. Distance-Based Phylogenetic Reconstruction (part II) Tutorial #11 © Ilan Gronau

. Distance-Based Phylogenetic Reconstruction ( part II ) Tutorial #11 © Ilan Gronau.
Dec 20, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

.
Distance-Based Phylogenetic
Reconstruction (part II)
Tutorial #11
© Ilan Gronau

.
Phylogenetic Reconstruction
We’d like to study the evolutionary history of species

.
Distance-Based Reconstruction
Given ML pairwise (evolutionary) distances between species,find the edge-weighted tree best describing this metric
The input: distance matrix – D– D(i,i) = 0– D(i,j) = D(j,i)– [ D(i,j) ≤ D(i,k) + D(k,j) ]
The Output: edge-weighted tree – T
• If D is additive, then DT = D
• Otherwise, return a tree best ‘fitting’ the input – D.
Note: Usually ML-estimated pairwise distances are not additive, but they are ‘close’ to some additive metric
metric
Bear Raccoon Weasel Seal Dog
Bear 0 26 34 29 32
Raccoon 26 0 42 44 48
Weasel 34 42 0 44 51
Seal 29 44 44 0 50
Dog 32 48 51 50 0
Bear
RaccoonWeasel
Seal
Dog
13
13
25.25
20
5.25
18.25
1.75

.
Neighbor-Joining Algorithms
Agglomerative approach: (bottom-up)
1. Find a pair of taxa neighbors – i,j2. Connect them to a new internal vertex – v (Define edge
weights)3. Remove i,j from taxon-set, and add v (Define distances from
v)4. Return to (1)
When only 2 taxa are left, connect them
Consistency: Given an additive metric DT:
- We always choose a pair of neighbors in T (stage 1)
- The reduced distance-matrix is consistent with the reduced tree (stage 3)
Neighbors: taxa connectedby a 2-edge path
By induction:We eventually reconstruct T

.
UPGMA (Unweighted Pair Group Method with Arithmetic-Mean)
UPGMA algorithm:1. Find a pair of taxa of minimal distace– i,j2. Connect them to a new internal vertex v3. Remove i,j from taxon-set, and add v (D(v,k) = αD(i,k) +(1- α)D(j,k))4. Return to (1)
When only 2 taxa are left, connect them
Consistency ? - Given an additive metric DT, do we always choose a pair of neighbors in T ?
a b c d
a 0 14 15 27
b 0 3 15
c 0 14
d 0
c
13
1
13
1
1
a
b
d
UPGMA chooses b,c
Closest taxon is notnecessarily a neighbor
α, 1- α – proportional to the number of ‘original’ taxa i,j represent

.
Ultrametric Trees
• Edge-weighted trees which have a point (root) equidistant from all leaves
• Additive metrics consistent with an ultrametric tree are called ultrametrics
A distance-matrix is ultrametric iff it obeys the 3-point condition:“ Any subset of three taxa can be labelled i,j,k such that
d(i,j) ≤ d(j,k) = d(i,k) ”
66.5
3.5 4 32 2
3.5
tim
e

.
UPGMA
Additional notes:
• In the reduction formula D(v,k) can be set to any value within the interval defined by D(i,k) and D(j,k).
In particular: D(v,k) = ½(D(i,k) + D(j,k)) (WPGMA algorithm) If we use: D(v,k) = min {D(i,k) , D(j,k)} we get the ‘closest’
ultrametric from below (unique subdominant ultrametric)
Run-time analysis:― Naïve implementation: Θ(n3)― By keeping a sorted version of each row in D: Θ(n2log(n))― Third variant can be executed in: Θ(n2)
1. Find a pair of taxa of minimal distace– i,j
2. Connect them to a new internal vertex v
3. Remove i,j from taxon-set, and add v (D(v,k) = αD(i,k) +(1- α)D(j,k))

.
Consistent distance-based reconstruction:
Given an additive metric D, find the unique tree T, s.t. DT =
T. Reminder: A metric is additive iff it obeys the 4-point condition:
“Any subset of four taxa can be labelled i,j,k,l such that
d(i,j) + d(k,l) ≤ d(i,l) + d(j,k) = d(i,k) + d(j,l)”
Next Time …Distance matrices
Additive matrices
Ultrametric matrices

