Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010 Page 1 of 142 CONFIDENTIAL WORKING PROTOCOL WORKING PROTOCOL NUMBER: 02 WORKING PROTOCOL DATE: 06 DECEMBER 2010 COMBINATION OF THE FOLLOWING APPROVED FINAL DOCUMENTS: PROTOCOL 27 APRIL 2010 PROTOCOL ADMINISTRATIVE CHANGE NUMBER 01; 09 SEPTEMBER 2010 PROTOCOL AMENDMENT 01, 06 DECEMBER 2010 Note: This document does not require formal approval as it is a working document containing a combination of the above two documents, which comprise the formal approved documents. Protocol Title: A Phase II Trial to Evaluate the Early Bactericidal Activity, Safety and Tolerability of the following: TMC207 alone, TMC207 plus pyrazinamide, TMC207 plus PA-824, PA-824 plus pyrazinamide and PA-824 plus pyrazinamide and moxifloxacin, in Adult Patients with Newly Diagnosed, Smear-Positive Pulmonary Tuberculosis. Protocol Number: NC-001-(J-M-Pa-Z)

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 1 of 142
CONFIDENTIAL
WORKING PROTOCOL
WORKING PROTOCOL NUMBER: 02 WORKING PROTOCOL DATE: 06 DECEMBER 2010
COMBINATION OF THE FOLLOWING APPROVED FINAL DOCUMENTS:
PROTOCOL 27 APRIL 2010 PROTOCOL ADMINISTRATIVE CHANGE NUMBER 01; 09 SEPTEMBER 2010
PROTOCOL AMENDMENT 01, 06 DECEMBER 2010
Note: This document does not require formal approval as it is a working document containing a combination of the above two documents, which comprise the formal approved documents.
Protocol Title: A Phase II Trial to Evaluate the Early Bactericidal Activity, Safety and Tolerability of the following: TMC207 alone, TMC207 plus pyrazinamide,
TMC207 plus PA-824, PA-824 plus pyrazinamide and PA-824 plus pyrazinamide and moxifloxacin, in Adult Patients with Newly Diagnosed, Smear-Positive
Pulmonary Tuberculosis.
Protocol Number: NC-001-(J-M-Pa-Z)

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 2 of 142
Development Phase: II Sponsor: Global Alliance for TB Drug Development
40 Wall Street, 24th Floor New York, NY 10005 United States of America
Clinical Director/Medical Director Christo van Niekerk; MD, FCPaed
Senior Director, Clinical Development Global Alliance for TB Drug Development Enterprise Building, 1st Floor The Innovation Hub Pretoria 0087 South Africa
Immediately Reportable Events Christo van Niekerk; MD, FCPaed
Senior Director, Clinical Development Global Alliance for TB Drug Development Enterprise Building, 1st Floor The Innovation Hub Pretoria 0087 South Africa Telephone: +27 (0)12 844 0955 Mobile: +27 (0)82 550 3856 Facsimile: + 27 (0)12 844 0959

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 3 of 142
Table of Contents
1. PROTOCOL SYNOPSIS .................................................................................................................... 11
1.1. Synopsis ................................................................................................................................. 11
1.2. Study Flow Chart ................................................................................................................... 14
1.3. PK and ECG Chart .................................................................................................................. 17
1.3.1. TMC207 (excluding TMC207 plus PA-824) and Rifafour e275® Containing Treatment
Arms 17
1.3.2. PA-824 (excluding TMC207 plus PA-824) Containing Treatment Arms ........................ 18
1.3.3. TMC207 (J) plus PA-824 (Pa) Containing Treatment Arms ............................................ 19
2. INTRODUCTION ............................................................................................................................ 20
2.1. Background Information ....................................................................................................... 20
2.2. Preclinical Studies .................................................................................................................. 24
2.2.1. TMC207 .......................................................................................................................... 24
2.2.2. PA-824 ............................................................................................................................ 26
2.2.3. Moxifloxacin ................................................................................................................... 29
2.3. Clinical Trials .......................................................................................................................... 29
2.3.1. TMC207 .......................................................................................................................... 29
2.3.2. PA-824 ............................................................................................................................ 35
2.3.3. Moxifloxacin ................................................................................................................... 44
2.4. Known and Potential Risks and Benefits of the Investigational Medicinal Product (IMP)
used in the Study .............................................................................................................................. 47
2.4.1. TMC207 .......................................................................................................................... 47
2.4.2. PA-824 ............................................................................................................................ 47
2.4.3. Moxifloxacin ................................................................................................................... 47
2.4.4. Pyrazinamide .................................................................................................................. 48
2.4.5. Standard, first-line TB treatment ................................................................................... 48
3. TRIAL RATIONALE AND OBJECTIVES ............................................................................................. 48
3.1. Trial Rationale ....................................................................................................................... 48
3.2. Trial Objectives ...................................................................................................................... 50
3.3. Trial Endpoints ....................................................................................................................... 50
3.3.1. Primary Endpoint ........................................................................................................... 50

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 4 of 142
3.3.2. Secondary Endpoints ..................................................................................................... 51
3.3.2.1. Efficacy ....................................................................................................................... 51
3.3.2.2. Safety and Tolerability................................................................................................ 51
3.3.2.3. Pharmacokinetics ....................................................................................................... 51
3.3.2.4. Pharmacokinetics-Pharmacodynamics (PK-PD) ......................................................... 52
3.3.2.5. Extent of QT Prolongation .......................................................................................... 52
3.4. Mycobacteriological Characterization .................................................................................. 52
3.5. Participant Inclusion/Exclusion Criteria ................................................................................ 53
3.5.1. Inclusion Criteria ............................................................................................................ 53
3.5.2. Exclusion Criteria............................................................................................................ 54
4. STUDY DESIGN AND DURATION ................................................................................................... 58
4.1. Summary of Study Design ..................................................................................................... 58
4.2. Treatment Plan ...................................................................................................................... 59
4.2.1. Pre-treatment Visits 1 to 4 (Days -9 to -1) ..................................................................... 59
4.2.2. Treatment Period (Visits 5 to 19 [Days 1 to 15]) ........................................................... 62
4.2.3. Follow-Up Period ........................................................................................................... 68
4.2.4. Early Withdrawal ............................................................................................................ 69
4.3. Participant Withdrawal Criteria ............................................................................................ 71
4.4. Trial Treatment Discontinuation ........................................................................................... 71
4.5. Stopping Rules ....................................................................................................................... 72
4.6. Participant Progress Definitions ............................................................................................ 72
4.6.1. Screening Failure ............................................................................................................ 72
4.6.2. Completed Treatment.................................................................................................... 72
4.6.3. Early Withdrawal ............................................................................................................ 72
4.6.4. Completed Trial .............................................................................................................. 72
4.6.5. Lost to Follow-up ........................................................................................................... 72
4.7. Restrictions ............................................................................................................................ 72
4.7.1. Foods and Beverages ..................................................................................................... 72
4.7.2. Prior and Concomitant Medications .............................................................................. 73
4.7.3. Activity ........................................................................................................................... 74
5. INVESTIGATIONAL MEDICINAL PRODUCT .................................................................................... 74
5.1. Trial Treatments .................................................................................................................... 75

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 5 of 142
5.2. Methods of Assigning Participants to Treatment Groups .................................................... 75
5.3. IMP Administration ............................................................................................................... 76
5.3.1. TMC207 and PA-824 Treatment Groups ....................................................................... 76
5.3.2. First-line standard TB treatment per SA national guidelines (Rifafour e-275 tablets)
Treatment Group .......................................................................................................................... 77
5.3.3. Participant Compliance .................................................................................................. 77
5.4. Blinding and Procedures for Breaking the Blind ................................................................... 77
5.5. IMP Packaging and Labeling .................................................................................................. 78
5.5.1. PA-824 and TMC207 Containing Treatment Arms ........................................................ 80
5.5.2. Standard, first line TB treatment ................................................................................... 81
5.6. Storage .................................................................................................................................. 81
5.7. Dispensing and Accountability .............................................................................................. 81
5.8. Returns and Destruction ....................................................................................................... 82
6. TRIAL VARIABLES .......................................................................................................................... 82
6.1. Demographic and Background Variables .............................................................................. 82
6.2. Efficacy Variables................................................................................................................... 83
6.3. Pharmacokinetic Variables .................................................................................................... 83
6.4. Safety Variables ..................................................................................................................... 83
6.5. Mycobacterial Characterization ............................................................................................ 84
7. TRIAL PROCEDURES ...................................................................................................................... 84
7.1. Sputum Sampling .................................................................................................................. 84
7.1.1. Spot Sputum ................................................................................................................... 84
7.1.2. Overnight Sputum .......................................................................................................... 85
7.2. Clinical Laboratory Tests ....................................................................................................... 86
7.2.1. Safety Laboratory Tests ................................................................................................. 86
7.3. Plasma Pharmacokinetics (PK) .............................................................................................. 88
7.4. Electrocardiography (ECG) .................................................................................................... 89
7.5. Ophthalmology Examinations ............................................................................................... 90
7.6. Physical Examinations and Vital Signs ................................................................................... 91
7.7. Chest X-ray ............................................................................................................................ 91
7.8. Retention Sample Collection ................................................................................................. 92
7.8.1. Plasma and Serum ......................................................................................................... 92

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 6 of 142
7.8.2. Sputum ........................................................................................................................... 92
7.8.3. Urine ............................................................................................................................... 92
8. ADVERSE EVENTS .......................................................................................................................... 93
8.1. Definitions ............................................................................................................................. 93
8.1.1. Adverse Event (AE) ......................................................................................................... 93
8.1.2. Serious Adverse Event (SAE) .......................................................................................... 93
8.1.3. Unlisted (Unexpected) Adverse Event ........................................................................... 94
8.1.4. Life Threatening ............................................................................................................. 94
8.1.5. Associated With the Use of the Drug............................................................................. 94
8.2. Attribution Definitions .......................................................................................................... 94
8.3. Reporting ............................................................................................................................... 95
8.3.1. Adverse Event Reporting ............................................................................................... 95
8.3.2. Serious Adverse Event Reporting................................................................................... 95
8.3.3. Follow-up of Adverse Events ......................................................................................... 96
8.3.4. Follow-up of Post Trial Adverse Events ......................................................................... 96
8.3.5. Clinical Laboratory Adverse Events ................................................................................ 97
8.3.6. Pregnancy ....................................................................................................................... 97
8.3.7. Disease Under Study ...................................................................................................... 98
9. MONITORING AND SAFETY FOR SPECIFIC TOXICITIES .................................................................. 98
9.1. ALT and AST ........................................................................................................................... 98
9.2. Amylase elevation ................................................................................................................. 99
9.3. Pancreatic amylase and/or lipase elevation ......................................................................... 99
9.4. Trypsin-like immunoreactivity ≥ 1.5 times ULN .................................................................... 99
9.5. Musculo-skeletal System and Cardiac Muscle .................................................................... 100
9.6. LDH and LDH-isoenzymes .................................................................................................... 100
9.7. Cardiac Rhythm Disturbances ............................................................................................. 100
9.8. Gastrointestinal System ...................................................................................................... 101
9.9. Other toxicities .................................................................................................................... 101
10. STATISTICAL ANALYSIS ................................................................................................................ 101
10.1. Sample Size ...................................................................................................................... 101
10.2. Primary Efficacy Analysis ................................................................................................. 102
10.3. Secondary Efficacy Analysis ............................................................................................. 102

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 7 of 142
10.4. Pharmacokinetic Analysis ................................................................................................ 103
10.5. Pharmacokinetic and Pharmacodynamic Statistical Analysis ......................................... 104
10.6. Safety and Tolerability Analyses ...................................................................................... 104
10.7. Mycobacteriology Characterization ................................................................................ 105
11. RECORDS MANAGEMENT ........................................................................................................... 106
11.1. Data Collection ................................................................................................................ 106
11.2. Source Documents ........................................................................................................... 106
11.3. File Management at the Trial Center .............................................................................. 106
11.4. Records Retention at the Trial Center ............................................................................. 106
12. QUALITY CONTROL AND QUALITY ASSURANCE ......................................................................... 107
12.1. Site Procedures ................................................................................................................ 107
12.2. Monitoring ....................................................................................................................... 107
12.3. Auditing............................................................................................................................ 108
13. ETHICS AND RESPONSIBILITY ...................................................................................................... 108
13.1. Basic Principles ................................................................................................................ 108
13.2. Independent Ethics Committee/Institutional Review Board (IEC/IRB) Review .............. 109
13.3. Regulatory Authorities ..................................................................................................... 109
13.4. Informed Consent ............................................................................................................ 109
13.5. Confidentiality ................................................................................................................. 110
13.6. Publication Policy ............................................................................................................. 110
13.7. Protocol Amendment Policy ............................................................................................ 111
13.8. Financial Aspects, Insurance and Indemnity ................................................................... 111
14. REFERENCES ................................................................................................................................ 113
APPENDIX 1: PROTOCOL SIGNATURE PAGES ..................................................................................... 116
APPENDIX 2: RIFAFOUR E-275 PACKAGE INSERT............................................................................ 117
APPENDIX 3: THE IUATLD SCALE ........................................................................................................ 120
APPENDIX 4: DIVISION OF MICROBIOLOGY AND INFECTIOUS DISEASES (DMID) ADULT TOXICITY
TABLE ................................................................................................................................................. 121
APPENDIX 5: CARDIOVASCULAR SAFETY ........................................................................................... 132
APPENDIX 6: PYRAZINAMIDE PACKAGE INSERT ................................................................................ 133
APPENDIX 7: MOXIFLOXACIN PACKAGE INSERT ................................................................................ 134

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 8 of 142
Table 1: Overview of the TMC207 Clinical Program ............................................................................ 30
Table 2: Overview of PA-824 Clinical Program .................................................................................... 36
Table 3 Summary of Phase IIb studies in which moxifloxacin was administered as part of a four-drug regimen during the intensive phase of treatment (total treatment duration: 2 months) .................. 45
Table 4 : Investigational Medicinal Product Details ............................................................................ 79
Table 5: Pyrazinamide dosing per weight ............................................................................................ 80
Table 6: Expected Standard Errors of Group mean log CFU and confidence intervals ..................... 102
Figure 1: PA-824-CL-007 Mean Group logCFU Over Time ................................................................... 39

