterstein1932, 1933a, b, Day and Erdmann 1963, Mulik andErdmann 1963, McKeown 1965). These and otherstudies were reviewed by Onyewu et al. (1982),who also reported on the formation of two non-volatile colourless carotene-like compounds in thethermal degradation of b-carotene. In a later studyon b-carotene degradation (Onyewu et al. 1986), over70 non-volatile compounds were observed.
Marty and Berset (1988) have reported 15 coloureddegradation products arising from the degradation oftrans-b-carotene during extrusion cooking. Factorsaffecting the thermal degradation of b-carotene werediscussed in a subsequent report (Marty and Berset1990). This study showed that prolonged heating ina model extrusion-cooking system at 180�C causedonly limited breakdown, but the presence of commonfood constituents, such as water or starch, combinedwith mechanical mixing favouring the diffusion ofoxygen, led to much higher losses. Several hypothesesconcerning the sequence of reactions involved inoxidative degradation of b-carotene were proposed,in which reversible stereoisomerization was importantin the formation of both non-oxidized volatiles andoxidation products.
A mechanism for the formation of volatile com-pounds by thermal degradation of carotenoids inaqueous medium has been proposed (Kanasawudand Crouzet 1990). The formation of a- and b-ionones by the thermal decomposition of b-carotenepresent in distilled alcoholic beverages in addition tothese aromatic hydrocarbons, has also been reported(LaRoe and Shipley 1970). The effects of differenttypes of oxidation-autoxidation at 20 and 80�C(photosensitized and chemical) on the degradationof b-carotene have been reported (Gloria et al.1993). Although similar volatile degradation productswere found for the different types of oxidation,their relative concentrations varied. The oxidationof capsanthin is reported to involve primarily theoxidation of hydroxyl groups, followed by scissionof the chain at the carbon–carbon bond a- to the
in-chain carbonyl group (Philip and Francis 1971).Since many of the non-volatile degradation productsof b-carotene possess chemically reactive groups suchas carbonyls, they are likely to react with amino acidsand/or their degradation products during the thermalprocessing of foods. Four major (yellow) colouredproducts were isolated by HPLC from methanolextracts of model systems comprising mixtures ofb-carotene and phenylalanine, following heating inliquid paraffin at 210�C (Papadopoulou and Ames1993).
Following on from the degradation studies on bixinreported by McKeown (1965), characterization ofthe coloured thermal degradation products of bixinfrom annatto food colouring material, and a revisedmechanism for their formation has been reported(Scotter 1995a). The main product was confirmedas 4,8-dimethyltetradecahexaenedioic acid and itsproduction was shown to be accompanied by theformation of m-xylene and (to a lesser extent) toluene.m-Xylene has been shown to be present in annattofood colour preparations up to a level of 200mgkg�1
(Scotter et al. 2000) and low levels of both m-xyleneand toluene have been detected in the headspace ofmodel systems and foods containing annatto, heatedin situ (Scotter 1995b).
Sulfur (IV) species
SO2 and sulfites
Wedzicha (1984, 1991, 1992) has comprehensivelyreviewed the chemistry of sulfur dioxide in foods.Detailed accounts of the safety aspects of SO2 andsulfites have been given by Taylor et al. (1986) andby Rose and Pilkington (1989), who reported amechanism for the antimicrobial action of this groupof preservatives. Most of what follows has been takenfrom these works.
The terms sulfur dioxide and sulfites are used com-monly to describe the oxo-species of sulfur oxidationstate (IV). The chemical nature of each chemicalspecies is well documented. In the normal pH rangeof food (about pH 3–6), the principal S(IV) species isHSO3
� in equilibrium with small but pH-sensitiveamounts of SO2�H2O and SO3
2�. The minor speciesare responsible for the preservative effect and thechemical reactivity of the additive. However, the pK
105Chemical interactions between additives in foodstuffs

values of SO2�H2O are sensitive to other composi-tional factors, such as salt concentration. This is inaccordance with theoretical predictions based onthe variation of solute activity coefficients with highionic strength. Certain non-electrolytes such as etha-nol have a very marked effect, increasing pK values byup to 2 units, whereas high concentrations of sucrosehave very little effect (Wedzicha and Goddard 1988,1991); hence, there are no simple rules.
Chemical reactivity of sulfur (IV)
The main cause of reactions between S(IV) speciesand food components is the nucleophilicity of sulfite,which can act as both a carbon and sulfur nucleophile(Davis 1968, Wedzicha 1984, 1987). Sulfite is the morenucleophilic, metabisulfite showing no significantnucleophilic activity because its action involves S-Sbond breaking. The pKa of bisulfite is similar to thatof H2PO4
�, so it is reasonable to expect that S(IV)species can act as general acid–base catalysts as dophosphate species. The potential for such activity isillustrated by the effect of S(IV) on the rate of muta-rotation of glucose, the hydrolysis of gluconolactoneand, most particularly, in the hydrolysis of p-nitro-phenyl acetate, for which it is some 1600 times moreeffective than, for example, phosphate — anothercommonly used food additives. This is attributed tothe ability of sulfite to initiate the hydrolysis bynucleophilic attack at the carboxyl carbon atom.Thus, whilst the implications of this reactivity arenot fully appreciated, it is possible that esters thatare either naturally present or added (as flavours forinstance) could be destabilized by S(IV). It is alsopossible that the rate of degradation of ascorbic acid,which proceeds by hydrolysis of the lactone moiety,is increased by S(IV) (Davies and Wedzicha 1992).This may therefore be relevant to preserved fruitbeverages. The metabisulfite ion is potentially a betteracid–base catalyst, although the tendency for it tocatalyse reactions between food components has notbeen confirmed.
The most significant homolytic reactions of S(IV) arethe result of oxidation by molecular oxygen catalysedby transition metal ions (Yang 1984). Complexingagents such as EDTA or citrate may act as antioxi-dants. Free-radical scavengers such as alcoholsare effective antioxidants. Caramel colour can actas a pro-oxidant at low concentration, presumablyby providing initiating free radicals, but acts as an
antioxidant at high concentrations (Wedzicha andClayton 1994). It is generally recognized that antiox-idants can become pro-oxidants under certain con-ditions. High (>30%) concentrations of ethanolfor instance allow rapid oxidation of S(IV), providedthat transition metal ions and amino compounds(including EDTA) are also present. These form redox-active complexes in solution that are oxidized bymolecular oxygen and in turn oxidize S(IV). Despitethe fact that many foods contain numerous knownantioxidants to the oxidation of S(IV), it is well recog-nized that some (5–20%) of the additive becomesoxidized in food (Wedzicha and Herrera-Viloria1991). The nature of the oxidizing agent, the catalystand the type of oxidation taking place have not beenestablished and so no hard and fast rules exist.
Oxidation of sulfur (IV)
There are several implications of S(IV) oxidation forfood quality. The depletion of S(IV) after exposure toair can lead to an increased risk of microbial spoilage,so benzoate is often used in combination with sulfite.Free-radical intermediates may be formed whichare capable of initiating oxidation of other foodcomponents in the same way that �OH or �O2
� do.The accepted role of S(IV) in foods with respect tooxidation is that of an antioxidant, despite evidencethat the species can easily mediate the oxidation ofunsaturated organic compounds in model systems.Such implications may be speculative and it is inter-esting to note that Til and Feron (1992) observed thatan oxidized lipid fraction, which is peculiar to sulfitedanimal diets, has some toxicological effect againstrats. Low levels of sodium sulfite have been impli-cated in causing oxidative-reductive depolymerizationof polysaccharides (i.e. cassava starch), which can beprevented by the addition of the antioxidant propylgallate (Mat Hashim et al. 1992).
Free and bound sulfur (IV)
The reversible binding of S(IV) is the reaction betweencarbonyl components of the food and S(IV) to formhydroxysulfonates (HS). The rate of decompositionof the HS depends upon the ease of proton removal,which in turn increases with pH. Furthermore, theease with which the initial sulfite attack takes placealso increases with pH. The two effects are almost in
106 M. J. Scotter and L. Castle

balance over the pH range of foods. However, thisis only representative of a simple model and the casein foodstuffs is far more complex; here reactivity isdependent upon the nature of other functional groupsin close proximity to the carbonyl moiety.
Inhibition of non-enzymic browning (NEB)
It is generally observed that the levels of free andbound S(IV) in food decrease with time. Thus, theadditive is converted by reaction to products thatdo not release S(IV) under conditions of analysis.Apart from sulfate, these products are organicsulfonates and can arise from the inhibition of NEB(i.e. Maillard and ascorbic acid browning). S(IV) isthe only permitted additive known to be effectiveagainst NEB and consequently this property is ofconsiderable technological value.
Inhibition of enzymic browning (EB)
EB is the result of oxidation of phenolic and o-diphe-nolic compounds of plant material to o-quinones,catalysed by polyphenol oxidase. Whilst the enzymeis not inhibited by S(IV), the latter reacts with thequinone intermediates to give substituted dihydroxy-benzenesulfonic acids. In foods, the substratesfor polyphenol oxidase are often 4-substituted cate-chols, e.g. dihydroxyphenylalanine, which forms 3- or5-sulfo derivatives.
Other sulfur (IV)–food component interactionsof interest
The nucleophilic behaviour of sulfite renders it able toreact with any electron-deficient chemical present infood such as in the following areas:
. Cleavage of thiamine (see also below).
. Cleavage of protein disulfide bonds, which hasbeen suggested as a reason for the observed inhi-bition of a large number of enzymes (and thencemicroorganisms) by sulfite, which is reversibleand subject to steric effects of P and P0, andcharged groups in the vicinity of –S–S–:
P�S�S�P0 þ SO2�3 Ð P0�S�SOþ
3 þ P�SH: ð2Þ
. Use of S(IV) as an additive for biscuit flour.
. In the non-reversible hydrolysis of S-sulfonates:
R�S�SO�3 þH2O ! RSHþHSO�
4 : ð3Þ
. Interaction with hydroperoxides via nucleophilicdisplacement, i.e. SO3
2�þROOH SO42�þROH.
At lower pH, the situation is more complex withthe formation of ketones, olefins and ethers inwhich there is evidence for the involvement offree radicals in solution.
. Addition to Schiff bases. The most likely reac-tion between a Schiff base and S(IV) oxoanionsis addition to the C¼N bond:
RCH ¼ NR0 �HSO�3 ! RCHðSO�
3 ÞNHR0
ÐH2O
RCHðSO�3 ÞOHþRNH2, ð4Þ
where the reaction product of the correspondingaromatic amine appears to be stable to hydrolysisand exists as an amine salt.
. Addition to olefinic groups. The reaction ofS(IV) oxoanions with carbon–carbon doublebonds may proceed by both heterolytic andhomolytic routes, depending upon the natureof the unsaturated compound. The reactionis reported to proceed over a wide range ofpH. For example, the sulfonation of the sodiumsalt of crotonic acid takes place in weakly acidicmedia and is unaffected by the presence ofoxygen or antioxidants.
RCH ¼ CH�COOH
��!SðIVÞ
RCðSO�3 Þ�CH2�CðOHÞ ¼ O: ð5Þ
. Interaction with isothiocyanates, e.g. as found inBrassicas and in some food flavourings, wheresubsequent oxidation of the intermediate speciesproduces a carbylamine and thiosulfate:
RNCSþKHSO3 ! RNHCS:SO3K: ð6Þ
. Formation of anionic g-adducts with aromaticcompounds such as anthocyanins.
. Addition to pyridine compounds, e.g. NADH�
readily undergoes nucleophilic addition of S(IV)oxoanion at position 4. NADH also reacts withS(IV) by both free radical and ionic mechanismsto give a variety of products. Other reactions of
107Chemical interactions between additives in foodstuffs