.
Saitou & Nei’s Neighbor Joining
S&N algorithm:1. Find a pair of taxa maximizing Q(i,j) = r(i) + r(j) – (n-2)D(i,j)
2. Connect them to a new internal vertex v with edges of weights:
3. Remove i,j from taxon-set, and add v - D(v,k) = ½(D(i,k) +D(j,k) -D(i,j))4. Return to (1)
When only 2 taxa are left, connect them (with edge of length D(i,j))
If D is additive (consistent with some tree T ):
•
• Q(i,j) is maximized for neighbor-pairs
• If i,j are neighbors then stages (2,3) are consistent
ik
kiDir ),()(
2
)()(),(
2
1),( ;
2
)()(),(
2
1(
n
irjrjiDvjw
n
jrirjiDi,v)w
jik
jipathkDjiDjiQ,
)),(,(),(2),( k
i j
v
n – current #taxa
shown in class
Conclusion: In such a case, given D, NJ returns T

.
Saitou & Nei’s Neighbor JoiningComplexity analysis
Run-time analysis:
• In each iteration we need to recalculate r(∙) for all taxa
• Q(∙,∙) values are ‘scrambled’ in each iteration
• Stage (1) takes O(n2)
• Total complexity - O(n3)
• No known way to speed this up significantly
ik
kiDir ),()(
2
)()(),(
2
1),( ;
2
)()(),(
2
1(
n
irjrjiDvjw
n
jrirjiDi,v)w
S&N algorithm:1. Find a pair of taxa maximizing Q(i,j) = r(i) + r(j) – (n-2)D(i,j)
2. Connect them to a new internal vertex v with edges of weights:
3. Remove i,j from taxon-set, and add v - D(v,k) = ½(D(i,k) +D(j,k) -D(i,j))
Note: There are consistent reconstruction algorithmswhich run in O(n2) or even O(n∙log(n)) time.

.
S&N’s NJ on Non-Additive Data
Example:
Bear Raccoon Weasel Seal Dog
Bear 0 26 34 29 32
Raccoon 26 0 42 44 48
Weasel 34 42 0 44 51
Seal 29 44 44 0 50
Dog 32 48 51 50 0
D:
D(B,R) + D(W,S) ; D(B,W) + D(R,S) ; D(B,S) + D(R,W)
26 + 44 (68) ; 34 + 44 (78) ; 29 + 42 (71)
D is not additive

.
S&N’s NJExample: 1st iteration
B R W S D
B 0 26 34 29 32R 0 42 44 48W 0 44 51S 0 50D 0
D:
Bear Dog Raccoon Weasel Seal
B-D
6 26
B R W S D
B 0 203 190 201 206R 0 205 195 197W 0 206 199S 0 198D 0
Q:
),(),(),(),(.3
2
)()(),(),(.2
),()(;),()2()()(),(.1
21
21
jiDkjDkiDkvD
n
jrirjiDviw
kiDirjiDnjrirjiQik
B R W S D
121 160 171 167 181r :

.
S&N’s NJExample: 2nd iteration
B-D R W S
B-D 0 21 26.5 23.5R 0 42 44W 0 44S 0
D:
Bear Dog Raccoon Weasel Seal
B-D
6 26
B-D R W S
B-D 0 136 130.5 135.5R 0 135.5 130.5W 0 136S 0
Q:
),(),(),(),(.3
2
)()(),(),(.2
),()(;),()2()()(),(.1
21
21
jiDkjDkiDkvD
n
jrirjiDviw
kiDirjiDnjrirjiQik
B-D R W S
71 107 112.5 111.5r :
B-D-R1.519.5
Calculate difference from oldvalues to new ones

.
S&N’s NJExample: 3rd iteration
B-D-R W S
B-D-R 0 23.75 23.25W 0 44S 0
D:
Bear Dog Raccoon Weasel Seal
B-D
6 26
Q:
),(),(),(),(.3
2
)()(),(),(.2
),()(;),()2()()(),(.1
21
21
jiDkjDkiDkvD
n
jrirjiDviw
kiDirjiDnjrirjiQik
B-D-R W S
47 67.75 67.25r :
B-D-R1.519.5
B-D-R W S
B-D-R 0 91 91W 0 91S 0
Reconstruct the uniquetree over 3 taxa
1.5
W-S
22.25 21.75