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 9 of 142
LIST OF ABBREVIATIONS AND DEFINITIONS OF TERMS
AE Adverse Event AFB acid fast bacilli AIDS acquired immune deficiency syndrome ALP alkaline phosphatase ALT alanine aminotransferase APTT activated partial thromboplastin time ART antiretroviral therapy AST aspartate aminotransferase AUC area under the plasma concentration time curve AUC(0-24) area under the plasma concentration time curve from zero to end of dosing
interval AV Atrioventricular BMI body mass index CFU colony forming units Cmax maximum observed plasma concentration
Cmin minimum observed plasma concentration at the end of the dosing interval CPK creatine phosphokinase CPK-MB creatine phosphokinase of myocardial band CRF case report form CRO Contract Research Organization
CYP3A4 cytochrome P450 3A4 DBP diastolic blood pressure DMID Division of Microbiology and Infectious Diseases DOTS Internationally agreed strategy for TB control E Ethambutol EBA early bactericidal activity ECG Electrocardiogram GGT gamma-glutamyltransferase H isoniazid HDL high density lipoprotein HIV Human Immunodeficiency Virus
IB Investigator Brochure
ICF informed consent form ICH GCP International Conference on Harmonization Good Clinical Practice IMP Investigational Medicinal Product INR international normalized ratio IUATLD International Union Against Tuberculosis and Lung Disease J TMC207 Kel terminal elimination rate constant LDH lactate dehydrogenase m Meters M2 N-monodesmethyl metabolite of TMC207
MBD minimum bactericidal dose MDR-TB multidrug resistant-tuberculosis

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 10 of 142
MED minimum effective dose MIC minimum inhibitory concentration MGIT mycobacterial growth indicator tube MTB Mycobacterium tuberculosis PCR polymerase chain reaction PD pharmacodynamic PK Pharmacokinetic PT prothrombin time PTT partial thromboplastin time QRS electrocardiographic QRS interval QT electrocardiographic QT interval
QTc corrected QT interval QTcB QT interval corrected by Bazett’s method QTcF QT interval corrected by Fridericia’s method R Rifampicin R Pearson’s correlation coefficient SAE Serious Adverse Event SA GCP Guidelines for Good Practice in the Conduct of Clinical Trials in Human Patients
in South Africa 2006 SAP Statistical Analysis Plan SBP systolic blood pressure T Time
t½ apparent terminal elimination phase half-life TB Tuberculosis TEAEs treatment-emergent adverse events Tmax time at which Cmax is observed TTP time to sputum culture positivity ULN upper limit of normal WBA whole blood assay
WHO World Health Organization Z Pyrazinamide
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 11 of 142
1. PROTOCOL SYNOPSIS
1.1. Synopsis
Name of Sponsor/Company: Global Alliance for TB Drug Development For National Authority Use Only
Name of Finished Products: TMC207 tablets; PA-824 tablets; pyrazinamide tablets; moxifloxacin tablets.
Protocol Title: A Phase II Trial to Evaluate the Early Bactericidal Activity, Safety and Tolerability of the following: TMC207 alone, TMC207 plus pyrazinamide, TMC207 plus PA-824, PA-824 plus pyrazinamide and PA-824 plus pyrazinamide and moxifloxacin, in Adult Participants with Newly Diagnosed, Smear-Positive Pulmonary Tuberculosis.
Treatment Indication: Pulmonary tuberculosis (TB).
Trial Objective: To evaluate the early bactericidal activity (EBA), safety, tolerability and pharmacokinetics of TMC207 alone, TMC207 plus pyrazinamide (Z), TMC207 plus PA-824, PA-824 plus pyrazinamide and PA-824 plus pyrazinamide and moxifloxacin, administered orally as once daily doses for 14 consecutive days, in adult participants with newly diagnosed, smear positive pulmonary tuberculosis, in order to help select appropriate combination therapies for later stage clinical development.
Trial Design: A two-center, partially double-blinded, randomized clinical trial in six parallel groups. The first five treatment arms are: TMC207 alone, TMC207 plus pyrazinamide, TMC207 plus PA-824, PA-824 plus pyrazinamide and PA-824 plus pyrazinamide plus moxifloxacin. The sixth arm will receive standard first line TB treatment as per the
South African TB guidelines (Rifafour e-275) and is included as a control for the EBA quantitative mycobacteriology. The following blinding will occur:
TMC207 alone treatment arm blinded against TMC207 plus pyrazinamide treatment arm;
PA-824 plus pyrazinamide treatment arm blinded against PA-824 plus pyrazinamide plus moxifloxacin treatment arm;
TMC207 plus PA-824 treatment arm and Rifafour e-275 treatment arm will be open-label.
Patient Population: A total of 85 male or female participants (5 groups of 15 participants receiving a TMC207 and/or PA-824 containing treatment arm and one group of 10 participants
receiving Rifafour e275) aged between 18 and 65 years (inclusive) with newly diagnosed, smear-positive, pulmonary TB.
Test Product, Dose and Mode of Administration:
TMC207 will be supplied as 100-mg tablets and PA-824 will be supplied as 200-mg tablets. Pyrazinamide will be supplied as 500-mg tablets or placebo. Moxifloxacin will be supplied as 400-mg tablets or placebo. Treatment will be administered orally once daily for 14 consecutive days in the following dosing schemes:
1. TMC207 700 mg Day 1; 500 mg Day 2; 400 mg Days 3-14; + pyrazinamide placebo (dosed by weight) Days 1-14;
2. TMC207 700 mg Day 1; 500mg Day 2; 400 mg Days 3-14 + pyrazinamide (dosed by weight) Days 1-14;
3. TMC207 700 mg Day 1; 500 mg Day 2; 400 mg Days 3-14 + PA-824 200 mg Days 1-14;
4. PA-824 200 mg + pyrazinamide (dosed by weight) + moxifloxacin 400 mg placebo Days 1-14;
5. PA-824 200 mg + pyrazinamide (dosed by weight) + moxifloxacin 400 mg Days 1-14.
Pyrazinamide and pyrazinamide placebo will be dosed based on the participant’s weight as follows: <55 kg: 1000 mg; >55kg – 75 kg: 1500 mg; > 75 kg: 2000 mg.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 12 of 142
Name of Sponsor/Company: Global Alliance for TB Drug Development For National Authority Use Only
Name of Finished Products: TMC207 tablets; PA-824 tablets; pyrazinamide tablets; moxifloxacin tablets.
Positive Control Product, Dose, and Mode of Administration:
Rifafour e-275 will be supplied as tablets and administered orally once daily for 14
days as per South African National TB Treatment Guidelines. The daily dose is dependent on the participants’ weight as follows: 30-37kg: 2 tablets; 38-54kg: 3 tablets; 55 – 70kg: 4 tablets; 71kg and over: 5 tablets.
Criteria for evaluation: Primary Endpoints: The EBA (EBA0-14) as determined by the rate of change in logCFU per ml sputum over the period Day 0 to Day 14 which may be described with linear, bi-linear or non-linear regression of logCFU on time. These data will be presented as descriptive analyses, and no inferential tests will be carried out. Secondary Endpoints: Efficacy: The secondary efficacy endpoints are as follows:
The EBA (EBA0-2, EBA2-14 and EBA7-14) as determined by the rate of change of logCFU in sputum over the period Day 0 to Day 2, Day 2 to Day 14 and Day 7 to 14 which may be described with linear, bi-linear or non-linear regression of logCFU on time.
The time to sputum culture positivity (TTP) (TTP0-2, TTP0-14, TTP2-14, and TTP7-14) in the Mycobacterial Growth Indicator Tube (Bactec MGIT960) system as determined by the rate of change in TTP in sputum over the periods Day 0 to Day 2, Day 0 to Day 14, Day 2 to Day 14 and Day 7 to Day 14 in participants, which may be described with linear, bi-linear or non-linear regression of TTP on time.
These data will be presented as descriptive analyses, and no inferential tests will be carried out. Safety and Tolerability: Proportion of participants with adverse events and proportion of participants who discontinue due to an adverse event in each experimental arm. These data will be presented as descriptive analyses, and no inferential tests will be carried out. Pharmacokinetics (PK): For TMC207 (excluding TMC207 plus PA-824 containing treatment arm) containing study arms: The following PK parameters will be estimated for TMC207, TMC207 metabolite M2 and pyrazinamide on Day 14 only: the maximum observed plasma concentration (Cmax), time to reach Cmax (Tmax), the minimum observed plasma concentration (Cmin) 24 hours following the last dose, area under the plasma concentration time (t) curve from zero to 24 hours (AUC(0-24)). These data will be presented as descriptive analyses, and no inferential tests will be carried out. For PA-824 (excluding TMC207 plus PA-824 containing treatment arm) containing study arms: The following PK parameters will be estimated for PA-824, pyrazinamide and moxifloxacin on Days 1, 8 and 14: the maximum observed plasma concentration (Cmax), time to reach Cmax (Tmax), the minimum observed plasma concentration (Cmin) 24 hours following the last dose, area under the plasma concentration time (t) curve from zero to 24 hours (AUC(0-24)). These data will be presented as descriptive analyses, and no inferential tests will be carried out. For TMC207 plus PA-824 containing study arm: The following PK parameters will be estimated for PA-824, TMC207 and TMC207 metabolite M2 on Day 14 only: the maximum observed plasma concentration (Cmax), time to reach Cmax (Tmax), the minimum observed plasma concentration (Cmin) 24 hours following the last dose, area under the plasma concentration time (t) curve from zero to 24 hours (AUC(0-24)). These data will be presented as descriptive analyses, and no inferential tests will be carried out. Pharmacokinetics-Pharmacodynamics (PK-PD): For TMC207 and PA-824 containing treatment arms, the following will be estimated for TMC207, PA-824, pyrazinamide and moxifloxacin as appropriate per the above PK analyses performed: The EBAs vs. the following PK variables (Day 14 only) will be presented:
Cmax;
AUC(0-24);
Time over Minimum inhibitory concentrations (MIC) (for TMC207, PA-824 and moxifloxacin). These data will be presented as descriptive analyses, and no inferential tests will be carried out.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 13 of 142
Name of Sponsor/Company: Global Alliance for TB Drug Development For National Authority Use Only
Name of Finished Products: TMC207 tablets; PA-824 tablets; pyrazinamide tablets; moxifloxacin tablets.
Criteria for evaluation (cont): Extent of QT prolongation For TMC207 and PA-824 containing treatment arms, administered for 14 days, the potential correlations between the plasma concentration of IMP and the change from baseline of QT interval corrected by Fridericia’s method (QTcF) and change from baseline of QT interval corrected by Bazett’s method (QTcB) with respect to time for the different treatment groups will be explored. These data will be presented as descriptive analyses, and no inferential tests will be carried out.
Mycobacteriology Characterization: Cultures grown from the overnight sputum collections from Day -2 and Day 14 will be assessed as follows:
MIC of TMC207, PA-824 and moxifloxacin;
Drug susceptibility testing of the M. tuberculosis isolates with the MGIT system for sensitivity to isoniazid, rifampicin, ethambutol and pyrazinamide;
Speciation of the infecting organism by polymerase chain reaction (PCR).
Statistical Methods: This is a descriptive study with no inferential statistics or hypothesis testing. The planned sample size of 15 participants per treatment group is in keeping with other Phase II trials of this type and accounts for the possibility of up to 3 drop-outs per arm, which, based on previous studies of this type conducted at these sites, represents a conservative estimate of the expected drop-out rate. It will allow estimation of the study endpoints with good accuracy and low variability. The Statistical Analysis Plan (SAP) will be developed before the database is locked.
Trial Duration: Estimated date of first participant enrolled: Quarter 3 2010 Estimated date of last participant enrolled: Quarter 1 2011 Estimated date of last participant completed: Quarter 3 2011 Duration of study: 41 Weeks (25-week enrolment period plus 1-week pre-treatment plus 2-week treatment period plus 13 weeks of post-treatment follow-up).
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 14 of 142
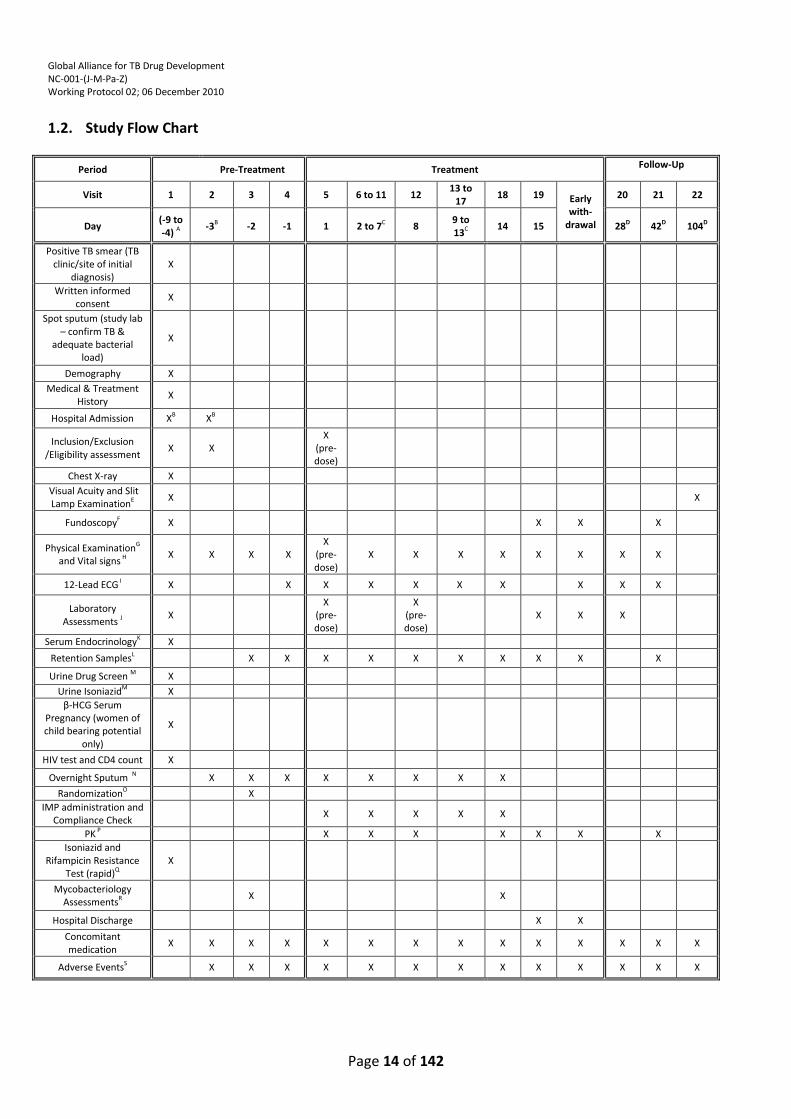
1.2. Study Flow Chart
Period Pre-Treatment Treatment Follow-Up
Visit 1 2 3 4 5 6 to 11 12 13 to
17 18 19 Early
with-drawal
20 21 22
Day (-9 to -4) A
-3B -2 -1 1 2 to 7C 8 9 to 13C
14 15 28D 42D 104D
Positive TB smear (TB clinic/site of initial
diagnosis) X
Written informed consent
X
Spot sputum (study lab – confirm TB &
adequate bacterial load)
X
Demography X
Medical & Treatment History
X
Hospital Admission XB XB
Inclusion/Exclusion /Eligibility assessment
X X
X
(pre-dose)
Chest X-ray X
Visual Acuity and Slit Lamp ExaminationE
X
X
FundoscopyF X X X X
Physical ExaminationG and Vital signs H X X X X
X (pre-dose)
X X X X X X X X
12-Lead ECG I X X X X X X X X X X
Laboratory Assessments J
X X
(pre-dose)
X
(pre-dose)
X X X
Serum EndocrinologyK X
Retention SamplesL X X X X X X X X X X
Urine Drug Screen M X
Urine IsoniazidM X
β-HCG Serum Pregnancy (women of child bearing potential
only)
X
HIV test and CD4 count X
Overnight Sputum N X X X X X X X X
RandomizationO X
IMP administration and Compliance Check
X X X X X
PK P X X X X X X X
Isoniazid and Rifampicin Resistance
Test (rapid)Q X
Mycobacteriology AssessmentsR
X X
Hospital Discharge X X
Concomitant medication
X X X X X X X X X X X X X X
Adverse EventsS X X X X X X X X X X X X X