S(IV) that have been studied are those withpyrimidines and disulfides.
. Quinines, such as vitamins K1 (natural product)and K3 (synthetic) (figure 8).
Uses of sulfur (IV) in food: general considerations— sulfite-mediated oxidations (Wedzicha 1984)
The pro-oxidant action of the sulfite ion in systems isusually discussed in the context of lipid browning,which can be described basically as the interactionof carbonylic oxidation products of unsaturated fatswith amines. An important step is the autoxidation ofthe sulfite ion and interaction between oxidizingradical intermediates and the unsaturated compound.Other organic compounds affected in this way includemethionine, which in the presence of Mn2þ formsmethionine sulfoxide (Yang 1970), tryptophan, whichforms four different products (Yang 1973), and chloro-phyll (Peiser and Yang 1977, 1978). Chlorophyllwas rapidly destroyed in the presence of bisulfiteand linoleic acid hydroperoxide, both of which werenecessary, but there was no oxygen requirement.Chlorophyll destruction occurred most readily atacidic pH, with little destruction occurring abovepH 8. However, chlorophyll loss was monitoredspectrophotometrically at 665 nm, thus subtle changesin porphyrin structure caused by the action of acid(loss of magnesium and phytyl side chain) andalkali (E-ring cleavage) were not taken into account.b-Carotene has been similarly studied (Peiser andYang 1977, 1979, Wedzicha and Lamikanra 1983).The products of the reaction between b-carotene and
sulfite (at least 10) are not well-defined, but arethought to be highly oxygenated, i.e. they have arelatively low sulfur content. The final degree ofS(IV)-mediated oxidation of b-carotene is similar tothat resulting from its autoxidation (Neto et al. 1981).
Catalytic amounts of Mn2þ and glycine promoteoxidation of S(IV) species in media high in alcohol,but at a slower rate than comparable aqueoussystems. The S(IV)-mediated oxidation of food com-ponents is possible both in aqueous solution andin dispersions. However, the probability of suchreactions taking place must be considered. It is likelythat the success of the S(IV) oxoanion as an anti-oxidant (in foods) arises from the physical state of thesubstrate in contact with the aqueous medium andthe presence of natural or added antioxidants in theaqueous and non-aqueous phases. The second pos-sibility is demonstrated by the observation that whenb-carotene is solvent-extracted from carrots, the crudeproduct is less easily oxidized in the presence of S(IV)oxoanion than is pure b-carotene (B. L. Wedzicha andO. Lamikanra, unpublished results, 1982).
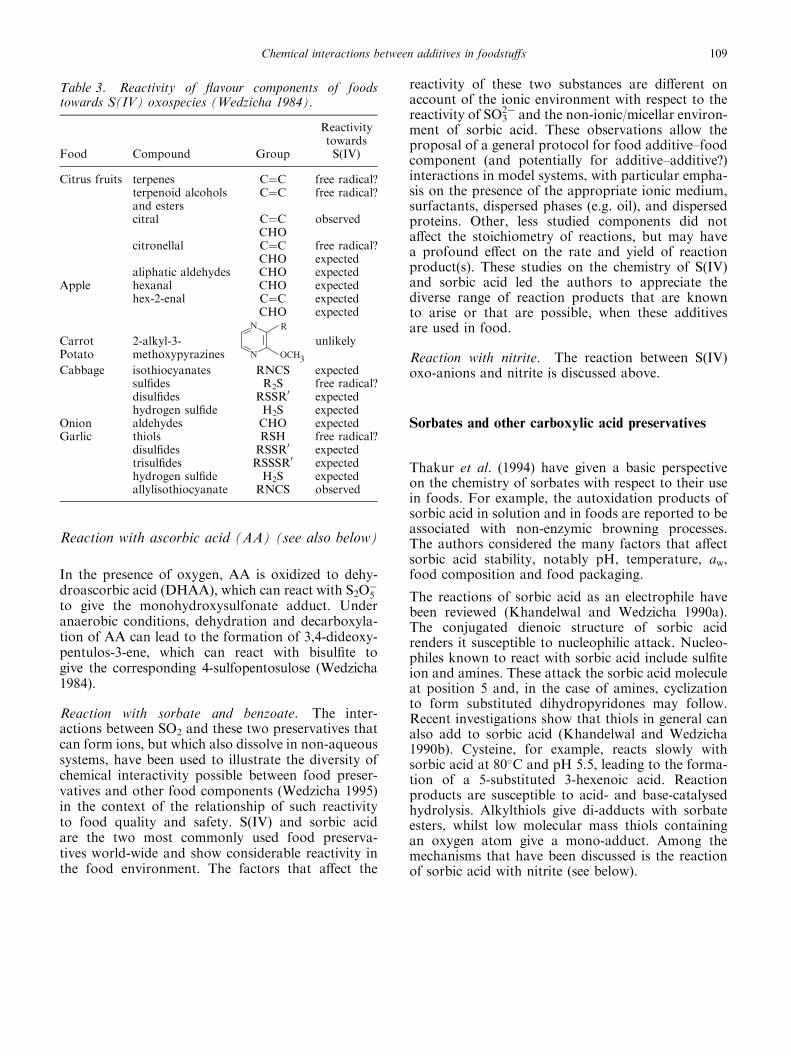
Effects of sulfur (IV) on flavourings
The most important effects of S(IV) on flavour are theresults of interactions with the volatile components.A wide range of functional groups is encounteredwith these substances; an indication of the knownand possible reactivity of flavour compounds towardsS(IV) oxospecies is given in table 3. For instance,the reaction between bisulfite ion and carbonylsis reported to suppress the formation of oxidizedflavour in beer (Hashimoto 1972) and allyl isothio-cyanate (from mustard) forms a non-volatile odour-less sulfonate (Griffiths et al. 1980, Frijters et al.1981).
The interactions of S(IV) species and anthocyaninshave already been discussed (see above). More specifi-cally, in wines, bisulfite combines with anthocyanins,with pyruvic acid, and with 2-ketoglutaric acid.The product of the reaction with acetaldehyde ispractically undissociable and the combined SO2 istotally ineffectual as a preservative. A similar effectis observed when SO2 is combined with pyruvic acidwhen the acid concentration is high, because in thiscase the rate of dissociation is low. The reactionbetween SO2 and 2-ketoglutaric acid is more dissoci-able and can be perceived as a reservoir of free andactive SO2 in wine (Usseglio-Tomasett 1992).
108 M. J. Scotter and L. Castle
O
O
O
O
or
free radicalmechanism
O
O
O
O H
H
Thermodynamic product
SO3 NaSO3 Na
Kinetic product
Vitamin KVitamin K 1 3
Figure 8. Reactions of S(IV) with K vitamins.
Reaction with ascorbic acid (AA) (see also below)
In the presence of oxygen, AA is oxidized to dehy-droascorbic acid (DHAA), which can react with S2O
�5
to give the monohydroxysulfonate adduct. Underanaerobic conditions, dehydration and decarboxyla-tion of AA can lead to the formation of 3,4-dideoxy-pentulos-3-ene, which can react with bisulfite togive the corresponding 4-sulfopentosulose (Wedzicha1984).
Reaction with sorbate and benzoate. The inter-actions between SO2 and these two preservatives thatcan form ions, but which also dissolve in non-aqueoussystems, have been used to illustrate the diversity ofchemical interactivity possible between food preser-vatives and other food components (Wedzicha 1995)in the context of the relationship of such reactivityto food quality and safety. S(IV) and sorbic acidare the two most commonly used food preserva-tives world-wide and show considerable reactivity inthe food environment. The factors that affect the
reactivity of these two substances are different onaccount of the ionic environment with respect to thereactivity of SO3
2� and the non-ionic/micellar environ-ment of sorbic acid. These observations allow theproposal of a general protocol for food additive–foodcomponent (and potentially for additive–additive?)interactions in model systems, with particular empha-sis on the presence of the appropriate ionic medium,surfactants, dispersed phases (e.g. oil), and dispersedproteins. Other, less studied components did notaffect the stoichiometry of reactions, but may havea profound effect on the rate and yield of reactionproduct(s). These studies on the chemistry of S(IV)and sorbic acid led the authors to appreciate thediverse range of reaction products that are knownto arise or that are possible, when these additivesare used in food.
Reaction with nitrite. The reaction between S(IV)oxo-anions and nitrite is discussed above.
Sorbates and other carboxylic acid preservatives
Thakur et al. (1994) have given a basic perspectiveon the chemistry of sorbates with respect to their usein foods. For example, the autoxidation products ofsorbic acid in solution and in foods are reported to beassociated with non-enzymic browning processes.The authors considered the many factors that affectsorbic acid stability, notably pH, temperature, aw,food composition and food packaging.
The reactions of sorbic acid as an electrophile havebeen reviewed (Khandelwal and Wedzicha 1990a).The conjugated dienoic structure of sorbic acidrenders it susceptible to nucleophilic attack. Nucleo-philes known to react with sorbic acid include sulfiteion and amines. These attack the sorbic acid moleculeat position 5 and, in the case of amines, cyclizationto form substituted dihydropyridones may follow.Recent investigations show that thiols in general canalso add to sorbic acid (Khandelwal and Wedzicha1990b). Cysteine, for example, reacts slowly withsorbic acid at 80�C and pH 5.5, leading to the forma-tion of a 5-substituted 3-hexenoic acid. Reactionproducts are susceptible to acid- and base-catalysedhydrolysis. Alkylthiols give di-adducts with sorbateesters, whilst low molecular mass thiols containingan oxygen atom give a mono-adduct. Among themechanisms that have been discussed is the reactionof sorbic acid with nitrite (see below).
109Chemical interactions between additives in foodstuffs
Table 3. Reactivity of flavour components of foodstowards S(IV) oxospecies (Wedzicha 1984).
Food Compound Group
ReactivitytowardsS(IV)
Citrus fruits terpenes C¼C free radical?terpenoid alcoholsand esters
C¼C free radical?
citral C¼CCHO
observed
citronellal C¼CCHO
free radical?expected
aliphatic aldehydes CHO expectedApple hexanal CHO expected
hex-2-enal C¼CCHO
expectedexpected
CarrotPotato
2-alkyl-3-methoxypyrazines N
N R
OCH3
unlikely
Cabbage isothiocyanates RNCS expectedsulfides R2S free radical?disulfides RSSR0 expectedhydrogen sulfide H2S expected
Onion aldehydes CHO expectedGarlic thiols RSH free radical?
disulfides RSSR0 expectedtrisulfides RSSSR0 expectedhydrogen sulfide H2S expectedallylisothiocyanate RNCS observed