.
How Good Is The Tree?
Bear Dog Raccoon Weasel Seal
B-D
6 26
B-D-R1.519.5
1.5
W-S
22.25 21.75
We observe the perturbations from the input matrixto the one implied by the output tree
B R W S D
B 0 26 34 29 32
R 0 42 44 48
W 0 44 51
S 0 50
D 0
D:
B R W S D
B 0 27 31.25 30.75 32
R 0 43.25 42.75 47
W 0 44 51.25
S 0 50.75
D 0
DT :
),(),(max,),(
),(),(,),(
,
,
jiDjiDDDDDL
jiDjiDDDDDL
Tji
TT
p
ji
p
TpTTp
B R W S D
B 0 1 2.75 1.75 0
R 0 1.25 1.25 1
W 0 0 0.25
S 0 0.75
D 0
|D-DT|:
How good is this?
.
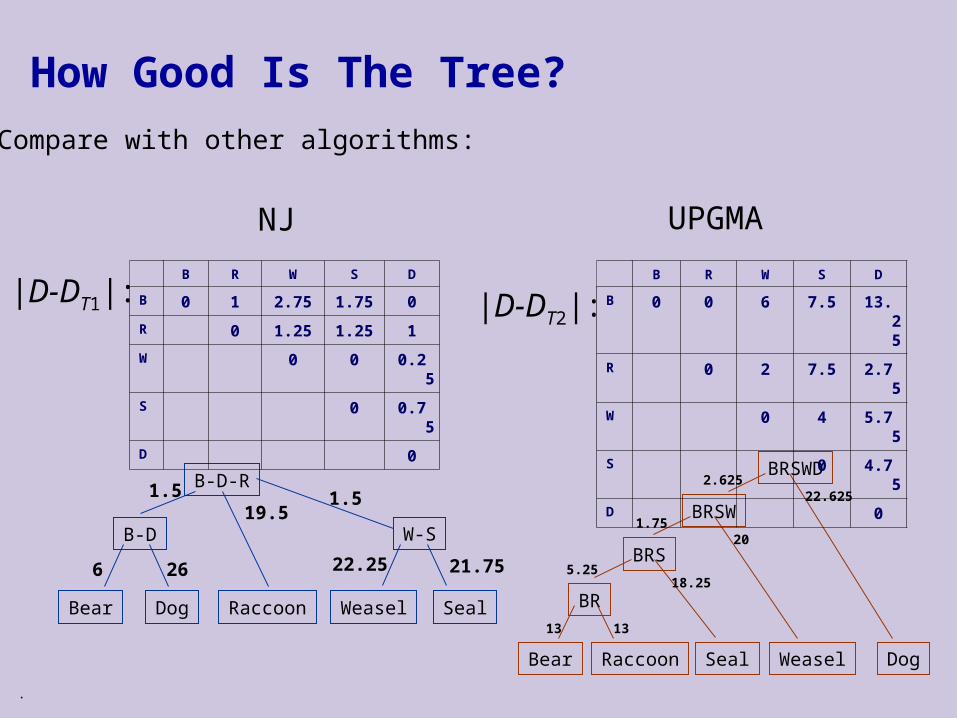
How Good Is The Tree?
Bear Dog Raccoon Weasel Seal
B-D
6 26
B-D-R1.519.5
1.5
W-S
22.25 21.75
Compare with other algorithms:
B R W S D
B 0 1 2.75 1.75 0
R 0 1.25 1.25 1
W 0 0 0.25
S 0 0.75
D 0
|D-DT2|:
Bear Raccoon WeaselSeal Dog
BR1313
BRS
18.255.25
BRSW20
1.75
BRSWD22.625
2.625
|D-DT1|:
NJ UPGMA
B R W S D
B 0 0 6 7.5 13.25
R 0 2 7.5 2.75
W 0 4 5.75
S 0 4.75
D 0

.
Can we do better?
Given a distance-matrix D, find an edge-weighted tree T,which minimizes ||D,DT||p
• For p = 1,2,∞ this task was shown to be NP-hard
• For p = 1,2 this task was shown to be NP-hard for ultrametric trees as well
• For p = ∞:― this task is easy (O(n2) algorithm) for ultrametric trees― 3-approximation algorithm for general trees
No algorithm which gives any good guarantees for non-additive data
Related Documents