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 15 of 142
A: The Visit 1 (Day -9 to -4) time period will be up to a maximum of 6 days. Should a site’s ethics committee request that this be modified, the agreed upon time period will be implemented for that site.
B: Participants can proceed with the Visit 2 (Day -3) assessments as soon as their Visit 1 (Day -9 to -4) assessments have been completed i.e. visits 1 and 2 may occur on the same day as long as the screening results are available in time for randomization. Participants may be hospitalized during the entire pre-treatment period if the investigator considers it advisable.
C: Except for ECGs, retention sample, and PK collection (as noted below), all events listed as occurring on Visit 6 (Day 2) to Visit 11 (Day 7) and Visit 13 (Day 9) to Visit 17 (Day 13) will be conducted each day during these times.
D: Follow-up visits will be performed at the site: (1) for participants in TMC207 containing arms (excluding TMC207 plus PA-824 containing treatment arm) at: Visit 20
(Day 28)(± 1 day) and Visit 21 (Day 42)(± 3 days); (2) for participants in PA-824 containing arms (excluding TMC207 plus PA-824 containing treatment arm) at Visit 20 (Day 28)(± 1 day) and Visit 22 (Day 104)(±14 days); (3) for participants in the TMC207 plus PA-824 and Rifafour e275® containing arms at Visit 20 (Day 28)(± 1 day), Visit 21 (Day 42)(± 3 days) and Visit 22 (Day 104)(±14 days).
E: Ophthalmologic Medical History will be obtained; Visual Acuity and Slit Lamp examination will be performed on all participants at Visit 1 (Day -9 to -4)(anytime during the pre-treatment period, however it must be performed before randomization on Visit 3 (Day -2). Visit 22 (Day 104): Visual Acuity and Slit Lamp examinations done on the PA-824 (including TMC207 plus PA-824) containing and the Rifafour e275® treatment arms.
F: Fundoscopy will be performed on all participants at Visit 1 (Day -9 to -4)(anytime during the pre-treatment period, however it must be performed before randomization on Visit 3 (Day -2). Visit 19 (Day 15) or early withdrawal and Visit 21 (Day 42): Eye exam with fundoscopy done on the TMC207 (including TMC207 plus PA-824) containing and the Rifafour e275® treatment arms.
G: Includes weight and height (m). Height (m) will only be collected once at Visit 1 (Day -9 to -4). To be performed within 2 hours before dosing on Days 1 through 14 and within 2 hours before the time dosing would have occurred on Day 15. For the TMC207 plus PA-824 treatment arm these are to be performed within 2 hours before the PA-824 dose.
H: Systolic blood pressure (SBP) and diastolic blood pressure (DBP) (mmHg), heart rate (beats per minute [bpm]), and body temperature (°C; axillary). To be performed within 2 hours before dosing on Days 1 through 14 and within 2 hours before the time dosing would have occurred on Day 15. For the TMC207 plus PA-824 treatment arm these are to be performed within 2 hours before the PA-824 dose.
I: 12-lead ECGs will be performed as noted in the ECG/PK flow charts. J: Laboratory Assessments: Hematology (hemoglobin, hematocrit, red blood cell count, white blood cell count with
differential), platelet count, Coagulation (activated partial thromboplastin time (APTT), prothrombin time (PT), international normalized ratio (INR)). Clinical Chemistry (albumin, urea, creatinine, direct, indirect and total bilirubin, uric acid, cholesterol (total), triglycerides, total protein, lipase, total amylase, alkaline phosphatase, creatine phosphokinase, aspartate aminotransferase (AST), alanine aminotransferase (ALT), gamma-glutamyl transferase (GGT), lactic dehydrogenase (LDH), phosphate, sodium, potassium, calcium (corrected for albumin), chloride, random/fasting glucose, bicarbonate/CO2). Urinalysis (pH, specific gravity, protein, glucose, micro-albumin, ketones, bilirubin, creatinine, nitrite, sodium, urobilinogen, blood, leukocytes, microscopy). If total amylase results are Grade 3 or higher, further testing of pancreatic amylase and trypsin like immunoreactivity should be considered after consultation with the Sponsor Medical Monitor. For the TMC207 plus PA-824 treatment arm at Visit 5/Day 1 and Visit 12/Day 8 these are to be performed pre the PA-824 dose.
K: Serum Endocrinology: testosterone, luteinizing hormone (LH), follicle-stimulating hormone (FSH). These will be performed on male participants only. The serum endocrinology sampling (in males, only) may be repeated in the morning (ideally 8 am) if the original results show an isolated abnormal value (i.e., only 1 of the 3 hormones is abnormal). Only the item which was abnormal in the original testing needs to be repeated.
L: Retention Sample collection: Plasma samples will be collected for: (1) all participants in parallel with the pre-dose blood draws planned for Visit 5 (Day 1), Visit 12 (Day 8) and with the laboratory samples at the Hour 0 draws on Visit 19 (Day 15) or early withdrawal. For the TMC207 plus PA-824 treatment arm at Visit 5/Day 1 ), Visit 12 (Day 8) and with the lab samples at the hour 0 draw scheduled for Visit 19 (Day 15), these are to be performed pre the PA-824 dose/dosing time; (2) for the TMC207(including TMC207 plus PA-824) and Rifafour e275® containing treatment arms, Visit 21 (Day 42) samples will also be collected. Serum samples (males only) will be collected for the PA-824 (including TMC207 plus PA-824) and Rifafour e275® containing treatment arms, in parallel from the pre-dose blood draws scheduled for Visit 5 (Day 1), Visit 12 (Day 8) and with the lab samples at the hour 0 draw scheduled for Visit 19 (Day 15) or early withdrawal. For the TMC207 plus PA-