The C-3 site of sorbic acid has the lowest electrondensity (using NMR) and is thus the more likelysite for nucleophilic attack by HþNu�, where Nu�
represents the nucleophile. Strong nuclophiles, suchas amines and thiols (represented by Nu:), will attackat C-5 also and without the need for the initialprotonation (figure 9). In this case, the addition ofHþ takes place as the final stage of the mechanism.Simple 1,2- or 1,4-addition of a strong nucleophileis generally under thermodynamic control, i.e. due tothe relative stability of carbonium ion intermediates.
The much greater extent to which the charge on theintermediate arising from attack on position 5 isdelocalized, suggests that this should be preferreddespite the lower electron density at position 3 ofsorbic acid. The reaction is completed by additionof Hþ to positions 2 or 4. The products thus formeddepend upon the relative rates of Hþ addition todifferent resonance forms of the carbonium ion inter-mediates (and somewhat on solvent polarity). Theconsequence of 1,2-addition is the possibility of add-ing a second nucleophile. Foods contain numerousnucleophiles; those most studied in reaction withsorbic acid are sulfite and nitrite. The reactionbetween sulfite ion and sorbic acid is well documented(e.g. Hagglund and Ringbom 1926, Heintze 1976).Only the 1,4-addition product is formed as a labile 1:1adduct.
Reaction with amines
Sorbic acid reacts with ammonia, alkylamines, aro-matic amines, and benzylamine at high temperature(about 150–200�C) to form (reportedly) the aminoacids (I) and dihydropyridones (i.e. lactams II and III)as shown in figure 10, lactam II being thermodyna-mically favoured.
There are important processing and toxicologicalconsiderations in relation to the use of sorbic acidin foods. For instance, in the grilling of cheese, sorbicacid may be exposed to high temperatures. Moreover,a large number of 3,4-unsaturated lactones have beenreported to be physiologically active (Haynes 1948).There is no evidence to suggest that sorbic acid reacts
with simple amines, such as butylamine or glycine,even after prolonged heating at 100�C in water atpH 5–7.
Reactions with thiols
Sorbic acid reacts slowly with thiols in aqueous solu-tion at 80�C and pH 3.7–5.7 (Wedzicha and Brook1989). The reaction of sorbic acid with mercapto-ethanol,mercaptoacetic acid, cysteine, and glutathioneare all second order. The adducts were difficult toisolate due to decomposition. Structural analysiswas achieved using synthesized methyl and ethylesters of the adducts. Under similar conditions, methyl2-mercaptoacetate, ethyl 2-mercaptoacetate, andmercaptoethanol, all give the corresponding mono-adducts. Alkylthiols (e.g. ethanethiol, 2-methyl-2-propanethiol, butanethiol) react with sorbic acidesters to give only di-adducts, the proposed mecha-nisms being somewhat speculative. The situation iscomplicated by the competing thermodynamic,kinetic and steric factors implying a range of potentialproducts.
pH effects
Undissociated sorbic acid is more susceptible toautoxidation than dissociated sorbic acid and theprocess is thus influenced by pH. The nature of otheracids present is also important; HCl, HOAc, TFAand H2SO4 all exert a strong catalytic effect on thedegradation of sorbic acid. This may be due to thereaction of the terminal hydroxyl group of sorbicacid with acid to form peroxy esters, which decom-pose to yield radicals (Pekkarinen and Rissanen1966). However, there is no appreciable effect ofvarious acids on sorbic acid stability in aqueous solu-tion at 60�C (Petropavlovski and Usinova 1967).Note that different pH and temperature conditions
110 M. J. Scotter and L. Castle
OH
ONHR N O N O
and
I II III
R R
Figure 10. Products of the high-temperature reaction ofsorbic acid with ammonia and other amines (Khandelwaland Wedzicha 1990a).
OH OHor
Nu: Nu:
53
O O
Figure 9. Sites of nucleophilic attack on sorbic acid(Khandelwal and Wedzicha 1990a).

were used in these studies. Vidyasagar and Arya(1983) investigated the role of various acids found infoodstuffs, where all the acids studied were reportedto exhibit a catalytic effect, except citric and malicacids. It was suggested that acid specificity had agreater effect than overall pH.
Antioxidants
The degradation of sorbic acid is affected by antioxi-dants such as BHA, dodecyl gallate and nordihydro-guaiaetic acid, which act by inhibiting free-radicalmechanisms.
Transition metal ions
Those transition metal ions that possess two or morevalency states with suitable redox potential betweenthem are found generally to accelerate the rate ofautoxidation. However, with sorbic acid, the effectis concentration dependent. At low concentration,autoxidation is enhanced, since it proceeds via thedecomposition of peroxides to form radicals (or viathe formation of free radicals following olefinic bondcleavage). Conversely, at high concentration, autoxi-dation is retarded, due to the ability of the metalions to complex with the peroxy radicals.
Other salts
The stability of sorbic acid in aqueous solutionis enhanced by the presence of NaCl and KCl, parti-cularly as their concentration increases. Conversely,stability is reduced in the presence of LiCl. Sulfatesexhibit a strong pro-oxidant effect, but the effectsof phosphate are more complex. The addition ofNa2HPO4 increases the pH and hence the stabilityof sorbic acid, but, when the pH falls below thenatural pH of sorbic acid (3.3), phosphate has a pro-oxidant effect. Bisulfites and sulfur dioxide acceleratesorbic acid degradation.
Sugars and glycerol
At low concentration (<5%), sucrose does notexert any significant effect on sorbic acid stability,but, at high concentration, the rate of sorbic aciddegradation is significantly reduced in aqueous solu-
tion. Differential rates of sorbic acid degradationhave been observed in sucrose and glycerol solutions;a ‘protective’ effect may be due to the fact that inviscous solutions, diffusion of oxygen through theaqueous layer becomes the rate-determining step inautoxidation.
Ascorbic acid
AA has been observed to exert an inhibitory effect onsorbic acid degradation in fruit preserves. In aqueoussystems, AA alone did not exert any inhibitory effect,but, in the presence of Fe3þ/Cu2þ, degradation wasaccelerated. In complex food systems, AA may com-pete with sorbic acid for available oxygen.
Amino acids and proteins
Amino acids and proteins, especially those containingfree sulfhydryl groups, are found to interact withsorbic acid.
Reaction of sorbic acid and analogousfood components with nitrite
Sorbic acid has been proposed as a partial replace-ment for nitrite in curing, since it inhibits Clostridiumbotulinum and also reduces the formation of nitrosa-mines (Ivey 1977). However, this practice may lead toother toxicological problems, since sorbic acid reactswith nitrite to yield mutagenic products (see below).According to Walker (1990), however, the conditionsunder which these compounds were shown to beformed (60–90�C and high equimolar concentration)do not represent those which obtain in curing brines,and negative results have been recorded in mutagenicassays on curing brines containing sorbic acid.Nevertheless, the following investigations are worthnoting.
Osawa and Namiki (1982) examined the formationof mutagens by the reaction of sodium nitrite withsome sorbic acid analogues using microbial mutageni-city tests (rec-assay and Ames test), together withchemical analysis using TLC. The conjugated dienoiccarbonyl structure was reported to be essentialfor mutagen formation by the reaction with nitrite.For instance, the reaction of 2-furanacrylic acid
111Chemical interactions between additives in foodstuffs

and b-carotene with nitrite, both showed less than20% of the starting material reacted, whereas piperineshowed 20–50% and methyl sorbate over 50%reacted. From a large-scale reaction of NaNO2
with methyl sorbate, 5-nitro-2,4-hexadienoic acidmethyl ester and ethylnitrolic acid were isolated andidentified as the main mutagens. The results impliedthat a nitro group adjacent to a double bond isan important factor in the development of mutageniccharacter.
In a similar study, the development of mutagenicityby the reaction between sorbic acid and nitrite wasreported to be highly dependent upon the reactionconditions, especially pH, reaching a maximum at pH3.5, but being negative at pH 5 6 (Namiki et al.1983). Most of the isolated mutagens were C-nitroand C-nitroso compounds. It was also concluded thatthe conjugated dienoic carbonyl structure is essentialfor mutagen formation, though details of the mechan-ism of formation were not clear. Among the foodconstituents tested was piperine, which formedstrong mutagens. Ascorbic acid and cysteine wereshown to inactivate the ‘effective’ mutagenicities ofthe C-nitro compounds, which were reported to bemore susceptible to being inactivated than N-nitrosomutagens.
Osawa et al. (1982) proposed a mechanism for muta-gen formation from the reaction between piperine(a) and nitrite. Initially, piperic acid (b) and piperi-dine (c) are formed, which both react with nitrite togive hitherto unknown C-nitro mutagens, including(d), along with N-nitrosopiperidine (e) (figure 11).
The products of sorbate-nitrite interactions areamong the putative mutagens and carcinogens infoods reviewed by Hartman (1983). Mutagen forma-tion was reported to be blocked by ascorbate at lowpH. Ascorbate at eightfold molar led to inactivationof 1,4-dinitro-2-methylpyrrole (DNMP) near neutralpH, but did not destroy the mutagenic nitro com-pound at low pH. It was concluded that the combina-tion of sorbate with nitrite represents a potentialhealth risk in the absence of adequate levels ofascorbate (cf. the comments made by Walker 1990).
Osawa et al. (1986) reported further on the desmuta-genic action of certain food components on DNMP.Whilst the results were somewhat inconclusive, it wasproposed that (1) ascorbic acid specifically reducesthe 3-nitro or 4-nitro groups on pyrrole or furan ringsto corresponding amino groups, (2) the mechanismfor inactivation by cysteine involves direct attack on
the aromatic ring of DNMP, followed by eliminationof the C-nitro group and (3) reducing substancesresponsible for desmutagenic action against the prod-ucts of the sorbic acid/nitrite reaction are present invegetable juices.
The principal nitrite-derived reactant is reported to beN2O3, since it is well established that it may bepolarized as NO2
þNO� for electrophilic nitration.Such species are known to add across doublebonds (Park and Williams 1976). Evidence for theinitial product (a) and its subsequent isomerizationto the oxime (b) is the isolation of the furoxan (e).The formation of structures, which may eventuallydegrade to nitrolic acid-type moieties, may only beconceived via a Beckmann rearrangement of (b).Product (c) is ethylnitrolic acid and product (d) isDNMP (figure 12).
Ascorbic acid
The interaction between AA and azo dyes, naturalcolours, sulfur (IV) species and sorbic acid havebeen discussed. Reaction with nitrite has also beendiscussed briefly.
112 M. J. Scotter and L. Castle
NO/NO2
O
OO
O
H+
NO/NO2
NNO
N
O
O
CHO
O2
+ unknowns
O
O
O
N(a)
(b) (c)
(d)(e)
NH
Figure 11. Proposed mechanism for mutagen formationfrom the reaction between piperine (a) and nitrite(Osawa et al. 1982).

Reaction with nitrite during curing
According to Fox et al. (1981) and Sebranek and Fox(1985), during the curing of meat, nitrite can reactwith AA under certain pH conditions to form dini-trosylascorbate. Under anaerobic conditions, thedecarboxylation of AA can lead to the formationof 4,5-dihydroxy-2-oxopentanal, which can poten-tially react with nitrite (Wedzicha et al. 1982).
Reaction with intense sweeteners
Since artificial sweeteners are commonly used inbeverages containing AA, the potential for inter-action is of interest. The degradation of aspartameand acesulfam K in acidic aqueous solution at hightemperatures is accelerated by the presence of AA(Kroyer et al. 1993). Under such high temperatureconditions, AA degradation was reported to takeplace first. Hsieh and Harris (1991) reported thatthe cupric ion-catalysed oxidation of AA occurs morerapidly in the presence of aspartame and concludedthat this was presumably due to the formation ofa complex between cupric ion and aspartame, whichis a more effective oxidation catalyst than cupric ion
alone. However, no information was given on thedegradation of aspartame under the given conditions.
Reaction with benzoate
Following the reports in 1990 of bottled water con-taminated with benzene, test laboratories stumbledover the finding of benzene in other beverages.McNeal et al. (1993) examined the potential forbenzoate and AA to react to produce benzene.Less than 2 mg kg�1 benzene was found in beverageswithout added benzoate, whereas with added benzo-ate the levels were up to 38mg kg�1. A model beveragesystem containing 0.04% potassium benzoate and0.025% AA was investigated. Exposure to UV lightfor 20 h at 45�C produced 300mg kg�1 benzene, whichwas constant after 8 days. At room temperature inthe dark, 4 mg kg–1 benzene were produced after 20 h,which increased to 266mg kg�1 after 8 days. Ina separate series of experiments, 0.025% AA wascombined with 0.04% of various compounds toproduce the yields of benzene given in table 4.
The decarboxylation of benzoate is a well-knownprobe for hydroxyl radicals (Winston and Cedarbaum1982) and it seems likely that this is the mechanismfor benzene formation in the beverages (Gardner and
113Chemical interactions between additives in foodstuffs
OR
O
OR
O
ON
NO2
(a)
OR
O
N
NO2
OH
N
NO2
NO2
CH3 (b)
NO
N+
O-
CH3
OH
OCH3 NO2
NOH
(d)
(c) (e)
Figure 12. Products formed from the interaction of nitrite and sorbate (Osawa et al. 1986).

Lawrence 1993). The role of AA is probably as asource of reducing equivalents in the Udenfriend-type generation of hydroxyl radicals from molecularoxygen and a transition metal-chelate catalyst (Castle1980). Even traces of metals in potable water, or inother ingredients used to make beverages, are suffi-cient when combined with AA, to produce the levelsof hydroxyl radicals responsible for formation of suchlow levels of benzene.
Other additives
Intense sweeteners (see also above)
Information on the chemical interactions betweenintense sweeteners and other food additives/foodcomponents is sparse. Much of the published workshas focused on stability and degradation studies infoods and model systems, including identificationof reaction products. Here, these additives are dis-cussed mostly from the point of view of the degrada-tion products produced and their potential reactivity.
Aspartame. Aspartame is the methyl ester of thedipeptide, aspartylphenylalanine, which is susceptibleto degradation under a wide range of conditions(heat, moisture and pH). The stability of aspartamehas been examined by Homler (1984), who showedthat a cola-based soft drink, containing aspartameand stored at 20�C for 6 months, remained acceptablysweet. The levels of aspartame and its condensationand degradation products were not reported. Husseinet al. (1984) showed that aspartame is quite reactivetowards aldehydes, which are among the principalflavour compounds used in chewing gums and certainsoft drinks.
Upon heating, aspartame cyclizes to 5-benzyl-3,6-dioxo-2-piperazineacetic acid (DKP) with one mole-cule of methanol liberated in the process. Slowformation of DKP may also be observed when aspart-ame is dissolved in aqueous solution. Up to 2% DKPwas reported to be formed from an aqueous solutionof aspartame after standing at 22�C for 4 h (Tsanget al. 1985). Other degradation pathways involvethe formation of phenylalanine methyl ester andaspartylphenylalanine, which may undergo furtherhydrolysis to aspartic acid and phenylalanine.According to Vetsch (1985), the stability of aspartameis affected by time, temperature, and pH — the latterbeing of particular importance, optimum stabilitybeing at pH 4.3. At elevated temperatures, such asthose found in short-time pasteurization (80�C),aspartame becomes less stable, notably in the pHrange 6–7. It was suggested that the decompositionof aspartame by ultrahigh temperature (UHT) pro-cessing, as used in aseptic processing, can be ignoredsince the time involved is too short for substantialdegradation to take place. This view may or may notbe correct, depending on the activation energy forthe degradation process(es).
Other intense sweeteners. Acesulfam K is reported tobe highly stable in aqueous solution and at tempera-tures encountered during food processing, andno indications of undesired reactions with foodingredients have been reported (Von RymonLipinski 1985, 1995, Mayer and Kemper 1991).Neohesperidin dihydrochalcone (NHDC) has beenreported (Borrego 1995) to show similarly goodstability, no degradation being detected by HPLC inunflavoured yoghurt fermentation (42�C for 6 h),blackcurrant jam manufacture (pH 3.0, 102–106�Cfor 35–40min), and pasteurization of soft drinks,based on apple, pineapple, orange, or lemon juice(90�C for 30min). HPLC data from stability studieson NHDC in aqueous buffers from pH 1 to 7, withstorage for up to 20 weeks at temperatures of between30 and 60�C, as determined by Canales et al. (1993),have been examined by Lindley (1996). He showedthat the breakdown of NHDC can be represented as apseudo-first-order reaction at all temperatures andpHs studied. Maximum stability was observed ata pH of about 4.0. Along with other stability dataderived from model food systems, this led to NHDCbeing considered to pose no significant stabilityproblems for food manufacturers, even though thereis a theoretical possibility of hydrolytic cleavageof the glycosidic linkage.
114 M. J. Scotter and L. Castle
Table 4. Summary of benzene yields resulting from thereaction between ascorbic acid and various compounds(McNeal et al. 1993).
Compound Benzene yielda
Benzaldehyde 74 ng g�1
Benzoic anhydride 4 ng g�1
Acetophenone 5 ng g�1
Toluene not detectedBenzyl alcohol not detectedBHT not detectedBHA not detected
aExperimental conditions given in the text.

Saccharin and cyclamate have been reported to behighly stable in both bulk form and in solution.Stability has been proven over a wide range ofpHs and temperatures (Beck 1983, Salminen andHallikainen 1990, Mikuschka 1995). According toBeck (1983), the low solids content of some foodproducts sweetened with non-nutritive sweetenersmay render them susceptible to microbial spoilage.Consequently, it becomes especially important touse preservatives, such as sorbate or benzoate, or toemploy thermal processes to achieve adequate shelflife. However, there is some controversy over thestability of saccharin and cyclamate. Kroyer andWashuettl (1982) have reported on the possible inter-actions of these sweeteners with food ingredients,particularly water-soluble vitamins and essentialamino acids. Stability studies in model aqueoussystems at ambient temperature and at 80�C showedno significant losses of sweeteners over 28 days,except for systems containing phenylalanine andtryptophan, where reductions of up to 13 and 47%were observed for tryptophan in the presence ofsaccharin and cyclamate, respectively, at ambienttemperature. In the same experiment, correspondingreductions in phenylalanine of 10 and 17% wereobserved in saccharin and cyclamate systems, respec-tively. Control systems containing sucrose showed upto 3% reduction of the amino acids.
Mixtures of vitamins B2, B6 and B12 with sucrose,saccharin and cyclamate in solid form indicated nosignificant losses after heating for 1 h at temperaturesof 120 and 150�C, but distinct changes of the vitaminsB1 and C were observed, particularly in the presenceof saccharin (33% reduction of B1 and 93% reductionof C at 150�C). Under extreme conditions, cyclamateis known to break down to cyclohexylamine, smallquantities of which were detected in model systemsheated between 150 and 200�C. Further studies onsaccharin and cyclamate stability during the prepara-tion of foods, such as cake, jam, punch, coffee andvinegar dressing, again showed various amounts ofsweetener loss. These ranged between 0% (punch)and 64% (cake) for saccharin, and between 1%(punch) and 17% (vinegar dressing) for cyclamate.However, there was no direct evidence for the inter-action of the sweeteners with other food components.
Starch-based sweeteners
It has been shown that all common carbohydrates,including glucose syrups, have the ability to complex
with iron and particularly with ferric iron. Kearsleyand Birch (1985), considered it likely that the pH-dependent chelation of inorganic species is notrestricted to iron, but that all metal ions will be com-plexed to a greater or lesser extent.Whilst the complex-ing of metal ions with carbohydrate may amelioratethe deleterious effects of certain metal species, it isalso possible that the bioavailability of certain traceelements such as ironmay be compromised.Moreover,addition of carbohydrate (hydrogenated glucose) hasbeen implicated in the inhibition of copper-catalysedoxidation of AA (Cross et al. 1984).
Interactions between micronutrients andfood additives
Cort (1987), reviewing the effects of food additives onnutrition, reported the following points.
Sulfites
Sulfites cause a nucleophilic displacement on themethylene bridge of thiamine, which is thus cleavedto form the free thiazole and the pyrimidylmethane-sulfuric acid. The reaction occurs faster at neutral pH.According to DeRitter (1969), SO2 will also cleavefolic acid to pteridine and p-aminobenzoylglutamicacid moieties, where the amino group of the latterundergoes further destruction.
Acids and alkalis
In liquid or semiliquid foods adjusted to pH 4 orbelow, vitamin A esters will de-esterify and isomerizeto the less bioactive cis-forms of retinol. Folic acid,pantothenic acid, and threonine would be expectedto decompose under acidic conditions. Cort (1987)cites the work of Moore and Folkers (1968), whoshowed that dilute acids cleaved vitamin B12 to theinactive corrin nucleus.
Aluminium sodium sulfate and the di- and tri-sodiumpotassium phosphates aid in the destruction ofthiamine. Several breakdown products have beenidentified from alkaline oxidation of thiamine(Dwivedi and Arnold 1973). These include thering-opened thiamine disulfide, which has thiamineactivity, along with thiochrome and thiothiazolone,
115Chemical interactions between additives in foodstuffs

which do not. In addition, thiamine may becleaved under these conditions to the pyrimidineand thiazole moieties, where the latter may furtherdegrade to a number of compounds, including3-mercaptopropanol, hydrogen sulfide, 2-methyl-furan, 2-methylthiophene, 2-methyl-4,5-dihydrothio-phene, and 2-methylthio-5-methylfuran.
Smoking and curing
Modern mild curing and smoking methods employsmall amounts of salt, sugar, nitrite, ascorbates,phosphates, and liquid smoke condensates in pro-cessed meat, poultry and fish products. Losses ofspecific nutrients due to curing appear to be verysmall. Thiamine is reported to suffer up to about 20%losses during heat treatment in the curing process,whereas riboflavin and niacin are very stable(Schweigert and Lushbough 1960).
Copper and iron
Other than from the use of copper complexes ofchlorophylls and chlorophyllins, the use of copper withrespect to food additives is very small. Conversely,iron is added to many foods. Because their reactionsare reported to be somewhat similar and more detri-mental to other nutrients than is generally recognized,they are here discussed together.
Ascorbic acid is known to be destroyed by copper. AAis first converted to DHAA which itself is unstable,degrading further to diketogulonic acid, which hasno bioactivity. Conversely, Cu2þ is reduced by AA tolower oxidation states, as is Fe3þ to Fe2þ (Laurenceand Ellis 1972). According to Jukes (1974), Cuþ canreduce peroxides (and hydroperoxides) to form acopper Cu2þ compound and an oxygen-containingfree radical, which can abstract hydrogen froma variety of compounds to form carbon freeradicals. The carbon free radicals can then be oxidizedby Cu2þ compounds to liberate Cuþ. In addition,alkyl radicals can be generated directly from thereaction between Cuþ and diacylperoxides. Theinteractions of Fe3þ and Cu2þ with phenolic anti-oxidants have also been examined (Cort 1974, Cortet al. 1974).
Cu2þ reacts with tocopherol to form the p-tocopherol-quinone, which has no vitamin E or antioxidantactivity. Reduction of Fe3þ to Fe2þ, with the
subsequent measurement of Fe2þ, has been the basisof the colorimetric assay for tocopherol for manyyears. Tocopherol will react to form the p-tocopherol-quinone under most conditions. However, whenEDTA is present, the dimeric keto-ether is formed(Cort et al. 1974). Fe2þ, Cuþ and Cu0 are reportednot to react with tocopherol. Cu2þ, as shown byDwivedi and Arnold (1973), also destroys thiamine(see above). In a separate review article, Borenstein(1971) reported that copper catalyses folic aciddestruction. Methionine and linolenic acid, as wellas a number of other substrates, have been reportedto be degraded to ethylene by AA and copper(Liebermann and Kunishi 1967).
Other reactions detrimental to nutrients
Feller and Macek (1955) have shown that thiazole,produced as a product of thiamine decomposition,promotes the breakdown of vitamin B12. This is aproblem in pharmaceutical preparations and, in fact,according to DeRitter (1969), most of the informationon vitamin interactions is to be found in pharmaceu-tical research publications. Fortunately, the thiaminelevels in most foods are so low that this is not amajor pathway for vitamin B12 destruction. Gambierand Rahn (1957) have shown that riboflavin aids theoxidation of thiamine to thiochrome in concentratedsolutions. Niacinamide and AA react together toform a yellow-coloured nicotinamide-AA complex.Amen (1973) reported that phosphates, phytates,and oxalates in spinach can complex iron and thuscompromise its bioavailability. Carbonyls present inflavour adjuncts can react with pyridoxamine toform inactive Schiff bases, and pyridoxal could reactwith amines similarly. Schroeder (1971) reports lossesof natural pyridoxal and pyridoxamine, as well asof pantothenic acid, biotin, folacin, choline, andinositol in food processing, where some of the vitaminB6 group losses were thought possibly to be due tointeractions.
Biological effects
The interactions of food additives and nutrients havebeen reviewed by Vanderveen (1988), who focussedlargely on food additives as defined by the US FDA,i.e. as ‘functional’ additives, and did not, for instance,include colouring materials. The review is a general
116 M. J. Scotter and L. Castle