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 16 of 142
824 treatment arm at Visit 5/Day 1 ), Visit 12 (Day 8) and with the lab samples at the hour 0 draw scheduled for Visit 19 (Day 15), these are to be performed pre the PA-824 dose/dosing time. Urine samples will be collected for all participants in parallel with urinalysis samples collected on Visit 5 (Day 1), Visit 12 (Day 8) and Visit 19 (Day 15) or early withdrawal. For the TMC207 plus PA-824 treatment arm at Visit 5/Day 1), Visit 12 (Day 8) and Visit 19 (Day 15), these are to be performed pre the PA-824 dose/dosing time. Sputum samples will be taken from the overnight sputum samples collected from all participants at Visit 3 (Day-2), Visit 4 (Day -1), Visit 5 (Day 1), Visit 6 (Day 2), Visit 7 (Day 3), Visit 8 (Day 4), Visit 10 (Day 6), Visit 12 (Day 8), Visit 14 (Day 10), Visit 16 (Day 12) and Visit 18 (Day 14).
M: Urine drug screen: cannabinoids, cocaine, amphetamines, opiates, benzodiazepines, barbiturates. Urine Drug screen and Urine Isoniazid test should be repeated at Visit 2 (Day -3) for patients who were not hospitalized at Visit 1 (Day -9 to -4).
N: On-study overnight sputum sampling will start on Visit 2 (Day -3) and will be collected from the afternoon and for 16 hours overnight. The 16-hour sputum sampling for each of the sampling days must be finished prior to the administration of the next day’s IMP. Sputum will be sampled starting on Visit 2 (Day –3) and will continue daily until Visit 18 (Day 14). Sputum sampling will stop on the morning of Visit 19 (Day 15). Overnight sputum collection will start at the latest on Visit 2 (Day -3), however may be collected for a number of more days if the screening results are delayed, or the mycobacterial testing on the first spot sputum shows an indeterminate result in which case the test may be repeated on a freshly collected spot sputum or an overnight sputum and that result used. It will not be collected for more than the pre-treatment time period. Visit 2 (Day -3) overnight sputum sampling will be used to ensure adequate volume and is not required for efficacy variable tests, retention sample collection and mycobacterial characterization testing
O: Randomization by the pharmacist/registered dispenser may occur once all the screening results are available and the investigator has determined that the participant is eligible for the trial. Randomization must occur prior to the Visit 4 (Day -1) ECGs.
P: Pharmacokinetics details are provided in the ECG/PK Flow charts. Q: Isoniazid and Rifampicin Resistance Test: Susceptibility testing for rifampicin and isoniazid, (rapid, sputum-based,
screening test). If the first spot sputum shows an indeterminate result, the test may be repeated on freshly collected spot sputum or overnight sputum and that result used.
R: The Mycobacterium tuberculosis (MTB) isolates from the Visit 3 (Day -2) and Visit 18 (Day 14) overnight sputum samples will be kept and later be batched and processed for (1) MIC against TMC207, PA-824 and moxifloxacin, (2) drug susceptibility testing for rifampicin, isoniazid, ethambutol and pyrazinamide with the MGIT system, (3) speciation of the infecting organism by polymerase chain reaction (PCR). If no Visit 18 (Day 14) pooled sputum sample is available these tests will be done on the last available sample. If the participant was treated with IMP for less than 9 days, mycobacteriology testing will be performed on the Visit 3 (Day -2) isolate only for these participants.
S: Adverse events will be collected by the Investigator from the time a participant signs the Informed Consent Form through to: (1) for TMC207 containing treatment arms (excluding TMC207 plus PA-824 containing treatment arm): - all AEs and SAEs – through to the end of Visit 21 (Day 42); (2) for PA-824 containing treatment arms (excluding TMC207 plus PA-824 containing treatment arm): - all AEs and SAEs – through to the end of Visit 20 (Day 28); - only ophthalmologic related adverse events and all serious adverse events - from Visit 20 (Day 28) through to the
end of Visit 22 (Day 104); (3) for TMC207 plus PA-824 and Rifafour e275® treatment arm: - all AEs and SAEs - through to the end of Visit 21 (Day 42); - only ophthalmologic related adverse events and all serious adverse events - from Visit 21 (Day 42) through to the
end of Visit 22 (Day 104).
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 17 of 142
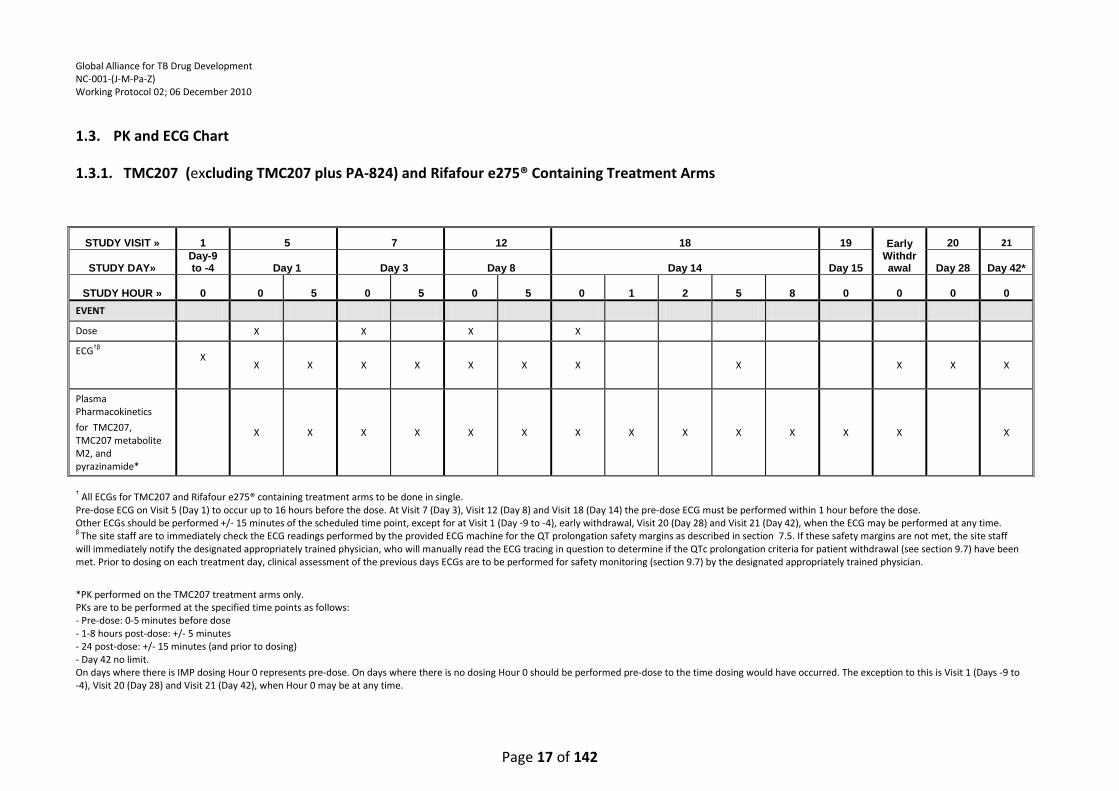
1.3. PK and ECG Chart
1.3.1. TMC207 (excluding TMC207 plus PA-824) and Rifafour e275® Containing Treatment Arms
† All ECGs for TMC207 and Rifafour e275® containing treatment arms to be done in single. Pre-dose ECG on Visit 5 (Day 1) to occur up to 16 hours before the dose. At Visit 7 (Day 3), Visit 12 (Day 8) and Visit 18 (Day 14) the pre-dose ECG must be performed within 1 hour before the dose. Other ECGs should be performed +/- 15 minutes of the scheduled time point, except for at Visit 1 (Day -9 to -4), early withdrawal, Visit 20 (Day 28) and Visit 21 (Day 42), when the ECG may be performed at any time. β The site staff are to immediately check the ECG readings performed by the provided ECG machine for the QT prolongation safety margins as described in section 7.5. If these safety margins are not met, the site staff will immediately notify the designated appropriately trained physician, who will manually read the ECG tracing in question to determine if the QTc prolongation criteria for patient withdrawal (see section 9.7) have been met. Prior to dosing on each treatment day, clinical assessment of the previous days ECGs are to be performed for safety monitoring (section 9.7) by the designated appropriately trained physician.
*PK performed on the TMC207 treatment arms only. PKs are to be performed at the specified time points as follows: - Pre-dose: 0-5 minutes before dose - 1-8 hours post-dose: +/- 5 minutes - 24 post-dose: +/- 15 minutes (and prior to dosing) - Day 42 no limit. On days where there is IMP dosing Hour 0 represents pre-dose. On days where there is no dosing Hour 0 should be performed pre-dose to the time dosing would have occurred. The exception to this is Visit 1 (Days -9 to -4), Visit 20 (Day 28) and Visit 21 (Day 42), when Hour 0 may be at any time.
STUDY VISIT » 1 5 7 12 18 19 Early Withdrawal
20 21
STUDY DAY» Day-9 to -4 Day 1 Day 3 Day 8 Day 14 Day 15 Day 28 Day 42*
STUDY HOUR » 0 0 5 0 5 0 5 0 1 2 5 8 0 0 0 0
EVENT
Dose X X X X
ECG†β X
X X X X X X X X X X X
Plasma Pharmacokinetics
for TMC207, TMC207 metabolite M2, and pyrazinamide*
X X X X X X X X X X X X X X
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 18 of 142
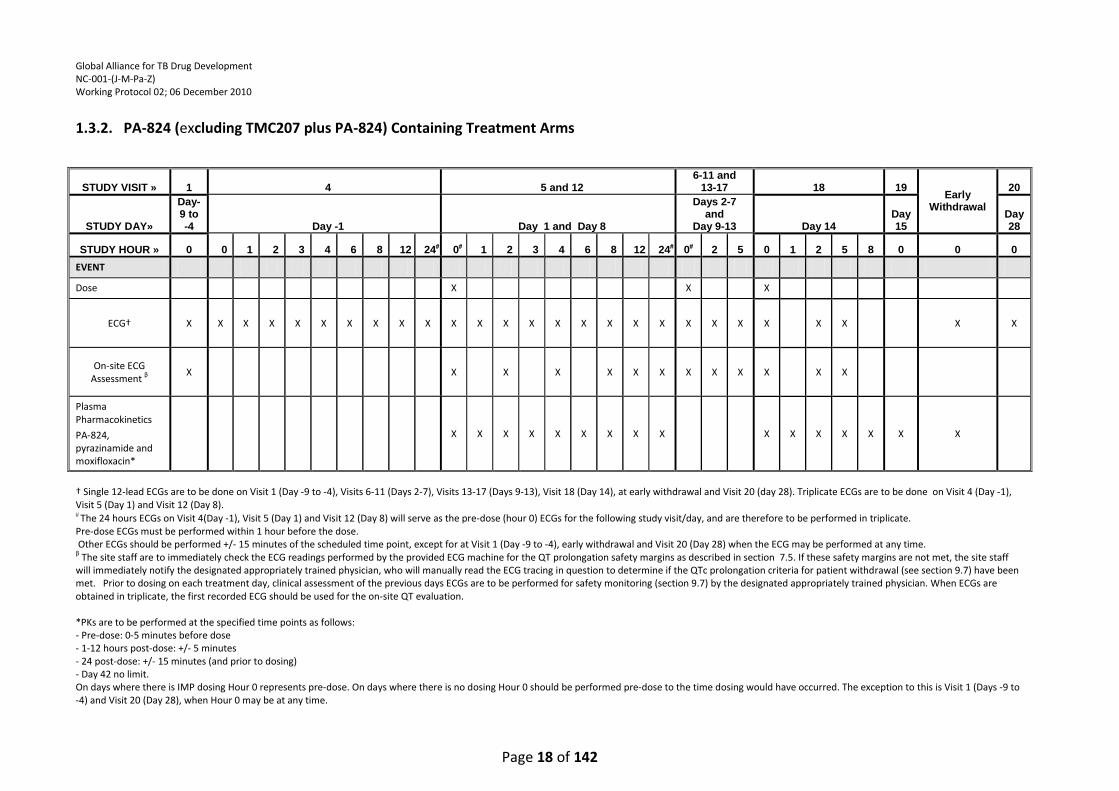
1.3.2. PA-824 (excluding TMC207 plus PA-824) Containing Treatment Arms
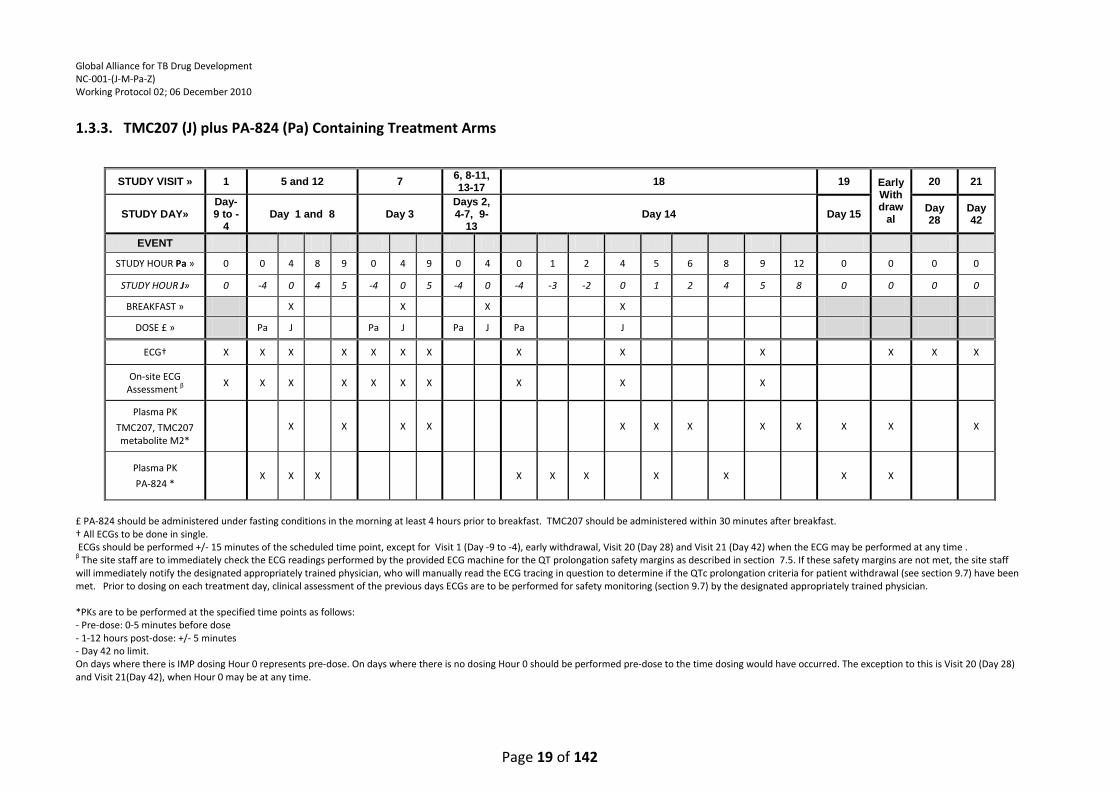
† Single 12-lead ECGs are to be done on Visit 1 (Day -9 to -4), Visits 6-11 (Days 2-7), Visits 13-17 (Days 9-13), Visit 18 (Day 14), at early withdrawal and Visit 20 (day 28). Triplicate ECGs are to be done on Visit 4 (Day -1), Visit 5 (Day 1) and Visit 12 (Day 8). The 24 hours ECGs on Visit 4(Day -1), Visit 5 (Day 1) and Visit 12 (Day 8) will serve as the pre-dose (hour 0) ECGs for the following study visit/day, and are therefore to be performed in triplicate. Pre-dose ECGs must be performed within 1 hour before the dose. Other ECGs should be performed +/- 15 minutes of the scheduled time point, except for at Visit 1 (Day -9 to -4), early withdrawal and Visit 20 (Day 28) when the ECG may be performed at any time. β The site staff are to immediately check the ECG readings performed by the provided ECG machine for the QT prolongation safety margins as described in section 7.5. If these safety margins are not met, the site staff will immediately notify the designated appropriately trained physician, who will manually read the ECG tracing in question to determine if the QTc prolongation criteria for patient withdrawal (see section 9.7) have been met. Prior to dosing on each treatment day, clinical assessment of the previous days ECGs are to be performed for safety monitoring (section 9.7) by the designated appropriately trained physician. When ECGs are obtained in triplicate, the first recorded ECG should be used for the on-site QT evaluation. *PKs are to be performed at the specified time points as follows: - Pre-dose: 0-5 minutes before dose - 1-12 hours post-dose: +/- 5 minutes - 24 post-dose: +/- 15 minutes (and prior to dosing) - Day 42 no limit. On days where there is IMP dosing Hour 0 represents pre-dose. On days where there is no dosing Hour 0 should be performed pre-dose to the time dosing would have occurred. The exception to this is Visit 1 (Days -9 to -4) and Visit 20 (Day 28), when Hour 0 may be at any time.
STUDY VISIT » 1 4 5 and 12 6-11 and
13-17 18 19 Early
Withdrawal
20
STUDY DAY»
Day-9 to -4 Day -1 Day 1 and Day 8
Days 2-7 and
Day 9-13 Day 14 Day 15
Day 28
STUDY HOUR » 0 0 1 2 3 4 6 8 12 24 0 1 2 3 4 6 8 12 24 0 2 5 0 1 2 5 8 0 0 0
EVENT
Dose X X X
ECG† X X X X X X X X X X X X X X X X X X X X X X X X X X X
On-site ECG Assessment β
X X X X X X X X X X X X X
Plasma Pharmacokinetics
PA-824, pyrazinamide and moxifloxacin*
X X X X X X X X X X X X X X X X
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 19 of 142
1.3.3. TMC207 (J) plus PA-824 (Pa) Containing Treatment Arms
£ PA-824 should be administered under fasting conditions in the morning at least 4 hours prior to breakfast. TMC207 should be administered within 30 minutes after breakfast. † All ECGs to be done in single. ECGs should be performed +/- 15 minutes of the scheduled time point, except for Visit 1 (Day -9 to -4), early withdrawal, Visit 20 (Day 28) and Visit 21 (Day 42) when the ECG may be performed at any time . β The site staff are to immediately check the ECG readings performed by the provided ECG machine for the QT prolongation safety margins as described in section 7.5. If these safety margins are not met, the site staff will immediately notify the designated appropriately trained physician, who will manually read the ECG tracing in question to determine if the QTc prolongation criteria for patient withdrawal (see section 9.7) have been met. Prior to dosing on each treatment day, clinical assessment of the previous days ECGs are to be performed for safety monitoring (section 9.7) by the designated appropriately trained physician. *PKs are to be performed at the specified time points as follows: - Pre-dose: 0-5 minutes before dose - 1-12 hours post-dose: +/- 5 minutes - Day 42 no limit. On days where there is IMP dosing Hour 0 represents pre-dose. On days where there is no dosing Hour 0 should be performed pre-dose to the time dosing would have occurred. The exception to this is Visit 20 (Day 28) and Visit 21(Day 42), when Hour 0 may be at any time.
STUDY VISIT » 1 5 and 12 7 6, 8-11, 13-17
18 19 Early Withdraw
al
20 21
STUDY DAY» Day-9 to -
4 Day 1 and 8 Day 3
Days 2, 4-7, 9-
13 Day 14 Day 15
Day 28
Day 42
EVENT
STUDY HOUR Pa » 0 0 4 8 9 0 4 9 0 4 0 1 2 4 5 6 8 9 12 0 0 0 0
STUDY HOUR J» 0 -4 0 4 5 -4 0 5 -4 0 -4 -3 -2 0 1 2 4 5 8 0 0 0 0
BREAKFAST » X X X X
DOSE £ » Pa J Pa J Pa J Pa J
ECG† X X X X X X X X X X X X X
On-site ECG Assessment β
X X X X X X X X X X
Plasma PK
TMC207, TMC207 metabolite M2*
X
X X X X X X X X X X X
Plasma PK
PA-824 * X X X X X X X X X X

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 20 of 142
2. INTRODUCTION
2.1. Background Information
Although some progress has been made in recent years in controlling TB globally, TB has remained
a persistent problem in the developing countries of Africa and Asia. TB is currently one of the top
three killer infectious diseases, and there is more TB in the world today than at any other time in
history. The current first-line antituberculosis agents have been in use for over 20 years and are
relatively ineffective in controlling TB as a public health problem. Although the current regimens
and drugs have been very successful in controlled clinical trials resulting in the permanent cure of
more than 95% of trial participants, treatment takes 6 months to complete. This, plus side effects,
result in poor compliance which is particularly likely to occur after the second month of treatment.
The full application of the DOTS strategy is becoming more and more difficult in the developing
countries of the world that are also battling to control the HIV-epidemic. As a result of poor
treatment compliance, drug resistance is becoming more common and fears of an epidemic with
virtually untreatable strains of TB are growing. Since the discovery of the rifamycins (1), and their
introduction into standard antituberculosis regimens, very few new classes of drugs have been
evaluated with a view to their registration as antituberculosis agents.
Following the declaration of TB as a global emergency by the World Health Organization (WHO) in
1993, there has been a resurgence of efforts to develop improved TB therapies and several
promising new agents are presently in or approaching clinical evaluation. New combination
regimens are desperately needed for two reasons; to shorten treatment to a duration more easily
manageable by patients and public health services and to provide more efficacious, safer and better
tolerated, affordable treatment for the growing number of patients suffering from multidrug-
resistant and extensively drug-resistant tuberculosis.
The bactericidal action of antituberculosis agents alone or in combination can be quantified by
studying the fall in the number of colony forming units (CFU) of M. tuberculosis (MTB) present in
the sputum of participants with microscopy smear-positive pulmonary TB. In the first
comprehensive evaluation of this technique all of the then available antituberculosis agents were
studied both alone and in combination during the first 14 days of treatment in small groups of
approximately 4 participants(2). This study showed significant differences between the different
drugs, but these differences were most obvious during the first 2 days of treatment and this period