one of the types of interactions between food addi-tives and nutrients that have been identified. Theauthor addresses the criteria by which food additivesshould be reviewed for approval. Additive nutrientinteractions were categorized in terms of the follow-ing biological effects, which in some cases may occursimultaneously:
. Decreased nutrient absorption, e.g. EDTAchelation of mineral elements, and the interac-tion of caffeine with riboflavin (Jusko and Levy1975).
. Enhanced nutrient absorption, e.g. the EDTA-mediated mobilization of Zn, Mn, Fe, the AA-enhanced bioavailability of Fe, and theenhanced (albeit conjectural) bioavailability ofCa by citric and malic acids.
. Altered nutrient metabolism, e.g. the metabo-lism of BHT by cytochrome P450 enzymes andimplication of its role in the inhibition of vitaminK absorption; the interaction of vitamin B6
with monosodium glutamate, and the basis ofChinese restaurant syndrome (Folkers et al.1984).
. Altered nutrient excretion, e.g. the enhancedexcretion of vitamin C following consumptionof hemicellulose (Keltz 1978).
. Nutrient destruction, e.g. the addition of pH-lowering substances which may cause thedestruction of the polyglutamate forms of fola-cin, whereas the addition of pH-raising sub-stances can increase the destruction of thiamine(Sauberlich 1985). Basu et al. (1984) reported onthe in vitro effect of AA on the spontaneousreduction of sodium nitrite concentration inreaction mixtures.
Main outcomes and recommendations
It is well established that food additives (and to alesser extent their degradation products) can interactwith one another and with food constituents to forman array of new products. In some instances, suchproducts have been identified, whereas, in others theyare poorly characterized or remain of unknownstructure. The consequences of such interactions arethe main consideration here, with food safety impli-cations being of particular concern. From the infor-mation and data in the key references and otherimportant articles relating to additive–additive andadditive–food component interactions, it is clear thatthe bulk of the research effort in this area has targetedfive main classes of food additive: the sulfur (IV)species of preservatives, synthetic food colouringmaterials, nitrate and nitrite, ascorbic acid, andsorbic acid, though the consequences of these inter-actions are by no means fully understood as yet. Theavailability of this information in the literature hasbeen summarized in table 5.
The effects of pH, temperature, light, reactive species,oxygen, and other parameters on the reactivity ofthese food additives have been discussed. Otherimportant aspects that have been addressed includethe formation of toxic reaction products and muta-genic compounds, protective effects and inhibition,kinetics and mechanisms, and where appropriatethe likelihood and significance of additive reactionsin food and model systems. The amount of availableinformation on the interactions of certain additives,which may have the potential for interaction is sparse,e.g. intense sweeteners, preservatives, gallate esters,
117Chemical interactions between additives in foodstuffs
Table 5. Summary of published literature on additive–additive interactions.
Ascorbicacid
Nitrates/nitrites
Sorbic acid/sorbates
Sulfur (IV)oxo-anions
Syntheticdyes
Natural foodcolours
Otheradditives
Ascorbicacid
intense sweeteners, benzoate,metal cations
Nitrates/nitrites þþ vitamins, phenols, antioxidants,amines, thiols
Sorbic acid/sorbates þþ þþSulfur (IV)oxo-anions
þþ þ þþ vitamins, olefins, amino acids
Synthetic dyes þþ þ – þþ acids, alkalis, preservatives, sugars
Natural foodcolours
þ – – þ – metal cations, preservatives,antioxidants, sugars, acids, alkalis
– None found; þ sparse; þþ significant.

sorbitol, mannitol, polyoxyethylene stabilizers, andmonosodium glutamate. (Note: because of the con-siderable amount of complex information availableon the chemistry of caramels, this group of foodadditives was not included, except where appropriateto discussions on other additives.)
The main conclusions to be drawn from this review interms of future requirements are the following:
. Research effort in this area has been largely tar-geted towards five main classes of food additive:the sulfur (IV) species of preservatives, syntheticfood colouring materials, nitrate and nitrite,ascorbic acid, and sorbic acid. Even so, the con-sequences of their interactions are not fullyunderstood.
. Information on certain additives, which have thepotential for interaction is sparse and these maymerit further studies, e.g.. intense sweeteners (saccharin, acesulfam K,aspartame, cyclamate, NHDC);
. preservatives ( p-hydroxybenzoates, biphenyl,thiabendazole, o-phenylphenol);
. gallate esters;
. sorbitol, mannitol;
. polyoxyethylene stabilizers;
. monosodium glutamate; and
. others, e.g. bulk additives, modified atmo-sphere gases.
. There is a clear requirement for a coordinatedapproach to research on food additive–additiveinteractions, especially for those additives thatmay be of concern.
. There is a need to develop our understanding ofthe important parameters that dictate the out-come of additive interactions. This might beachieved by using the literature data within theframework of the review essentially as a trainingset to evolve the framework into a diagnostic tool.
. There is a need to formalize our understanding ofthe type of information required to indicate wherefurther studies (if any) are required to allow astrategic assessment of additive interactions.
Other workers have sought to formalize schemesfor assessing priorities for safety (toxicity) testing offood additives, based upon a quantitative structure–activity relationship (QSAR) approach. Phillips et al.(1987), for instance, used a modified decision treescheme, based on that of Cramer, Ford and Hall,to allow more chemical structures to be consideredand to take into account advances in toxicology.
The majority of food additives (then) permitted ineither the UK, USA or Canada were processedthrough a modified decision tree questionnaire andtheir classification compared with available chronictoxicity data. Additives were then assigned to threetoxicity classes, low, moderate, or high. Althoughless discriminating than originally suggested, it wasconcluded that the decision tree approach remainsa useful method for classifying compounds in terms oftheir probable toxicity and that further modificationsto the tree could be made in the light of the occur-rence of incorrect assignments. This type of approachis therefore worthy of consideration, especiallyfor assessing the potential reactivity of groups ofadditives that have similar chemical functionality.
As mentioned above, it is feasible to regard the infor-mation summarized here as a training set to developour understanding of the important parameters thatdictate the outcome of additive interactions. Thiscould be achieved by assessing the data within aframework, which could develop into a diagnostictool. The framework (and the relational database inwhich it would operate) would formalize our under-standing of what information is needed and sowould automatically indicate where further studies(if any) are required to allow a strategic assessment ofadditive interactions. In order to predict and assessthe outcome of additive–additive or additive–foodencounters, a provisional set of reaction parametersmust be assembled. With a basic framework in place,the data obtained from the literature could be usedas a Teaching Set to identify any factors overlookedor, conversely, factors proposed that have in factnegligible consequence. Thus, a multilevel frame workoperating systematically may be developed along thefollowing lines:
Level 1. Description of food additives:
. Concentration (e.g. bulk additives, gums andbulk sweeteners versus trace additives, coloursand preservatives).
. Reactivity (e.g. electrophilic, nucleophilic,redox-active, free-radical precursor, chelator,light or heat-sensitive).
Level 2. Description of food components:
. Concentration (e.g. macro-constituents, lipids,proteins and carbohydrates versus micro consti-tuents, vitamins and minerals).
. Reactivity (e.g. electrophilic, nucleophilic,redox-active, free-radical precursor).
118 M. J. Scotter and L. Castle

Level 3. Likelihood of encounter:
. Permitted additive–food combinations (accord-ing to EU Directives):. Occurrence.. Concentration range.
. Actual food-additive combinations (accordingto Food Standards Agency [FSA] database onadditive usage):. Occurrence.. Concentration range.
Level 4. Likelihood of interaction during processingand storage:
. Chemical compatibility.
. Reactivity of the additive with the additive orfood component that it encounters, e.g. nucleo-phile-electrophile, oxidant-oxidizable substrate,free-radical precursor-autoxidizable substrate,nitrosating agent-nitrosatable substrate.
. Time, temperature, pH, water activity.
The types of food in which the additive is permittedgive the reaction conditions that should be consid-ered — with respect to pH and water activity. Thetime and temperature parameters can be taken fromthe foreseeable conditions of use (e.g. a chilled dairyproduct or an extrusion-formed and heat-dried snackproduct). It is likely that knowledge will be incom-plete in this area. The use of chemical modellingto predict the products that may be formed is aforeseeable requirement, as is the use of, for instance,NMR and radio-tracer experiments, to determinethe fate of reactive additives.
Level 5. Consequence of the interaction:
. Known or predictable products that can beassessed toxicologically (e.g. by QSAR).
. Unknown and unpredictable products thatrequire identification.
. Known or unknown products that neverthelessare bound to macromolecules and not absorbed.
Acknowledgement
Financial support for this work was provided by theUK Food Standards Agency.
References
ADAMS, J. B., 1973, Thermal degradation of anthocyanins with parti-cular reference to the 3-glycosides of cyanidin. I. In acidifiedsolution at 100�C. Journal of the Science of Food andAgriculture, 24, 747–762.
ADAMS, J. B., 1997, Food additive–additive interactions involvingsulphur dioxide and ascorbic and nitrous acids: a review.Food Chemistry, 59, 401–409.
ADAMS, J. B., and LANGLEY, F. M., 1995, Interaction between Additivesin Model Food Systems. MAFF Project No. 11371. Campdenand Chorleywood Food Research Association Report.
ADAMS, J. B., and WOODMAN, J. S., 1973, Thermal degradationof anthocyanins with particular reference to the 3-glycosidesof cyanidin. II. The anaerobic degradation of cyanidin-3-rutinoside at 100�C and pH 3.0 in the presence of sodiumsulphite. Journal of the Science of Food and Agriculture, 24,763–768.
AMEN, R. J., 1973, Trace minerals as nutrients. Food Production andDevelopment, 7, 74–78.
ASH, M., and ASH, I., 1995, Handbook of Food Additives (Aldershot:Gower).
BASU,T. K., WEISER, T., and DEMPSTER, J. F., 1984, An in vitro effect ofascorbic acid on the spontaneous reduction of sodium nitriteconcentration in a reaction mixture. International Journal ofVitamin and Nutrition Research, 54, 233–236.
BECK, K. M., 1983, Non-nutritive sweeteners: saccharin and cyclamate.CRC Handbook of Food Additives, 2nd edn, vol. II, edited byT. E. Furia (Boca Raton: CRC Press), pp. 125–185.
BIBEAU, T. C., and CLYDESDALE, F. M., 1978a, Thermal stability ofsubsidiary dyes in amaranth (FD&C Red No. 2). CanadianInstitute of Food Science and Technology Journal, 11, 173–176.
BIBEAU, T. C., and CLYDESDALE, F. M., 1978b, Thermal stability ofsubsidiary dyes associated with FD&C Yellow No. 6. Journalof Food Science, 43, 521–523.
BIRCH, G. G., and PARKER, K., 1974, Vitamin C: Recent Aspects of itsPhysiological and Technological Importance (London: Elsevier).
BIRCH, G. G., and PEPPER, T., 1983, Protection of vitamin C by sugarsand their hydrogenated derivatives. Journal of Agriculturaland Food Chemistry, 31, 980–985.
BOLEY, N. P., CROSBY, N. T., ROPER, P., and SOMERS, L., 1981,Determination of indigo carmine in boiled sweets and similarconfectionery products. Analyst, 106, 710–713.
BORENSTEIN, B., 1971, Rationale and technology of food fortification.CRC Critical Reviews in Food Technology, 2, 171–186.
BORREGO, F., 1995, Neohesperidin DC: properties and applications.Low Calorie Sweeteners: Harmonisation in Europe. Proceedingsof the 1995 International Sweeteners Association Seminar,Prague, May 2–3, 1995, edited by S. G. Lisansky and A. J.Corti (Newbury: CRC Press), pp. 80–86.
BOYLAND, E., and WALKER, S. A., 1974, Effect of thiocyanate onnitrosation of amines. Nature, 248, 601–602.
BRITTON, G., LIAANEN-JENSEN, S., and PFANDER, H., 1995, Carotenoids,vol. 1A: Isolation and Analysis (Basel: Birkhauser).
BUNNELL, R. H., 1968, Enrichment of fruit products and fruit juices.Journal of Agricultural and Food Chemistry, 16, 177–183.
CANALES, I., BORREGO, F., and LINDLEY, M. G., 1983, Neohesperidindihydrochalcone stability in aqueous buffer solutions. Journalof Food Science, 58, 589–591, 643.
CASTLE, L., 1980, A study of oxy-radicals as simple models for cyto-chrome P450-dependent monooxygenation. Thesis, Depart-ment of Chemistry, University of York.
CHICHESTER, C. O., 1972, The Chemistry of Plant Pigments (New York:Academic Press).
119Chemical interactions between additives in foodstuffs

CHRISTIANSEN, L. N., JOHNSON, R. W., KAUTTER, D. A., HOWARD, J. W.,and AUNAN, W. J., 1973, Effect of nitrite and nitrate on toxinproduction by Clostridium botulinum and on nitrosamineformation in perishable canned comminuted cured meat.Applied Microbiology, 25, 357–362.
COLEMAN, M. H., HANNAN, R. S., and OSBORNE, D. R. D., 1974, Curedmeats. British Patent 1377154.
CORT, W. M., 1974, Antioxidant activity of tocopherols, ascorbylpalmitate and ascorbic acid and their mode of action. Journalof the American Oil Chemists Society, 51, 321–325.
CORT, W. M., 1987, Effects of treatment with food additives onnutrients. Nutritional Evaluation of Food Processing, 3rd edn,edited by E. Karmas and R. S. Harris (New York: VanNostrand Reinhold), pp. 447–456.
CORT, W. M., SCOTT, J. W., MERGENS, W. J., OSADCA, M., and ARAUJO,M., 1974, Antioxidant activity and stability of a number ofchromans. Presented at the 65th Annual Spring meeting, Am.Oil Chem., Mexico, Apr. 27. Abstr. Journal of the American OilChemists Society, 51, 279A.
COULTATE, T. P., 2002, Food: The Chemistry of its Components, 4th edn(Cambridge: Royal Society of Chemistry).
CROSS, H. L., PEPPER, T., KEARSLEY, M. W., and BIRCH, G. G., 1985,Mineral complexing properties of food carbohydrates. Starke,37, 132–135.
CULIK, J., and KELLNER, V., 1995, Toxic compounds arising by inter-action of food constituents with food additives. Part A. Nitrosocompounds. Natural Toxic Compounds of Foods (Boca Raton:CRC Press), pp. 229–252.
DAMANT, A. P., 1990, An investigation into the stability of azo foodcolours to other food additives in soft drinks. PhD thesis,University of Reading.
DAMANT, A., REYNOLDS, S., and MACRAE, R., 1989, The structuralidentification of a secondary dye produced from the reactionbetween sunset yellow and sodium metabisulphite. FoodAdditives and Contaminants, 6, 273–282.
DAVIDEK, J., VELIsEK, J., and POKORNY, J., 1990, Chemical ChangesDuring Food Processing. Developments in Food Science 21(Oxford: Elsevier Science).
DAVIES, C. G. A., and WEDZICHA, B. L., 1992, Kinetics of the inhibitionof ascorbic acid browning by sulphite. Food Additives andContaminants, 9, 471–477.
DAVIES, R., DENNIS, M. J., MASSEY, R. C., and MCWEENY, D. J., 1978,Some effects of phenol and thiol-nitrosation reactions onN-nitrosamine formation. Environmental Aspects of N-nitrosoCompounds, edited by E. A. Walker, M. Castegnaro, L.Griciute and R. E. Lyle (Lyon: IARC), pp. 183–197.
DAVIS, R. E., 1968, Inorganic Sulfur Chemistry, edited by G. Nickless(Amsterdam: Elsevier), p. 85.
DAY, W. C., and ERDMANN, J. G., 1963, Ionene: a thermal degradationproduct of b-carotene. Science, 141, 808.
DERITTER, E., 1969, Vitamin liquid formulation. Unpublished datapresented at Meeting of Society of Pharmacists, St Louis, MO.
DOUGLASS, M. L., KABACOFF, B. L., ANDERSON, G. A. and CHENG, M.C., 1978, The chemistry of nitrosamine formation, inhibitionand destruction. Journal of the Society of Cosmetic Chemists,29, 581.
DWIVEDI, B. K., and ARNOLD, R. G., 1973, Chemistry of thiamindegradation in food products and model systems: a review.Journal of Agricultural and Food Chemistry, 21, 54–60.
EC, 1995a, European Parliament and Council Directive 95/2/EC of 20February 1995 on food additives other than colours and sweet-eners. Official Journal of the European Communities, No. L61
of 18 March 1995, 1–140.EC, 1995b, Commission Directive 95/45/EC, of 26 July 1995 laying
down specific criteria of purity concerning colours for usein foodstuffs. Official Journal of the European Communities,No. L226 of 22 September 1995, 1–44.
EISENBRANDT, J., and LANG, E., 1966, Uber die reduktive Spaltungvon Brillianschwarz BN durch Ascorbinsaure und uber dieautokatalytische Ausbleichung des Farbstoffes. DeutscheLebensmittel Rundschau, 62, 167–169.
FELLER, B. A., and MACEK, T. J., 1955, Effect of thiamin hydrochlorideon the stability of solutions of crystalline vitamin B12. Journalof the American Pharmaceutical Association, 44, 662–665.
FINOT, P. A., AESCHBACHER, H. V., HURRELL, R. F., and LIARDON, R.,1990, The Maillard Reaction in Food Processing, HumanNutrition and Physiology (Basel: Birkhauser).
FLEMING, M., PARKER, K. J., and WILLIAMS, J. C., 1968, Aspects ofthe chemistry of the browning fraction of reducing sugars.Proceedings of the International Society of Sugar CaneTechnologists, 13, 1781–1800.
FOGG, A. G., and SUMMAN, A. M., 1983, Differential-pulse polaro-graphic monitoring of permitted synthetic food colouring mat-ters and ascorbic acid in accelerated light degradation studiesand the spectrophotometric determination of ammonia andsimpler amines formed. Analyst, 108, 691–700.
FOGG, A. G., and SUMMAN, A. M., 1984, Further differential-pulsepolarographic and visible spectrophotometric studies of thedegradation of permitted synthetic food colouring matterswith and without the addition of ascorbic acid. Analyst, 109,743–747.
FOLKERS, K., SHIZUKUISHI, S., WILLIS, R., SCUDDER, S. L., TAKEMURA,K., and LONGENECKER, J. B., 1984, The biochemistry of vitaminB6 is basic to the cause of Chinese restaurant syndrome.Hoppe-Seyler’s Zeitschrift fur Physiologische Chemie, 365, 405–414.
FORTMANN, K. L., and JOINER, R. R., 1971, Wheat Chemistry andTechnology, edited by Y. Pomeranz (St Paul: AACC), p. 51.
FOX, J. B., FIDDLER, R. N., and WASSERMAN, A. E., 1981, Initialreaction intermediates in the oxidation of ascorbic acid bynitrous acid. Journal of Food Protection, 44, 28–32.
FRANCIS, F. J., 1989, Food colorants — anthocyanins. Critical Reviewsin Food Science and Nutrition, 28, 273–314.
FRIJTERS, J. E. R., GRIFFITHS, N. M., MATHER, A. M., REYNOLDS, J.,and FENWICK, G. R., 1981, Effect of sulphur dioxide on thechemical composition and odour of mustard paste. ChemicalSenses, 6, 33–43.
GAMBIER, A. S., and RAHN, E. P. G., 1957, The combination ofB-complex vitamins and ascorbic acid in aqueous solutions.Journal of the American Pharmaceutical Association ScientificEdition, 46, 134–140.
GARCIA-PRIETO, J. C., MATEOS, R., CALLE, E., and CASADO, J., 1998,Inhibition of nitrosation by the reaction medium. Journal ofAgricultural and Food Chemistry, 46, 3517–3520.
GARCIA-VIGUERA, C., and BRIDLE, P., 1999, Influence of structureon colour stability of anthocyanins and flavylium salts withascorbic acid. Food Chemistry, 64, 21–26.
GARDNER, L. K., and LAWRENCE, G. D., 1993, Benzene productionfrom decarboxylation of benzoic-acid in the presence of ascor-bic-acid and a transition-metal catalyst. Journal of Agriculturaland Food Chemistry, 41, 693–695.
GLORIA, M. B. A., GRULKE, E. A., and GRAY, J. I., 1993, Effect oftype of oxidation on beta-carotene loss and volatile productsformation in model systems. Food Chemistry, 46, 401–406.
GRAY, J. I., and DUGAN, L. R., 1975, Inhibition of N-nitrosamineformation in model food systems. Journal of Food Science, 40,981–984.
GRIFFITHS, N. M., MATHER, A. M., FENWICK, G. R., FRIJTERS, J. E. R.,and MERZ, J. H., 1980, An effect of sulphur dioxide on theodour of mustard paste. Chemistry and Industry (London),239–240.
GROTEN, J. P., BUTLER, W., FERON, V. J., KOZIANOWSKI, G., RENWICK,A. C., and WALKER, R., 2000, An analysis of the possibility forhealth implications of joint actions and interactions between
120 M. J. Scotter and L. Castle