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 21 of 142
of activity was consequently labeled EBA or “standard EBA”. Since this pioneering study, a number
of EBA studies have been carried out, many in the Western Cape Province of South Africa. These
studies have been carried out mainly over 2 days in view of the initial findings by Jindani et al (2).
More recently, EBA studies have been conducted over 5, 7 and 14 days and it has become apparent
that valuable additional information may be found by conducting EBA studies over a longer period
(“extended EBA”) (3-6).
TMC207 is a new agent being developed for TB treatment. As detailed in the Investigator’s
Brochure (7), TMC207 is a diarylquinoline investigational compound that offers a novel mechanism
of anti-TB action by specifically inhibiting mycobacterial adenosine triphosphate (ATP) synthase (8).
In vitro, TMC207 potently inhibits both drug-sensitive and drug-resistant MTB isolates (9, 10), and is
also bactericidal against non-replicating tubercle bacilli (11). In the murine model of TB, TMC207 was
as active as the triple combination of isoniazid (H), rifampin (R), and pyrazinamide (Z), while
addition of TMC207 to this drug regimen results in accelerated clearance of bacilli (7). There
appears to be a synergistic interaction with pyrazinamide, 100% of mice were culture negative after
8 weeks of treatment with TMC207 and pyrazinamide compared to 0% of mice treated with the
standard regimen of rifampicin, isoniazid and pyrazinamide (12). Collectively, these findings in the
mouse model have led to the suggestion that a regimen containing TMC207 and pyrazinamide
could be effective in the treatment of both drug-sensitive and multi-drug resistant (MDR) TB as well
as shorten treatment duration in patients. On the other hand, the combination of TMC207 and PA-
824 in the murine model of TB, while somewhat antagonistic relative to TMC207, alone, appeared
as active as the triple combination of HRZ (38). Thus a novel regimen with a TMC207 plus PA-824
core could be effective in the treatment of MDR-TB by providing two novel drugs for which there is
no known pre-existing resistance. Moreover, the underlying mechanism of the antagonism seen in
the mouse model is not known, and it is not clear whether it will also be seen in humans, or
whether an additive or synergistic effect might be seen instead.
TMC207 has been studied in one dose-ranging 7-day EBA trial to date (6). The results from the 7-day
EBA trial showed clinically relevant bactericidal activity only at the highest TMC207 dose (400 mg)
tested with a delay in onset of response (activity detected from Day 4 onwards). No clinically
relevant changes were seen with the lower doses of TMC207 (25 mg and 100 mg) during the 7 days
of treatment. The reason for this delayed onset of activity (400 mg) or lack of bactericidal activity
(at lower doses) is not yet clear but may be partly due to suboptimal plasma concentrations initially

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 22 of 142
or throughout the 7-day duration of treatment. Based on mouse data, the average target plasma
concentration of TMC207 for efficacy in humans is about 600ng/ml (IB, page 27) (7). In an ongoing
14-day dose-ranging EBA study (TMC207-CL001) being conducted in South Africa, different dosing
regimens are being studied to obtain average plasma concentrations ranging from 500 to 2000
ng/ml throughout most of the dosing period. Because combination, multidrug therapy is required
for treatment of TB, it is now important to determine the safety and efficacy of TMC207 in an EBA
study in combination with other antituberculosis drugs such as pyrazinamide and PA-824, to help
determine how TMC207 might be combined with other drugs to optimize TB treatment, prior to
evaluation of such a regimen in longer-term studies.
PA-824 is also a new agent being developed for TB treatment. As detailed in the Investigator’s
Brochure (13), PA-824 is a new chemical entity and a member of a class of compounds known as
nitroimidazo-oxazines, which possess significant antituberculosis activity and a unique mechanism
of action (14). Nonclinical studies of PA-824 highlighted important properties that may result in
significant improvements in TB treatment. PA-824 demonstrated in vitro activity against both drug-
sensitive and MDR-TB(15), and in vivo activity in a mouse model of tuberculosis (15,16).
PA-824 has been studied in two dose-ranging, 14-day EBA trials to date. In these studies, PA-824
monotherapy has demonstrated significant mycobactericidal activity. The efficacy data from study
PA-824-CL-007 indicated that all doses of PA-824 (200, 600, 1000 and 1200 mg) produced an
equivalent decrease in sputum CFU counts over the 14-day treatment period; no difference could
be discerned among the PA-824 treatment groups. In study PA-824-CL-010, an EBA study with a
similar design to study PA-824-CL-007 except for the use of lower doses of PA-824 (50, 100, 150 and
200 mg/day), preliminary results indicate that PA-824 treatment resulted in a measurable dose-
dependent mycobactericidal activity. As stated above for TMC207, it is also important to determine
the safety and efficacy of PA-824 in an EBA study in combination with other antituberculosis drugs
(e.g., moxifloxacin and pyrazinamide), to help determine how PA-824 might be combined with
other drugs to optimize TB treatment, prior to evaluation of such a regimen in longer-term studies.
Moxifloxacin (M) is an 8-methoxyquinolone whose oral formulation (which will be used in this
study) has been approved in South Africa and most other countries around the world for the
treatment of acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis,
community-acquired pneumonia, skin and soft tissue infections. It has enhanced activity against
Gram-positive pathogens, and anaerobes while retaining useful activity against Gram-negative

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 23 of 142
organisms. Moxifloxacin is not metabolized by the cytochrome P450 system, thus the risk of
clinically relevant drug interactions is reduced. It has a positive safety profile within the
fluoroquinolone class. Clinical studies have been carried out in additional indications (urinary tract
infections, pelvic infections, pharyngitis/tonsillitis, and tuberculosis).
Studies in murine models of TB and in TB patients have shown that moxifloxacin has significant
mycobactericidal activity(17-26). These include EBA studies as well as four 8-week treatment period
phase IIb studies in which moxifloxacin was substituted for either isoniazid or ethambutol in the
standard, first-line, TB treatment regimen (see Table 3). Currently, a phase III trial is enrolling in
which moxifloxacin replaces either isoniazid or ethambutol in first-line treatment and is
administered in combination with the other first-line drugs for four months, with the expectation
that total treatment duration of drug-sensitive TB can be shortened to four months. The dose of
moxifloxacin employed in these TB clinical trials is 400 mg daily, the registered dose (for other
indications as stated above) and the dose that is commonly used as second line therapy for TB
(e.g., in MDR-TB patients). This is the same dose we plan to use in the proposed study.
Pyrazinamide, the pyrazine analogue of nicotinamide, is an approved anti-tuberculosis agent.
Pyrazinamide is indicated for the initial treatment of active tubercuIosis in adults and children when
combined with other antituberculosis agents and it contributes significantly to the sterilization of
lesions and thus, treatment shortening (27).
The current study is being conducted as part of the clinical development of TMC207- and PA-824-
based combinations to treat TB. As noted above, in the mouse model of TB, 100% of mice treated
with the combination of TMC207 and pyrazinamide were culture negative after 8 weeks of
treatment compared to 0% in the group of mice treated with the first-line regimen of rifampicin,
isoniazid and pyrazinamide (12). In different studies using a similar mouse model of TB, the
combination of PA-824 and TMC207 as well as the combination of PA-824 and pyrazinamide
displayed mycobactericidal activity that was equivalent to that of the first-line regimen of
rifampicin, isoniazid and pyrazinamide (28), while the combination of PA-824, moxifloxacin and
pyrazinamide cured mice more rapidly than the first-line regimen (4 versus 6 months) (29). Thus,
these novel regimens, which contain neither isoniazid nor rifampicin, may have the potential to (1)
shorten the duration of TB chemotherapy and (2) treat both drug-sensitive and multidrug-resistant
TB.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 24 of 142
This study will be conducted in accordance with the Guidelines for Good Practice in the Conduct of
Clinical Trials in Human Participants in South Africa 2006 (SA GCP), International Conference on
Harmonization Good Clinical Practice (ICH GCP), the ethical principles that have their origin in the
Declaration of Helsinki and the U.S. Code of Federal Regulations, 21 CFR Parts 50, 56, and 312 (30,31).
The participant population will consist of adult males and females with newly diagnosed sputum
smear microscopy-positive pulmonary TB.
2.2. Preclinical Studies
2.2.1. TMC207
Full details of the preclinical studies are provided in the current TMC207 Investigator’s Brochure
(Pages 20-48) (7).
Microbiology
In vitro studies have demonstrated that the range of minimum inhibitory concentrations (MICs) for
M. tuberculosis H37Rv, the international reference strain, and 6 fully drug-susceptible clinical
isolates was 0.030 to 0.120 µg/ml. The activity of TMC207 appears to be specific for mycobacteria,
as the MICs for non-mycobacteria were at least 500-fold higher. The activity of the main
metabolite of TMC207, M2, was determined against M. tuberculosis H37Rv in both solid and liquid
media and its MIC was found to be 0.1 µg/ml. This MIC shows that M2 is active against M.
tuberculosis but 3-6 times less active than the parent compound TMC207 (MIC of 0.015-
0.025µg/ml). TMC207 demonstrated similar in vitro efficacy against M. tuberculosis clinical isolates
resistant to the known anti-TB drugs (isoniazid, rifampin, pyrazinamide, streptomycin, ethambutol,
or fluoroquinolones). As expected, from the lack of cross-resistance with currently used anti-TB
agents, TMC207 retained activity against MDR-TB isolates.
Nonclinical Safety Studies
The non-clinical safety evaluation of TMC207 includes pharmacology, pharmacokinetics, toxicology
and metabolism studies that were conducted in accordance with current ICH guidelines. Repeated
dose toxicity studies were performed with dosing durations up to 3 months in mice and up to 6
months in rats and in dogs. Recovery was studied in rats and dogs. For more detailed information
please refer to the IB (Pages 20-48) (7).

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 25 of 142
Respiratory parameters were unaffected by treatment. There were no effects suggestive of
neurological impairment or delayed neurotoxicity in rats. In single dose toxicity studies there were
no mortalities following oral doses of up to 200 mg/kg in mice and rats. No mutagenicity or
clastogenic effects were seen in a series of in vitro and in vivo genotoxicity tests. TMC207 was
evaluated for possible developmental toxicity effects in the rat and the rabbit. No teratogenic
effects were found.
In vitro, TMC207 slightly to moderately inhibited the delayed rectifier potassium current (IKr) in the
human ether-à-go-go-related gene (hERG) model but this effect was not manifest as a prolongation
of repolarization in subsequent in vivo studies. There were no relevant effects on the isolated right
atrium of guinea pigs in vitro, or in the isolated Langendorff-perfused rabbit heart. In vivo, positive
chronotropic effects were seen in the anesthetized guinea pig after i.v. administration, but not in
the conscious dog. In conscious, telemetered dogs, oral TMC207 had no relevant effects on cardio-
hemodynamic and electrocardiogram (ECG) parameters.
Prior to the use of PA-824 in combination with TMC207 in this clinical study, a preclinical
cardiovascular safety pharmacology study was conducted in unrestrained beagle dogs with both
drugs to explore the potential for additive effects on QT prolongation induced by the combination.
TMC207 was administered at 100 mg/kg, PO, for 7 days in order to allow for the accumulation of
TMC207 and its metabolites, M2 and M3. On Day 7, these animals were also given PA-824 at 100
mg/kg, SC. The dose and route of PA-824 chosen, 100 mg/kg, SC, was shown in preliminary PK
studies to achieve a Cmax ~3-4-fold higher than the clinical exposure at the 200-mg dose. To assess
the impact of PA-824 administered to dogs receiving TMC207, the QT interval for this group of
animals was compared on Day 6 (following 6 daily doses of TMC207) vs Day 7 (following 7 daily
doses of TMC207 and a single dose of PA-824). To assess the effect of TMC207 administration to
dogs receiving PA-824, the QT interval for the group of animals described above, on Day 7, was
compared to a separate group of animals receiving PA-824 only. A third group of animals receiving
both vehicles acted as control. The cardiovascular parameters recorded were systolic, diastolic, and
mean arterial pressures, heart rate and ECG (including QRS duration; PR, RR, and QT intervals; and
height of R wave). Monitoring periods were baseline prior to study start (Day -1), Day 6, and Day 7
for a full 24 hours each day. Preliminary results indicate that administration of 100 mg/kg TMC207
daily for 7 days causes a small increase in QTc interval by Day 6 in some animals that is not
influenced by the addition of 100 mg/kg PA-824 on Day 7. The effect of PA-824 dosing alone on QT

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 26 of 142
interval appeared to be due to discomfort related to the SC route of administration and not related
to the plasma exposure. Given these limited findings in this preclinical study, participants will not
be exposed to undue risk in the study, during which they will be closely monitored in the hospital
throughout the treatment period, including extensive ECG monitoring.
2.2.2. PA-824
Full details of the preclinical studies are provided in the current Investigator’s Brochure (Pages 34-
57) (13).
Microbiology
In vitro studies have demonstrated that the minimum inhibitory concentration (MIC) of PA-824
against a variety of drug-sensitive MTB isolates is similar to the MIC of isoniazid (MIC of PA-824,
0.015 to 0.25 g/mL; MIC of isoniazid, 0.03 to 0.06 g/mL). PA-824 was efficacious in vitro against
drug-resistant clinical isolates of MTB, with MIC values ranging from 0.03 to 0.53 µg/mL. The
minimum effective dose (MED) of PA-824 was 12.5 mg/kg/day in a mouse model of TB. The MED is
defined as the lowest dose able to prevent the development of macroscopic lung lesions and
splenomegaly. The minimum bactericidal dose (MBD) was 100 mg/kg/day in the same mouse
model. The MBD is defined as the lowest dose able to reduce the lung TB colony forming unit (CFU)
counts by 99%. The magnitude of CFU reduction by PA-824 at this dose is similar to that seen with
the highest dose of isoniazid tested in the same study (25 mg/kg/day).
Nonclinical Safety Studies
The non-clinical safety evaluation of PA-824 includes pharmacology, pharmacokinetics, toxicology
and metabolism studies that were conducted in accordance with current ICH guidelines.
Metabolite analyses in rats, monkeys, and humans indicate an overall similar metabolic profile in
these species with some differences among minor metabolites. These studies have confirmed that
rats and monkeys are appropriate species for the toxicology program.
PA-824 was negative in all genotoxicology studies performed. One of its metabolites (M50) that is
found in rat, monkey and human plasma was positive in a screening Ames assay. M50 is not a major
metabolite in humans and the exposure relative to parent drug is higher in the rat (24%) and
monkey (18%) than in humans (6%).
PA-824-induced effects in respiratory, CNS, and cardiovascular safety pharmacology studies were
generally mild and reversible; effects were most prominent at 450 mg/kg/day. PA-824 is a weak