food additives. Regulatory Toxicology and Pharmacology, 31,77–91.
GUTTERIDGE, J. M. C., and QUINLAN, G. J., 1986, Carminic acid-promoted oxygen radical damage to lipid and carbohydrate.Food Additives and Contaminants, 3, 289–293.
HARBORNE, J. B., MARBRY, T. J., and MARBRY, H., 1988, TheFlavanoids (London: Chapman & Hall).
HARTMAN, P. E., 1983, Review: Putative mutagens and carcinogensin foods. II. Sorbate and sorbate-nitrite interactions.Environmental Mutagenesis, 5, 217–222.
HASHIMOTO, N., 1972, Oxidation of higher alcohols by melanoidinsin beer. Journal of the Institute of Brewing, 78, 43–51.
HAVLIKOVA, L., 1985, PhD thesis. Prague, Institute of ChemicalTechnology.
HAYNES, L. J., 1948, Physiologically active unsaturated lactones.Quarterly Reviews (London), 2, 46–72.
HEINTZE, K., 1976, Uber die gegenseitige Beeinflussung vonSorbinsaure und schwefliger Saure. Die industrielle Obst- undGemuseverwert, 61, 555–556.
HENDRY, G. A. F., and HOUGHTON, J. D., 1992,Natural Food Colorants(London: Blackie).
HAGGLUND, E., and RINGBOM, A., 1926, Uber die Sulfitaddition anungesattigte Verbindungen. Zeitschrift fur Anorganische undAllgemeine Chemie, 150, 231–253.
HOMLER, B. E., 1984, Properties and stability of aspartame. FoodTechnology, 38, 50–55.
HSIEH, Y. P., and HARRIS, N. D., 1991, Destructive effect of aspartameon ascorbic acid in Cu-catalysed solutions. Journal of FoodScience, 56, 14–16, 20.
HUSSEIN, M. M., DAMELIA, R. P., MANZ, A. L., JACIN, H., and CHEN,
W. T. C., 1984, Determination of reactivity of aspartame withflavor aldehydes by gas-chromatography, HPLC and GPC.Journal of Food Science, 49, 520–524.
ISHIMITSU, S., OHMORI, N., TSUJI, S., and SHIBATA, T., 1995, Formationof a hydroxyl radical from tar dye by photoillumination.Chemical and Pharmaceutical Bulletin, 43, 1810–1812.
IVEY, F. J., 1977, Potassium sorbate as a partial replacement of nitrite.Food Additives: Competitive, Regulatory and Safety Problems.Hearings before the Select Committee on Small Business,Unites States Senate, 95th Congress, Part 2 (Washington,DC: US Government Printing Office).
JACKMAN, R. L., YADA, R. Y., TUNG, M. A., and SPEERS, R. A., 1987,Anthocyanins as food colorants — a review. Journal of FoodBiochemistry, 11, 201–247.
JUKES, A. E., 1974, The organic chemistry of copper. Advances inOrganometallic Chemistry, vol. 12, edited by F. G. A. Stoneand R. West (New York: Academic Press).
JURD, L., 1964, Reactions involved in sulfite bleaching of anthocya-nins. Journal of Food Science, 29, 16–19.
JUSKO, W. J., and LEVY, G., 1975, Absorption, protein binding andelimination of riboflavin. Riboflavin, edited by R. S. Rivilin(New York: Plenum), p. 99.
KALUS, W. H., MUNZNER, R., and FILBY, W. G., 1990, Isolationand characterization of some products of the BHA-nitritereaction: examination of their mutagenicity. Food Additivesand Contaminants, 7, 223–233.
KANASAWUD, P., and CROUZET, J. C., 1990, Mechanism of formationof volatile compounds by thermal degradation of carotenoidsin aqueous medium. 1. ß-Carotene degradation. Journal ofAgricultural and Food Chemistry, 38, 237–243.
KEARSLEY, M. W., and BIRCH, G. G., 1985, The chemistry andmetabolism of starch-based sweeteners. Food Chemistry, 16,
191–207.KELTZ, F., 1978, Urinary ascorbic acid excretion in the human
as affected by dietary fibre and zinc. American Journal ofClinical Nutrition, 31, 1167–1171.
KHANDELWAL, G. D., and WEDZICHA, B. L., 1990a, Nucleophilicreactions of sorbic acid. Food Additives and Contaminants, 7,685–694.
KHANDELWAL, G. D., and WEDZICHA, B. L., 1990b, Derivatives ofsorbic acid-thiol adducts. Food Chemistry, 37, 159–169.
KNOWLES, M. E., GILBERT, J., and MCWEENY, D. J., 1974, C-nitrosa-tion products in food. Abstracts of the 4th InternationalCongress of Food Science and Technology, 9a, 214.
KNOWLES, M. E., GILBERT, J., and MCWEENY, D. J., 1975, Thepotential formation of C-nitroso compounds in smoked, curedmeats. N-nitroso Compounds in the Environment, edited byP. Bogovski and E. A. Walker (Lyon: IARC), 9, pp. 115–120.
KROYER, G., PINGER, R., WASHUETTLL, J., and STEINER, I., 1993,Literature review on stability and interaction studies ofaspartame and acesulfame-K in food. Ernahrung/Nutrition,17, 546–549.
KROYER, G., and WASHUETTL, J., 1982, Interactions of artificialsweeteners with food additives. Recent Developments in FoodAnalysis, edited by W. Baltes, P. B. Czedik-Eysenberg andW. Pfannhauser (Weinheim: Verlag Chemie), pp. 428–433.
KRUKAR, R. M., 1980, Flavors and colors in extruded snack foods.Cereal Foods World, 25, 596–598.
KUHN, R., and WINTERSTEIN, A., 1932, Conjugated double bonds. PartXXV. Thermal degradation of carotene dyestuffs. Berichte derDeutschen Chemischen Gesellschaft, 65B, 1873–1880.
KUHN, R., and WINTERSTEIN, A., 1933a, Berichte der DeutschenChemischen Gesellschaft, 66, 429–432.
KUHN, R., and WINTERSTEIN, A., 1933b, Berichte der DeutschenChemischen Gesellschaft, 66, 1733–1741.
KURECHI, T., KIKUGAWA, K., and KATO, T., 1980, Effects of alcoholson nitrosamine formation. Food and Cosmetics Toxicology, 18,591–595.
LANCASTER, F. E., and LAWRENCE, J. F., 1986, Thermal decompositionof the food colours amaranth, sunset yellow FCF and tartrazinein the presence of sucrose and glucose. Food Additives andContaminants, 3, 295–304.
LAROE, E. G., and SHIPLEY, P. A., 1970, Whiskey composi-tion: formation of alpha- and beta-ionone by the thermaldecomposition of beta-carotene. Journal of Agricultural andFood Chemistry, 18, 174–175.
LATHIA, D., and BLUM, A., 1989, Role of vitamin E as nitrite scavengerand N-nitrosamine inhibition: a review. International Journalfor Vitamin and Nutrition Research, 59, 430–438.
LAURENCE, G. S., and ELLIS, K. J., 1972, The detection of a complexintermediate in the oxidation of ascorbic acid by ferricion. Journal of the Chemical Society, Dalton Transactions,1667–1670.
LEE, S. H., and CASSENS, R. G., 1976, Nitrite binding sites in myoglo-bin. Journal of Food Science, 41, 969–970.
LIEBERMANN, M., and KUNISHI, A. T., 1967, Propanal may be aprecursor of ethylene in metabolism. Science, 158, 938.
LIN, Y. D., CLYDESDALE, F. M., and FRANCIS, F. J., 1971, Organic acidprofiles of thermally processed, stored spinach puree. Journal ofFood Science, 36, 240–242.
LINDLEY, M. G., 1996, Neohesperidin dihydrochalcone: recent findingsand technical advances. Advances in Sweeteners, edited by T. H.Grenby (London: Blackie), pp. 240–252.
MABRY, T. J., 1980, Secondary Plant Products, edited by E. A. Bell andB. V. Charlwood (Springer-Verlag, Berlin).
MAFF, 1987, Survey of Colour Usage in Food. 19th Report of theSteering Group on Food Surveillance. The Working Party onFood Colours. Food Surveillance Paper No. 19 (London:HMSO).
MAGGIORA, L. L., PETKE, J. D., GOPAL, D., IWAMOTO, R. T., andMAGGIORA, G. M., 1985, Experimental and theoretical-studiesof Schiff-base chlorophylls. Photochemistry and Photobiology,42, 69–75.
121Chemical interactions between additives in foodstuffs

MAJEED, M., BADMAEV, V., SHIVAKUMAR, U., and RAJENDRAN, R.,2000, Curcuminoids antioxidant phytonutrients. SabsinaCorporation [http://www.curcuminoids.com].
MARKAKIS, P., 1982, Anthocyanins as Food Colors, edited byP. Markakis (New York: Academic Press).
MAROVATSANGA, L., and MACRAE, R., 1987, The determination ofadded azo dye in soft drinks via its reaction products. FoodChemistry, 24, 83–98.
MARTY, M., and BERSET, C., 1988, Degradation products of trans-ß-carotene produced during extrusion cooking. Journal of FoodScience, 53, 1880–1886.
MARTY, M., and BERSET, C., 1990, Factors affecting the thermaldegradation of all-trans-ß-carotene. Journal of Agriculturaland Food Science, 38, 1063–1067.
MASSEY, R. C., and LEES, D., 1992, Surveillance of preservativesand their interactions in foodstuffs. Food Additives andContaminants, 9, 435–440.
MAT HASHIM, D. B., MOORTHY, S. N., MITCHELL, J. R., HILL, S. E.,LINFOOT, K. J., and BLANSHARD, J. M. V., 1992, The effects oflow levels of antioxidants on the swelling and solubility ofCassava starch. Starke, 44, 471–475.
MAYER, D., and KEMPER, F., 1991, Acesulfame-K (New York: MarcelDekker).
MAZZA, G., 1997, Anthocyanins: structure, stability and analysis. FoodScience: Advances and Perspective in Latin America, edited byD. B. Rodriguez-Amaya and G. M. Pastore. Proceedings of the1st Latin-American Symposium on Food Science, 1995,Unicamp, Brazil.
MCCORMICK, R. D., 1971, Allura red: new food color offers greaterbrilliance and stability. Food Products Development, 5, 26–28.
MCKEOWN, G. G., 1965, Composition of oil-soluble annatto foodcolours. III. Structure of the yellow pigment formed by thethermal degradation of bixin. Journal of the Association ofOfficial Analytical Chemists, 48, 835–837.
MCNEAL, T. P., NYMAN, P. J., DIACHENKO, G. W., and KOLLIFIELD,
H. C., 1993, Survey of benzene in foods using headspaceconcentration techniques and capillary gas chromatography.Journal of the AOAC International, 76, 1213–1219.
MIKUSCHKA, G., 1995, Saccharin and cyclamate. Low CalorieSweeteners: Harmonisation in Europe. Proceedings of the 1995International Sweeteners Association Seminar, Prague, 2–3May 1995, edited by S. G. Lisansky and A. J. Corti(Newbury: CRC Press), pp. 87–90.
MOORE, H. W., and FOLKERS, K., 1968, Vitamin B12. II. Chemistry.The Vitamins, vol. 2, edited by W. H. Sebrell, Jr and R. H.Harris (New York: Academic Press).
MOORE, W. J., 1972, Physical Chemistry (London: Longman).MULIK, J. D., and ERDMANN, J. G., 1963, Genesis of hydrocarbons of
low molecular weight in organic-rich aquatic systems. Science,141, 806–807.
NAMIKI, M., OSAWA, T., KADA, T., TSUJI, K., and NAMIKI, K., 1983,Formation of C-nitro and C-nitroso mutagens by the reactionof nitrite with sorbic acid and its analogues and their inactiva-tion with food constituents. Carcinogens and Mutagens in theEnvironment, 3, 109–117.
NETO, R. O. T., KAREL, M., SAGUY, I., and MIZRAHI, S., 1981, Oxygenuptake and ß-carotene decoloration in a dehydrated foodmodel. Journal of Food Science, 46, 665–669, 676.
NURSTEN, H. E., and WILLIAMS, G., 1969, The stability of coal tarfood colours permitted in the UK. Chemistry and Industry,1798–1803.
O’BRIEN, J., NURSTEN, H. E., CRABBE, M. J. C., and AMES, J. M., 1998,The Maillard Reaction in Foods and Medicine. Proceedings ofthe 6th International Conference on the Maillard Reaction(Cambridge: Royal Society of Chemistry), pp. 141–146.
ONYEWU, P. N., DAUN, H., and HO, C.-T., 1982, Formation oftwo thermal degradation products of ß-carotene. Journal ofAgricultural and Food Chemistry, 30, 1147–1151.
ONYEWU, P. N., DAUN, H., and HO, C.-T., 1986, Characterizationof ß-carotene thermal degradation products in a model foodsystem. Journal of the American Oil Chemists Society, 63,
1437–1441.OSAWA, T., ISHIBASHI, H., NAMIKI, M., KADA, T., and TSUJI, K., 1986,
Desmutagenic action of food components on mutagens formedby the sorbic acid/nitrite reaction. Agricultural and BiologicalChemistry, 50, 1971–1977.
OSAWA, T., and NAMIKI, M., 1982, Mutagen formation in the reactionof nitrite with the food components analogous to sorbic acid.Agricultural and Biological Chemistry, 46, 2299–2304.
OSAWA, T., NAMIKI, M., and NAMIKI, K., 1982, Mutagen formation inthe nitrite-piperic acid reaction. Agricultural and BiologicalChemistry, 46, 3105–3108.
OZAKI, A., KITANO, M., ITOH, N., KURODA, K., FURUSAWA, N.,MASUDA, T., and YAMAGUCHI, H., 1998, Mutagenicity andDNA-damaging activity of decomposed products of foodcolours under UV radiation. Food and Chemical Toxicology,36, 811–817.
PAPADOPOULOU, K., and AMES, J. M., 1993, Separation of thecoloured reaction products formed in ß-carotene and/orphenylalanine model systems. Food Chemistry, 46, 65–71.
PARK, J. R., and WILLIAMS, D. L. H., 1976, Reaction of dinitrogentrioxide with 2-methylpropene and other alkenes. Journal of theChemical Society, Perkin Transactions, 2, 828–832.
PEISER, G. D., and YANG, S. F., 1977, Chlorophyll destruction by thebisulphite-oxygen system. Plant Physiology, 60, 277–281.
PEISER, G. D., and YANG, S. F., 1978, Chlorophyll destruction inthe presence of bisulphite and linoleic acid hydroperoxide.Phytochemistry, 17, 79–84.
PEISER, G. D., and YANG, S. F., 1979, Sulphite mediated destructionof ß-carotene. Journal of Agricultural and Food Chemistry, 27,446–449.
PEKKARINEN, L., and RISSANEN, P., 1966, Suomen kemistilehti B, 39, 50;Chemical Abstracts 1966, 65, 7016.
PENSABENE, J. W., FIDDLER, W., DOERR, R. C., LAKRITZ, L., andWASSERMAN, A. E., 1975, Formation of dimethylnitrosaminefrom commercial lecithin and its components in a modelsystem. Journal of Agricultural and Food Chemistry, 23,
979–980.PETROPAVLOVSKI, E. I., and USINOVA, A. V., 1967, Izvestiya Vysshikh
Uchebnykh Zavedenii Pishchchevaya Tekhnologiya, 6, 36;Chemical Abstracts 1968, 68, 72241.
PHILIP, T., and FRANCIS, F. J., 1971, Oxidation of capsanthin. Journalof Food Science, 36, 96–97.
PHILLIPS, J. C., PURCHASE, R., WATTS, P., and GANGOLLI, S. D., 1987,An evaluation of the decision tree approach for assessingpriorities for safety testing of food additives. Food Additivesand Contaminants, 4, 109–123.
ROSE, A. H., and PILKINGTON, B. J., 1989, Mechanisms of Action ofFood Preservation Procedures, edited by G. W. Gould (London:Elsevier), pp. 201–223.
ROSENTHAL, I., YANG, G. C., BELL, S. J., and SCHER, A. L., 1988, Thechemical photosensitizing ability of certified colour additives.Food Additives and Contaminants, 5, 563–571.
ROSS, K. D., 1975, Reduction of the azo food dyes FD&C Red 2(amaranth) and FD&C Red 40 by thermally degraded D-fruc-tose and D-glucose. Journal of Agricultural and Food Chemistry,23, 475–478.
ROUSSEAU, B., and ROSAZZA, P. N., 1998, Reaction of ferulic acid withnitrite: Formation of 7-hydroxy-6-methoxy-1,2(4H)-benzoxa-zin-4-one. Journal of Agricultural and Food Chemistry, 46,
3314–3317.
122 M. J. Scotter and L. Castle