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 27 of 142
inhibitor (IC50=20uM) of the hERG channel. In a telemetry monkey study, in the dose range 50-450
mg/kg, there was no or minor prolongation of the QT interval, depending on the method of
correction. The weight of evidence suggests that there should be no effect on QT in the
concentration range being explored in the clinical studies.
Prior to the use of PA-824 in combination with moxifloxacin in the planned clinical study, a
preclinical cardiovascular safety study was conducted in unrestrained cynomolgus monkeys with
both drugs to explore the potential for PA-824 to alter the QT prolongation induced by
moxifloxacin. The doses of moxifloxacin chosen for this monkey study, 30 mg/kg and 100 mg/kg,
have been shown in the literature and historically by the research lab performing the study, to
induce at least a 20-msec increase in QTc (32). The dose of PA-824 chosen, 50 mg/kg, has been
shown in previous monkey studies to result in a free plasma concentration ~10-fold higher than the
clinical exposure at the 200 mg dose. This study design included 2 Latin squares with 4 animals in
each; the first group of animals receiving vehicle, low-dose moxifloxacin, PA-824, and PA-824
together with moxifloxacin. The second group followed a similar design with the high dose of
moxifloxacin. All animals received a 1-week washout period between doses. The cardiovascular
parameters recorded were systolic, diastolic, and mean arterial pressures, heart rate and ECG
(including QRS duration; PR, RR, and QT intervals; and height of R wave). Monitoring periods were
baseline prior to study start, 24 hours pre-dose and 24 hours post dose. Statistical evaluations will
be made comparing all dose intervals to vehicle as well as comparing the combination PA-
824/moxifloxacin dose to the moxifloxacin-alone dose. Preliminary results indicate that
administration of 50 mg/kg PA-824 in combination with 100 mg/kg moxifloxacin resulted in
increases in QT and QTc intervals in 1 animal (out of 4 animals tested) that were ~10% higher than
administration of 100 mg/kg moxifloxacin alone. Given this limited finding with the relatively high
doses of PA-824 and moxifloxacin employed in the preclinical study, participants will not be
exposed to undue risk in the proposed clinical study, during which they will be closely monitored in
the hospital throughout the treatment period, including extensive ECG monitoring.
Repeat-dose toxicology studies with PA-824 have been conducted in male and female rats (14 days
to 6 months) and in male and female monkeys (7 days to 3 months). In all studies, dose-dependent
reduced food consumption and reduced weight gain or weight loss were noted. In addition,
testicular atrophy was observed in rats while cataracts were observed by indirect ophthalmoscopy
in both rats and monkeys. In general, toxicity in both rat and monkey was significantly greater when

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 28 of 142
exposures exceeded approximately 300 μg·hr/mL in the 14-day studies and approximately
200 μg·hr/mL in the 3-month studies.
Reproductive toxicology studies show that PA-824 is not teratogenic in rats or rabbits. In the rat
fertility study, dose-dependent reduced fertility rates, due to decreased sperm numbers and
decreased motility, were observed at doses of 30 mg/kg and greater. This effect was partially
reversible. As in the 3-month rat toxicology study, irreversible testicular lesions were not observed
at 30 mg/kg, only at 100 and 300 mg/kg.
To more fully characterize the cataract and male reproductive system findings, a 3-month monkey
study in sexually mature males (0, 50, 150, 300 mg/kg/day) and a 6-month rat study (0, 30, 100,
300 mg/kg) in males and females were conducted. Ocular assessments were conducted in a much
more careful and systematic manner than in the initial 3-month toxicology studies described above.
In each of the later studies, all ophthalmologic examinations were conducted by a single
ophthalmologist, using both indirect ophthalmoscopy and slit-lamp biomicroscopy. Animals were
screened before dosing to ensure no animal had cataracts at baseline, and then monthly during
dosing and recovery. In this monkey study, although similar drug exposures were attained as in
the original 3-month monkey study, no cataracts or testes effects (semenology, organ weights,
histopathology, or hormones [testosterone, follicle-stimulating hormone, Inhibin-B]) were observed
at any point during dosing or during a 20-week recovery period. PA-824 does not appear to cause
cataracts or testicular toxicity in monkeys. In the 6-month rat study, PA-824 caused irreversible
cataracts at 100 mg/kg from Day 118 of the study in males and females. In contrast to the original
3-month rat report, rats in this more carefully conducted study developed cataracts while on drug
but not during recovery. The NOAEL was 30 mg/kg for cataracts and 10 mg/kg for testicular
toxicity. Rats that developed cataract and testicular toxicity also experienced marked decreases in
body weight gain and food consumption. The AUC safety multiples (relative to the exposures
obtained at the anticipated clinical dose of 200 mg/day) for cataract are ~1.5x in the rat; in the
monkey at the highest dose tolerated, where there were no cataracts in the second well conducted
study, the multiple is at least 3.7x.
To summarize, cataracts have been detected in multiple animals from two similar rat studies at
mid-to-high doses. In contrast, the finding of cataracts in one monkey study could not be
confirmed in a follow-up study. Thus, both cataracts and the testicular effects appear to be
species-limited.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 29 of 142
Refer to the TMC207 preclinical section for details on the PA-824 TMC207 combination preclinical
cardiovascular study. Given these limited findings in this preclinical study, participants will not be
exposed to undue risk in the proposed clinical amendment, during which they will be closely
monitored in the hospital throughout the treatment period, including extensive ECG monitoring.
2.2.3. Moxifloxacin
Moxifloxacin has been approved in South Africa and most other countries around the world for the
treatment of acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis,
community-acquired pneumonia, skin and soft tissue infections. An extensive preclinical program
supports the safety and efficacy of 400 mg moxifloxacin given once daily for 5 to 21 days in adults
for the above specified indications. For more detailed information, please refer to the IB for
Moxifloxacin (34).
2.3. Clinical Trials
2.3.1. TMC207
Six Phase I trials in healthy patients and 1 short-term Phase II trial in treatment-naïve patients with
sputum smear-positive pulmonary TB have been conducted. One long-term double-blind Phase II
trial consisting of 2 different stages and one long-term open label trial in patients infected with
newly diagnosed sputum smear-positive pulmonary MDR-TB are ongoing. In the former, Stage 1
has completed dosing with TMC207. The principal findings of these trials are summarized below
(Table 1). For more detailed information, please refer to the IB for TMC207 (Pages 53-83) (7).
A second short-term (14 days) Phase II trial in treatment-naïve patients with sputum smear-positive
pulmonary TB is being conducted in South Africa (Study Protocol TMC207-CL001). Preliminary
results indicate that all dosing regimens were well tolerated and produced measurable
mycobactericidal activity with a statistically significant linear trend over the dose groups. Thus, the
regimen chosen for this study (700mg on day 1, 500mg on day 2 and 400mg days 3-14) is
appropriate.
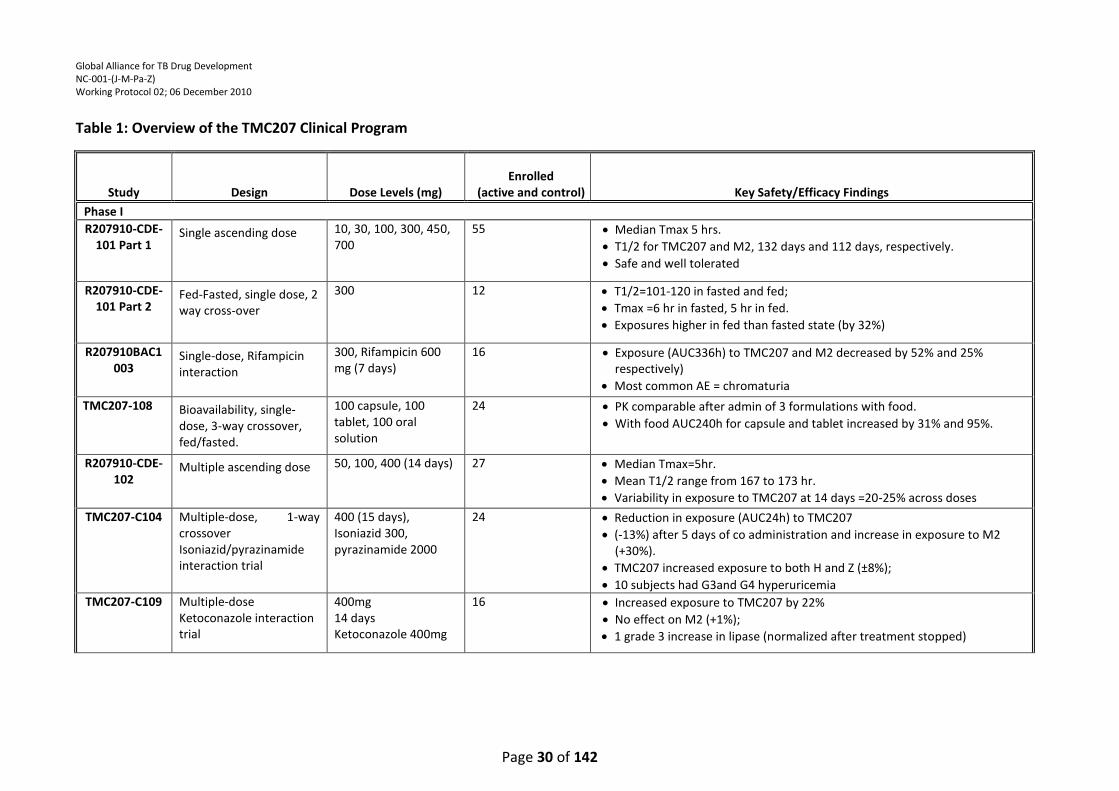
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 30 of 142
Table 1: Overview of the TMC207 Clinical Program
Study Design Dose Levels (mg) Enrolled
(active and control) Key Safety/Efficacy Findings
Phase I
R207910-CDE-101 Part 1
Single ascending dose 10, 30, 100, 300, 450, 700
55
Median Tmax 5 hrs.
T1/2 for TMC207 and M2, 132 days and 112 days, respectively.
Safe and well tolerated
R207910-CDE-101 Part 2
Fed-Fasted, single dose, 2 way cross-over
300 12
T1/2=101-120 in fasted and fed;
Tmax =6 hr in fasted, 5 hr in fed.
Exposures higher in fed than fasted state (by 32%)
R207910BAC1003
Single-dose, Rifampicin interaction
300, Rifampicin 600 mg (7 days)
16
Exposure (AUC336h) to TMC207 and M2 decreased by 52% and 25% respectively)
Most common AE = chromaturia
TMC207-108 Bioavailability, single-dose, 3-way crossover, fed/fasted.
100 capsule, 100 tablet, 100 oral solution
24
PK comparable after admin of 3 formulations with food.
With food AUC240h for capsule and tablet increased by 31% and 95%.
R207910-CDE-102
Multiple ascending dose 50, 100, 400 (14 days) 27
Median Tmax=5hr.
Mean T1/2 range from 167 to 173 hr.
Variability in exposure to TMC207 at 14 days =20-25% across doses
TMC207-C104 Multiple-dose, 1-way crossover Isoniazid/pyrazinamide interaction trial
400 (15 days), Isoniazid 300, pyrazinamide 2000
24
Reduction in exposure (AUC24h) to TMC207
(-13%) after 5 days of co administration and increase in exposure to M2 (+30%).
TMC207 increased exposure to both H and Z (±8%);
10 subjects had G3and G4 hyperuricemia
TMC207-C109 Multiple-dose Ketoconazole interaction trial
400mg 14 days Ketoconazole 400mg
16
Increased exposure to TMC207 by 22%
No effect on M2 (+1%);
1 grade 3 increase in lipase (normalized after treatment stopped)
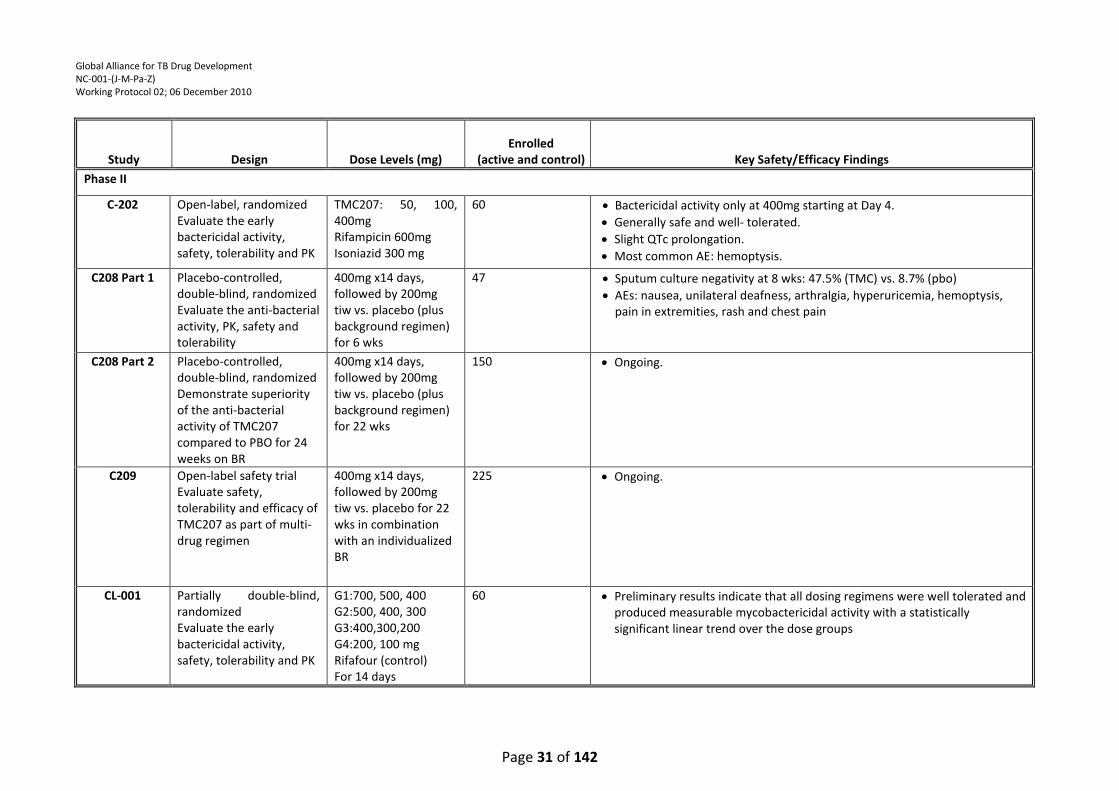
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 31 of 142
Study Design Dose Levels (mg) Enrolled
(active and control) Key Safety/Efficacy Findings
Phase II
C-202 Open-label, randomized Evaluate the early bactericidal activity, safety, tolerability and PK
TMC207: 50, 100, 400mg Rifampicin 600mg Isoniazid 300 mg
60
Bactericidal activity only at 400mg starting at Day 4.
Generally safe and well- tolerated.
Slight QTc prolongation.
Most common AE: hemoptysis.
C208 Part 1 Placebo-controlled, double-blind, randomized Evaluate the anti-bacterial activity, PK, safety and tolerability
400mg x14 days, followed by 200mg tiw vs. placebo (plus background regimen) for 6 wks
47
Sputum culture negativity at 8 wks: 47.5% (TMC) vs. 8.7% (pbo)
AEs: nausea, unilateral deafness, arthralgia, hyperuricemia, hemoptysis, pain in extremities, rash and chest pain
C208 Part 2 Placebo-controlled, double-blind, randomized Demonstrate superiority of the anti-bacterial activity of TMC207 compared to PBO for 24 weeks on BR
400mg x14 days, followed by 200mg tiw vs. placebo (plus background regimen) for 22 wks
150
Ongoing.
C209 Open-label safety trial Evaluate safety, tolerability and efficacy of TMC207 as part of multi-drug regimen
400mg x14 days, followed by 200mg tiw vs. placebo for 22 wks in combination with an individualized BR
225
Ongoing.
CL-001 Partially double-blind, randomized Evaluate the early bactericidal activity, safety, tolerability and PK
G1:700, 500, 400 G2:500, 400, 300 G3:400,300,200 G4:200, 100 mg Rifafour (control) For 14 days
60 Preliminary results indicate that all dosing regimens were well tolerated and produced measurable mycobactericidal activity with a statistically significant linear trend over the dose groups

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 32 of 142
Phase I
The Phase I trials have provided a basic understanding of TMC207’s pharmacokinetic
characteristics, drug-drug interaction potential, and short-term safety/tolerability profile in 173
healthy patients.
TMC207 was well absorbed with a median time to reach the maximum plasma concentration (tmax)
of about 5 hours after dosing. The maximum plasma concentration (Cmax) and area under the
plasma concentration-time curve (AUC) increased proportionally up to the highest doses studied
(700 mg single dose and 400 mg multiple q.d. doses). Accumulation from Day 1 to Day 14 was
approximately 2-fold expressed as increase in AUC, while trough concentrations increased up to 3.5
fold. The pharmacokinetics of TMC207 in patients with pulmonary TB were comparable to those in
healthy patients. The average terminal elimination half-life (t1/2,term) of TMC207 and its N-
monodesmethyl metabolite (M2) was about 132 and 112 days, respectively. The latter was found
to be active against M. tuberculosis, although less potent than TMC207 (7).
Administration of TMC207 as the tablet formulation with food increased the relative bioavailability
(by 95%) as compared to administration without food.
Drug-drug interaction studies confirmed the role of cytochrome P450 3A4 (CYP3A4) in the
metabolism of TMC207 to M2. A study with rifampicin (a CYP3A4 inducer) showed that the
exposure (AUC336h) to TMC207 as well as M2 was significantly reduced (by 52 and 25%,
respectively). Furthermore, co-administration of ketoconazole (a CYP3A4 inhibitor) increased
exposure (AUC24h) to TMC207 by 22%, without a significant effect on M2 (+1%). A drug-drug
interaction study with the combination of isoniazid/pyrazinamide showed a reduction in the
exposure (AUC24h) to TMC207 (-13%) after 5 days of co-administration, while exposure to M2
increased (+30%). TMC207 increased exposure (AUC24h) to both isoniazid and pyrazinamide (≤8%)
which is not considered clinically relevant. Thus, no significant drug-drug interaction between
TMC207 and pyrazinamide is anticipated in the proposed clinical study.
Phase II
In a Phase IIa study (6), efficacy (bactericidal activity) was measured by change in the amount of
bacilli in the sputum, estimated by changes in the number of CFU. The log10 of the CFU counts, the
changes from baseline in log10 sputum CFU counts (log10 fall) as well as the average daily change
in log10 sputum CFU counts (Early Bactericidal Activity [EBA]), over a period of 7 days, were

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 33 of 142
derived. Over a period of 7 days, mean values for extended EBA (eEBA) were -0.01, -0.04 and -0.11
log10 CFU/day for the TMC207 25 mg, 100 mg and 400 mg group, respectively. Corresponding
values for the control treatments were -0.24 log10 CFU/day for the rifampin group and -0.27 log10
CFU/day for the isoniazid group.
Over a period of 2 days, mean values for EBA were 0.01, -0.10 and 0.02 log10 CFU/day for the
TMC207 25 mg, 100 mg and 400 mg group, respectively. Corresponding values for the control
treatments were -0.44 log10 CFU/day for the rifampin group and -0.28 log10 CFU/day for the
isoniazid group.
On Day 7, changes from baseline in log10 sputum CFU count were -0.04, -0.26 and -0.77 for the
TMC207 25 mg, 100 mg and 400 mg groups, respectively. For the control treatments, the changes
versus baseline on Day 7 were -1.70 and -1.88 for the rifampin and isoniazid groups, respectively.
For the control treatments, the changes versus baseline on Day 2 were -0.88 and -0.57 for the
rifampin and isoniazid groups, respectively. Note that the Day 2 changes for the isoniazid group
were influenced by 2 outliers. All patients started standard TB therapy after collection of the
overnight sputum specimen on the morning of Day 7. All TMC207 dose groups had an additional
log10 fall of approximately 0.40 log10 CFU on Day 8.
The addition of TMC207 to a 5-drug MDR-TB treatment regimen in an ongoing Phase IIB trial
resulted in significantly shorter time to culture conversion compared to placebo. During the 8-
week treatment in Stage 1, 10 of 21 (47.6%) patients in the TMC207 group became sputum culture
negative in the Mycobacteria Growth Indicator Tube (MGIT) compared to 2 of 23 (8.7%) patients in
the placebo group. No differences in exposure to TMC207 and M2 in plasma or sputum between
patients with or without culture conversion were observed. The selected dosing regimen (400 mg
q.d. for the first 2 weeks and 200 mg t.i.w. for the following 6 weeks) successfully controlled
accumulation of TMC207 and M2 in plasma, with most patients achieving average concentrations
above the targeted 600 ng/mL throughout the dosing period. The results of Stage 1 supported the
extension of this dosing regimen up to 24 weeks of treatment in Stage 2 of the trial (33) which is
currently ongoing.
Clinical Safety
In the Phase I trials, TMC207 was generally safe and well tolerated. The most common adverse
events (AEs) were headache, nasopharyngitis, postural dizziness, and hyperuricemia.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 34 of 142
Hyperuricemia is a known side effect of pyrazinamide. There were no deaths or serious adverse
events (SAEs) reported.
In the short-term, 7-day Phase IIa EBA trial in 75 treatment-naïve TB-infected patients, the most
commonly reported AE was hemoptysis, a common complication of pulmonary TB (6). The most
commonly reported AE during treatment phases including TMC207 was hemoptysis, reported by 1
(6%) subject in the 100 mg group and 3 (21%) patients in the 400 mg group. All AEs were
considered not or doubtfully related to the trial medication, except for 2 AEs (diarrhea and rash) in
the 100 mg TMC207 group and 1 AE (somnolence) in the 400 mg TMC207 group, which were
considered possibly related to the study medication. All AEs were grade 1 or 2 in severity, except
for 1 subject in the isoniazid group, with a grade 4 hemoptysis. This event was serious and
considered not related to the study medication, but related to TB and led to premature
discontinuation. One additional subject in the isoniazid group prematurely discontinued treatment
due to an AE (hemoptysis; grade 3 in severity) that had already started during the screening period.
Two patients in the TMC207 400 mg group died during the follow-up period. These were a 25-year
old female who died due to retroviral infection and pulmonary TB, and a 41-year old male who was
prematurely withdrawn after 3 days of treatment, started standard TB therapy and eventually died
due to massive hemoptysis.
Changes in QTcF were seen in both the TMC207 and control groups. Average increases of more
than 10 ms in QTcF were seen on Day 7 for the TMC207 400 mg group and for both control groups.
On Day 7, effects were larger 5 hours post-dose compared to pre-dose for all treatment groups.
Increases by 30-60 ms in QTcF were observed for 1 and 6 subjects of the TMC207 100 and 400 mg
groups, respectively, and for 4 and 2 subjects of the rifampin and isoniazid groups, respectively. In
addition, 1 patient in the rifampin group had an increase >60 ms in QTcF.
In Stage 1 of the ongoing Phase IIb trial in 47 patients with newly diagnosed sputum smear-positive
pulmonary MDR-TB infection, the most common AEs (reported in more than 10% of patients in the
TMC207 group) were nausea, arthralgia, hyperuricemia, unilateral deafness, hemoptysis, dizziness,
and diarrhea (16). Except for nausea (26.1% versus 4.2%), the incidences of AEs in the TMC207
group were similar or lower than the incidences of AEs in the placebo group. Mean QTcF and QTcB
increased in both treatment groups but the increases were more pronounced in the TMC207 group.
With respect to mean QTcF, the difference from placebo fluctuated between 1.0 and 10.8 ms and