SAGUY, I., KOPELMAN, I. J., and MIZRAHI, S., 1978, Thermal kineticdegradation of betanin and betalamic acid. Journal ofAgricultural and Food Chemistry, 26, 360–368.
SALMINEN, S., and HALLIKAINEN, A., 1990, Sweeteners. Food Additives,edited by A. L. Branen, P. M. Davidson and S. Salminen(New York: Marcel Dekker), pp. 297–326.
SAUBERLICH, H. E., 1985, Bioavailability of vitamins. Progress in Foodand Nutrition Science, 9, 1–33.
SAXBY, M. J., and STEPHEN, M. A., 1981, MAFF Special Project M15,Part X (Leatherhead: Food Research Association).
SCHABER, P. M., HUNT, J. E., FRIES, R., and KATZ, J. J., 1984,High-performance liquid-chromatographic study of the chloro-phyll allomerization reaction. Journal of Chromatography, 316,25–41.
SCHROEDER, H. A., 1971, Losses of vitamins and trace minerals result-ing from processing and preservation of foods. AmericanJournal of Clinical Nutrition, 24, 562–573.
SCHWEIGERT, B. S., and LUSHBOUGH, C. H., 1960, Effects of processingon meat products. Nutritional Evaluation of Food Processing,edited by R. S. Harris and H. von Loesecke (New York: Wiley).
SCOTTER, M. J., 1983, A Study into the Fate of Black PN Used to StainMeat. MAFF Food Science Laboratory Report No. 83/17(London: Food Standards Agency).
SCOTTER, M. J., 1995a, Characterisation of the coloured thermaldegradation products of bixin from annatto and a revisedmechanism for their formation. Food Chemistry, 53, 177–185.
SCOTTER, M. J., 1995b, Thermal Degradation of Annatto in ModelSystems and Foods. MAFF Central Science LaboratoryReport No. FD 94/174 (London: Food Standards Agency).
SCOTTER, M. J., 2003, Properties and determination of syntheticpigments. Encyclopaedia of Food Sciences and Nutrition, editedby B. Caballero, L. Trugo and P. M. Finglas (London:Academic Press), pp. 1556–1567.
SCOTTER, M. J., WILSON, L. A., APPLETON, G. P., and CASTLE, L., 2000,Analysis of annatto (Bixa orellana) food colouringformulations. 2. Determination of aromatic hydrocarbonthermal degradation products by gas chromatography.Journal of Agricultural and Food Chemistry, 48, 484–488.
SEBRANEK, J. G., and FOX, J. B., Jr, 1985, A review of nitrite andchloride chemistry: interaction and implications for curedmeats. Journal of the Science of Food and Agriculture, 36,
1169–1182.SINGER, J. W., and VON ELBE, J. H., 1979, Degradation rates of
vulgaxanthine I. Journal of Food Science, 45, 489–491.SINGH, M., 1970, Stability of color additives: FD&C Red 2 in baked
goods. Journal of the Association of Official Analytical Chemists,53, 23–25.
SKOVGAARD, N., 1992, Microbiological aspects and technological need:technological needs for nitrates and nitrites. Food Additivesand Contaminants, 9, 391–397.
STANGE CO., 1970, Manual of Certified Food Colours (Chicago); citedin Lancaster and Lawrence (1986).
SUGIMOTO, N., KAWASAKI, Y., SATO, K., AOKI, H., ICHI, T., KODA, T.,YAMAZAKI, T., and MAITANI, T., 2002, Structure of acid-stablecarmine. Journal of the Food Hygienic Society of Japan, 43,
18–23.TARU, Y., and TAKAOKA, K., 1982, Thermal stability and mechanism of
the thermal degradation of the tarry dyes for food additives.Journal of the Japanese Society of Colour Material, 55, 537–545;cited in Ozaki et al. (1998).
TAYLOR, S. L., HIGHLEY, N. A., and BUSH, R. K., 1986, Sulfites infoods: uses, analytical methods, residues, fate, exposure assess-ment, metabolism, toxicity and hypersensitivity. Advances inFood Research, 30, 1–76.
THAKUR, B. R., SINGH, R. K., and ARYA, S. S., 1994, Chemistryof sorbates — a basic perspective. Food Reviews International,10, 71–91.
TIL, H. P., and FERON, V. J., 1992, Toxicology of sulphiting agents I:Animal studies. Food Additives and Contaminants, 9, 587–595.
TIMBERLAKE, C. F., and BRIDLE, P., 1968, Flavylium salts resistant tosulphur dioxide. Chemistry and Industry (London), 1489.
TONNESEN, H. H., ARRIETA, A. F., and LERNER, D., 1995, Studies oncurcumin and curcuminoids. Part XXIV: Characterization ofthe spectroscopic properties of the naturally occurring curcu-minoids and selected derivatives. Pharmazie, 50, 689–693.
TSANG, W. S., CLARKE, M. A., and PARRISH, F. W., 1985,Determination of aspartame and its breakdown products in softdrinks by reverse-phase chromatography with UV detection.Journal of Agricultural and Food Chemistry, 33, 734–738.
USSEGLIO-TOMASETT, L., 1992, Properties and use of sulphur dioxide.Food Additives and Contaminants, 9, 399–404.
VANDERVEEN, J. E., 1988, Interactions of food additives and nutrients.Nutrient Interactions, edited by C. E. Bodwell and J. W.Erdman, Jr (New York: Marcel Dekker), pp. 351–363.
VETSCH, W., 1985, Aspartame: technical considerations and predicteduse. Food Chemistry, 16, 245–258.
VIDYASAGAR, K., and ARYA, S. S., 1983, Stability of sorbic acidin orange squash. Journal of Agricultural and Food Chemistry,31, 1262–1264.
VON RYMON LIPINSKI, G.-F., 1985, The new intense sweetener acesul-fame K. Food Chemistry, 16, 259–269.
VONRYMONLIPINSKI, G.-F., 1995, Acesulfame-K— key properties andapplications. Low calorie sweeteners: harmonisation in Europe.Proceedings of the 1995 International Sweeteners AssociationSeminar, Prague, 2–3 May 1995, edited by S. G. Lisansky andA. J. Corti (Newbury: CRC Press), pp. 64–68.
WALKER, E. A., PIGNATELLI, B., and FRIESEN, M., 1982, The role ofphenols in catalysis of nitrosamine formation. Journal of theScience of Food and Agriculture, 33, 81–88.
WALKER, R., 1990, Toxicology of sorbic acid and sorbates. FoodAdditives and Contaminants, 7, 671–676.
WALTERS, C. L., 1992, Reactions of nitrate and nitrite in foods withspecial reference to the determination of N-nitroso compounds.Food Additives and Contaminants, 9, 441–447.
WALTERS, C. L., and TAYLOR, A., McM., 1964, Nitrite metabolism bymuscle in vitro. Biochimica et Biophysica Acta, 86, 448–458.
WEDZICHA, B. L., 1984, Chemistry of Sulphur Dioxide in Foods(Barking: Elsevier).
WEDZICHA, B. L., 1987, Chemistry of sulfur-dioxide in vegetabledehydration, International Journal of Food Science and Techno-logy, 22, 433–450.
WEDZICHA, B. L., 1991, Inhibition of browning reactions by sulfites.Nutritional and Toxicological Consequences of Food Processing,edited by M. Friedman (New York: Plenum), pp. 217–236.
WEDZICHA, B. L., 1992, Chemistry of sulphiting agents in food. FoodAdditives and Contaminants, 9, 449–459.
WEDZICHA, B. L., 1995, Interactions involving sulfites, sorbic acidand benzoic acid. Ingredient Interactions: Effects on FoodQuality, edited by A. G. Gaonkar (New York: MarcelDekker), pp. 529–559.
WEDZICHA, B. L., and BROOK, M. A., 1989, Reaction of sorbicacid with nucleophiles: preliminary studies. Food Chemistry,31, 29–40.
WEDZICHA, B. L., and CLAYTON, A. L., 1994, Oxidation of sulfite ina caramel-containing system, Food Chemistry, 51, 395–397.
WEDZICHA, B. L., and GODDARD, S. J., 1988, The dissociation-constant of hydrogen sulfite ion at high ionic-strength.Food Chemistry, 30, 67–71.
WEDZICHA, B. L., and GODDARD, S. J., 1991, The state of sulfur-dioxideat high-concentration and low water activity. Food Chemistry,40, 119–136.
WEDZICHA, B. L., GODDARD, S. J., and ZEB, A., 1993, Approach to thedesign of model systems for food additive–food componentinteractions. Food Chemistry, 47, 129–132.
123Chemical interactions between additives in foodstuffs

WEDZICHA, B. L., and HERRERA-VILORIA, J. C., 1991, Formationof sulphate ion during the dehydration of sulphited vegetables.Food Additives and Contaminants, 8, 683–692.
WEDZICHA, B. L., HILL, D., and COCKSHOTT, J. B., 1982, The reactivityof intermediates in non-enzymic browning reactions towardsnitrite ion. Journal of the Science of Food and Agriculture, 33,306–308.
WEDZICHA, B. L., and LAMIKANRA, O., 1983, Sulphite mediateddestruction of ß-carotene: the partial characterisation ofreaction products. Food Chemistry, 10, 275–283.
WEDZICHA, B. L., and RUMBELOW, S. J., 1981, The reaction of anazo food dye with hydrogen sulphite ions. Journal of theScience of Food and Agriculture, 32, 699–704.
WEDZICHA, B. L., and WEI, T., 1989, Kinetics of the reaction between3-deoxyhexosulose and nitrite ion. Food Chemistry, 31,
189–203.
WHITE, R. C., JONES, I. D., and GIBBS, E., 1963, Determinationof chlorophylls, chlorophyllides, phaeophytins and phaeophor-bides in plant material. Journal of Food Science, 28, 431.
WINSTON, G. W., and CEDARBAUM, A. I., 1982, Oxidative decarboxy-lation of benzoate to carbon dioxide by rat liver microsomes:a probe for oxygen radical production during microsomalelectron transfer. Biochemistry, 21, 4265–4270.
YANG, S. F., 1970, Sulphoxide formation from methionine or itssulphide analogues during aerobic oxidation of sulphite.Biochemistry, 9, 5008–5014.
YANG, S. F., 1973, Destruction of tryptophan during the aerobicoxidation of sulphite ions. Environmental Research, 6, 395–402.
YANG, S. F., 1984, Reactions of oxidation intermediates of sulfite spe-cies with some cellular components of plants, Food Chemistry,15, 113–124.
124 M. J. Scotter and L. Castle

Related Documents











![H20youryou[2] · 2020. 9. 1. · 65 pdf pdf xml xsd jpgis pdf ( ) pdf ( ) txt pdf jmp2.0 pdf xml xsd jpgis pdf ( ) pdf pdf ( ) pdf ( ) txt pdf pdf jmp2.0 jmp2.0 pdf xml xsd](https://static.cupdf.com/doc/110x72/60af39aebf2201127e590ef7/h20youryou2-2020-9-1-65-pdf-pdf-xml-xsd-jpgis-pdf-pdf-txt-pdf-jmp20.jpg)