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 35 of 142
did not change over time. There were 2 patients who reported an SAE, of which 1 patient was
randomized to the TMC207 group (a grade 4 diabetic ketoacidosis in a diabetic patient 42 days after
first intake of drug). The SAE was not considered related to study medication and the adverse
event resolved after treatment for ketoacidosis.
2.3.2. PA-824
Seven Phase I and one Phase II clinical trials with PA-824 have been completed. Both a Phase I
midazolam drug-drug interaction study and a Phase II EBA trial using lower doses than an earlier
study have completed the treatment phase and are in the follow-up phase. Table 1 provides a high-
level summary of the completed and ongoing studies. Full details of the clinical studies are
provided in the current Investigator’s Brochure (pages 68-103) (13).
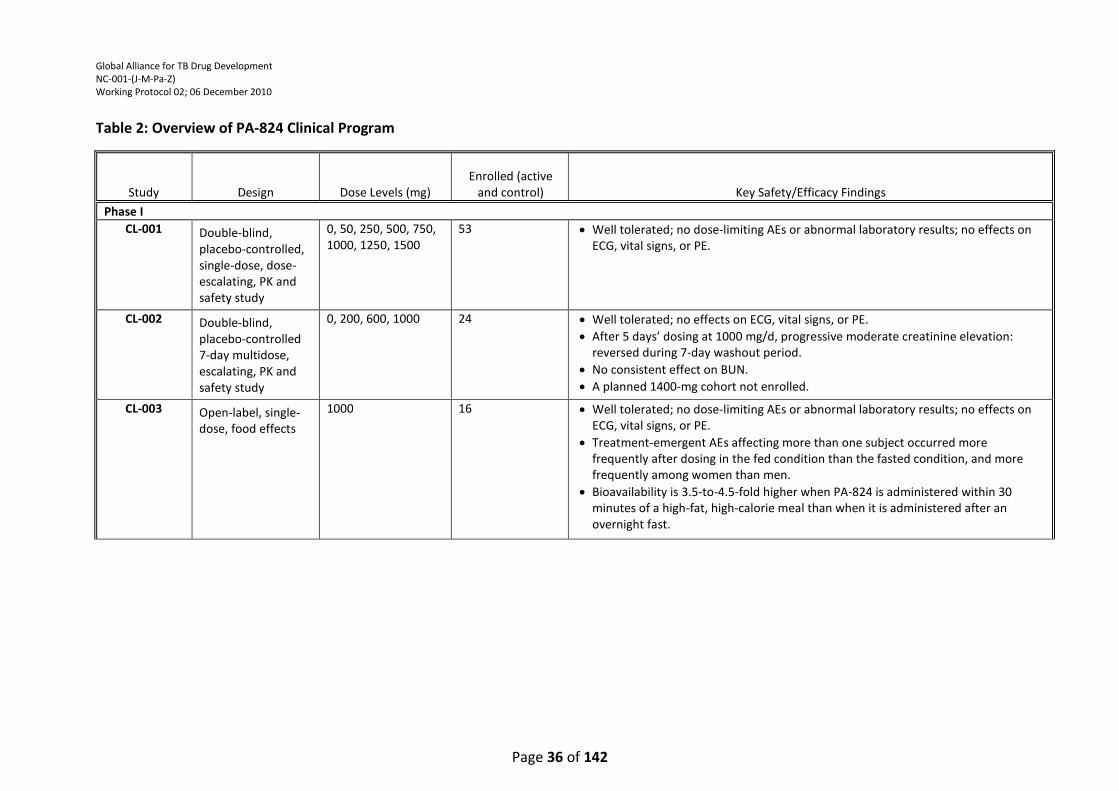
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 36 of 142
Table 2: Overview of PA-824 Clinical Program
Study Design Dose Levels (mg) Enrolled (active
and control) Key Safety/Efficacy Findings
Phase I
CL-001 Double-blind, placebo-controlled, single-dose, dose-escalating, PK and safety study
0, 50, 250, 500, 750, 1000, 1250, 1500
53
Well tolerated; no dose-limiting AEs or abnormal laboratory results; no effects on ECG, vital signs, or PE.
CL-002 Double-blind, placebo-controlled 7-day multidose, escalating, PK and safety study
0, 200, 600, 1000 24
Well tolerated; no effects on ECG, vital signs, or PE.
After 5 days’ dosing at 1000 mg/d, progressive moderate creatinine elevation: reversed during 7-day washout period.
No consistent effect on BUN.
A planned 1400-mg cohort not enrolled.
CL-003 Open-label, single-dose, food effects
1000 16
Well tolerated; no dose-limiting AEs or abnormal laboratory results; no effects on ECG, vital signs, or PE.
Treatment-emergent AEs affecting more than one subject occurred more frequently after dosing in the fed condition than the fasted condition, and more frequently among women than men.
Bioavailability is 3.5-to-4.5-fold higher when PA-824 is administered within 30 minutes of a high-fat, high-calorie meal than when it is administered after an overnight fast.
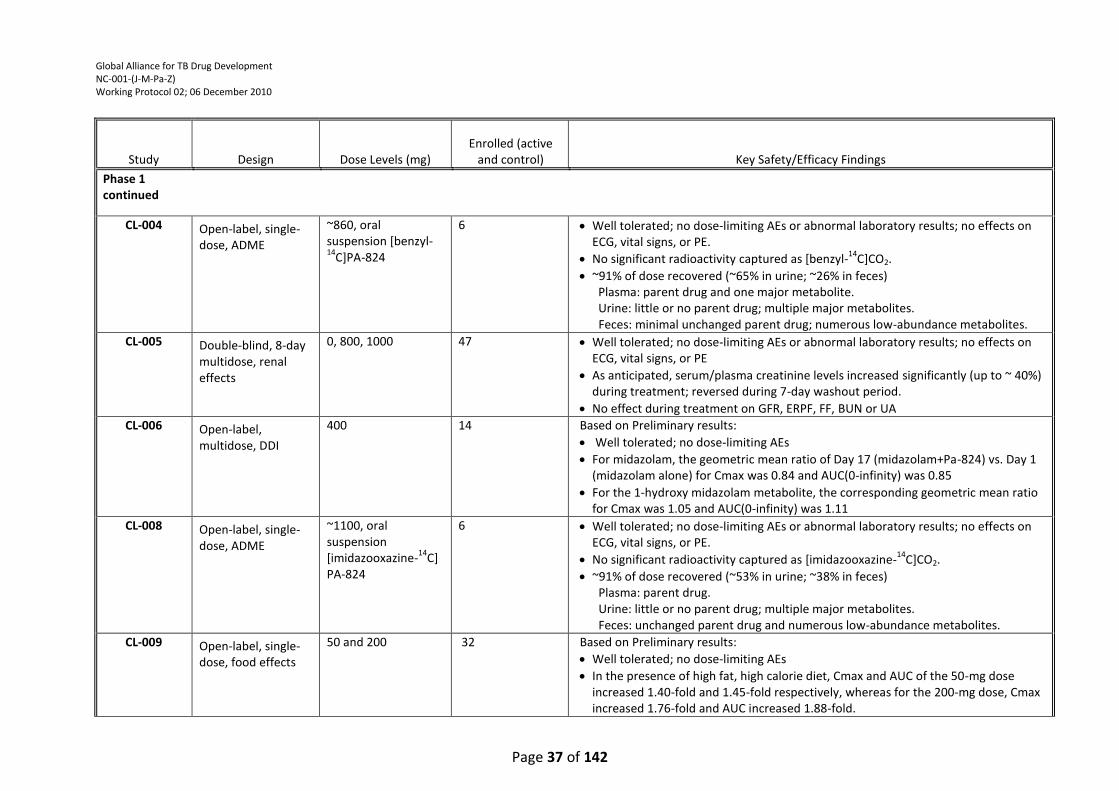
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 37 of 142
Study Design Dose Levels (mg) Enrolled (active
and control) Key Safety/Efficacy Findings
Phase 1 continued
CL-004 Open-label, single-dose, ADME
~860, oral suspension [benzyl-14
C]PA-824
6
Well tolerated; no dose-limiting AEs or abnormal laboratory results; no effects on ECG, vital signs, or PE.
No significant radioactivity captured as [benzyl-14
C]CO2.
~91% of dose recovered (~65% in urine; ~26% in feces) Plasma: parent drug and one major metabolite. Urine: little or no parent drug; multiple major metabolites. Feces: minimal unchanged parent drug; numerous low-abundance metabolites.
CL-005 Double-blind, 8-day multidose, renal effects
0, 800, 1000 47
Well tolerated; no dose-limiting AEs or abnormal laboratory results; no effects on ECG, vital signs, or PE
As anticipated, serum/plasma creatinine levels increased significantly (up to ~ 40%) during treatment; reversed during 7-day washout period.
No effect during treatment on GFR, ERPF, FF, BUN or UA
CL-006 Open-label, multidose, DDI
400 14
Based on Preliminary results:
Well tolerated; no dose-limiting AEs
For midazolam, the geometric mean ratio of Day 17 (midazolam+Pa-824) vs. Day 1 (midazolam alone) for Cmax was 0.84 and AUC(0-infinity) was 0.85
For the 1-hydroxy midazolam metabolite, the corresponding geometric mean ratio for Cmax was 1.05 and AUC(0-infinity) was 1.11
CL-008 Open-label, single-dose, ADME
~1100, oral suspension [imidazooxazine-
14C]
PA-824
6
Well tolerated; no dose-limiting AEs or abnormal laboratory results; no effects on ECG, vital signs, or PE.
No significant radioactivity captured as [imidazooxazine-14
C]CO2.
~91% of dose recovered (~53% in urine; ~38% in feces) Plasma: parent drug. Urine: little or no parent drug; multiple major metabolites. Feces: unchanged parent drug and numerous low-abundance metabolites.
CL-009 Open-label, single-dose, food effects
50 and 200 32
Based on Preliminary results:
Well tolerated; no dose-limiting AEs
In the presence of high fat, high calorie diet, Cmax and AUC of the 50-mg dose increased 1.40-fold and 1.45-fold respectively, whereas for the 200-mg dose, Cmax increased 1.76-fold and AUC increased 1.88-fold.
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 38 of 142
Study Design Dose Levels (mg) Enrolled (active
and control) Key Safety/Efficacy Findings
Phase II
CL-007 Partially double-blinded (blinded as to PA-824 dose), 14-day multidose, extended early bactericidal activity
200, 600, 1000, 1200 69 Overall well tolerated with relatively few AEs and no dose-limiting AEs or laboratory findings. No clinically significant effects on ECG, vitals, or PE noted.
Two SAEs occurred during study, both were considered possibly related to TB disease (hemoptysis).
PA-824 treatment produced a measurable decrease in log CFU, with the magnitude of effect equivalent for all doses.
CL-010 Partially double-blinded (blinded as to PA-824 dose), 14-day multidose, extended early bactericidal activity
50, 100, 150, 200 69 Based on Preliminary results:
Well tolerated.
PA-824 treatment produced a measurable decrease in log CFU with some evidence of dose dependence.
ADME = absorption, distribution, metabolism, excretion; AE = adverse events; BUN = blood urea nitrogen; ECG = electrocardiogram; ERPF = effective renal plasma flow; F = female; FF = filtration fraction; GFR = glomerular filtration rate; hr = hour; M = male; PD = pharmacodynamics; PE = physical exam; SAE = serious adverse event; UA = uric acid.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 39 of 142
Phase I
In these trials, PA-824 has been administered in doses ranging from 50 to 1500 mg as 50 or 200 mg
tablets or as an oral suspension. PK parameters have largely been consistent in each study and can
be summarized as follows:
PA-824 is moderately rapidly absorbed, with median Tmax values across subjects and dose
levels ranging from 4 to 7 hours.
The mean half-life for elimination (t½) across subjects and dose levels was approximately 16
- 25 hours.
Exposure increased approximately linearly but less than dose-proportionally, with increasing
doses up to approximately 600 – 1000 mg, while higher doses achieved minimal additional
increases in either Cmax or AUC.
Two studies using radiolabeled PA-824 in an oral-suspension formulation have been conducted to
investigate the metabolism and excretion patterns of PA-824 in humans: Study PA-824-CL-004,
which used [benzyl-14C] PA-824 and Study PA-824-CL-008, which used [imidazooxazine-14C]PA-824.
The mass balance results from the two studies were very similar. In each study, the majority (53-
65%) of radioactivity was excreted in the urine; an additional 26-38% was collected in the feces
such that approximately 91% of the administered dose was ultimately recovered in the excreta.
Radioprofiling and metabolite ID work have been completed on samples from the two human
studies as well as from analogous work in rat and monkey using both radiolabeled PA-824
preparations. The metabolism of PA-824 proceeds via a combination of reductive metabolism (~20
– 25% of the dose) and oxidative metabolism (remainder of the characterized metabolites). The
metabolic profile of PA-824 in vivo was qualitatively similar in the three species, with quantitative
differences being minor. No human unique metabolites were detected and there is no one single
metabolic path that can be considered major. Furthermore, there are no major metabolites in
human plasma.
Study PA-824-CL-006, a drug-drug interaction study with midazolam to assess the extent of CYP3A
inhibition by PA-824, also recently completed and a complete analysis is pending. Preliminary
results indicate that dosing of PA-824 400 mg once daily for 14 days did not have a major effect on
the exposure of midazolam or its major metabolite 1-hydroxy midazolam. For midazolam, the
geometric mean ratio of Day 17 (midazolam+PA-824) vs. Day 1 (midazolam alone) for Cmax was

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 40 of 142
0.84 and AUC was 0.85. For the 1-hydroxy midazolam metabolite, the corresponding geometric
mean ratio for Cmax was 1.05 and AUC was 1.11. The data suggests that PA-824 should not cause
clinically significant drugs interactions with drugs metabolized by CYP3A. No drug-drug interaction
is anticipated between PA-824 and TMC207 or pyrazinamide or moxifloxacin.
Study PA-824-CL-009, a food effects study using lower doses of PA-824 (200 mg and 50 mg),
recently completed and a complete analysis is pending. Preliminary results indicate that the food
effect observed is dependent on the PA-824 dose administered. When a single dose of PA-824 was
administered with a high fat, high calorie meal, Cmax and AUC of the 50 mg dose increased 1.40-
fold and 1.45-fold respectively, whereas for the 200 mg dose, Cmax increased 1.76-fold and AUC
increased 1.88-fold.
Phase II
Study PA-824-CL-007 examined the effects of 14 days’ treatment with PA-824 monotherapy at
doses ranging from 200 to 1200 mg/day on TB participants’ lung bacterial load measured as logCFU
(log of Colony Forming Units) in the sputum. Four groups of approximately 15 men and women
received 200, 600, 1000, or 1200 mg/day PA-824. An additional group of eight participants
received the standard South African 4-drug combination TB treatment (Rifafour® e-275, comprised
of a fixed-dose combination of isoniazid, rifampin, pyrazinamide and ethambutol). This group was
included to provide control for the microbiological techniques used to measure lung TB bacterial
burden. The efficacy data from this study indicated that all doses of PA-824 including the lowest
dose, 200 mg, produced a measureable and equivalent decrease in sputum CFU counts over the 14-
day treatment period; no difference could be discerned among the PA-824 treatment groups
(Figure 1).
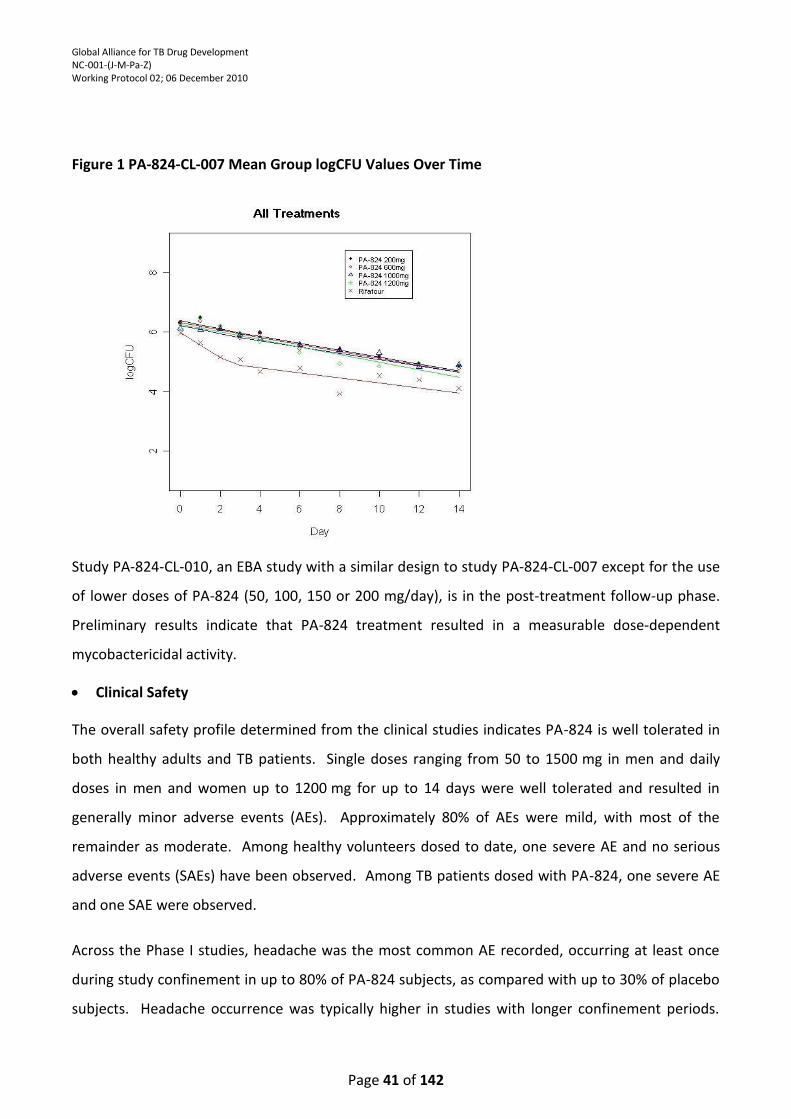
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 41 of 142
Figure 1 PA-824-CL-007 Mean Group logCFU Values Over Time
Study PA-824-CL-010, an EBA study with a similar design to study PA-824-CL-007 except for the use
of lower doses of PA-824 (50, 100, 150 or 200 mg/day), is in the post-treatment follow-up phase.
Preliminary results indicate that PA-824 treatment resulted in a measurable dose-dependent
mycobactericidal activity.
Clinical Safety
The overall safety profile determined from the clinical studies indicates PA-824 is well tolerated in
both healthy adults and TB patients. Single doses ranging from 50 to 1500 mg in men and daily
doses in men and women up to 1200 mg for up to 14 days were well tolerated and resulted in
generally minor adverse events (AEs). Approximately 80% of AEs were mild, with most of the
remainder as moderate. Among healthy volunteers dosed to date, one severe AE and no serious
adverse events (SAEs) have been observed. Among TB patients dosed with PA-824, one severe AE
and one SAE were observed.
Across the Phase I studies, headache was the most common AE recorded, occurring at least once
during study confinement in up to 80% of PA-824 subjects, as compared with up to 30% of placebo
subjects. Headache occurrence was typically higher in studies with longer confinement periods.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 42 of 142
Other common AEs associated with PA-824 treatment include elevated serum creatinine and
stomach discomfort (including nausea and other gastrointestinal symptoms such as flatulence
and/or diarrhea). Stomach discomfort AEs were the most commonly reported AEs in the PA-824-
CL-007 Phase IIa study of TB patients. For the multidose, placebo-controlled Studies PA-824-CL-
002, PA-824-CL-005 and PA-824-CL-007, overall AE frequency tended to be greater among PA-824
subjects than among placebo subjects, and tended to be higher in higher PA-824 dose groups.
Study PA-824-CL-005 was undertaken to determine the mechanism responsible for the elevation in
serum creatinine seen with PA-824 dosing in studies PA-824-CL-001 and PA-824-CL-002. This study
explored the effects of PA-824 on kidney function by measuring glomerular filtration rate (GFR),
effective renal plasma flow (ERPF), filtration fraction (FF, calculated as GFR/ERPF), and creatinine
clearance. Subjects were dosed in blinded fashion with placebo, 800 mg PA-824, or 1000 mg
PA-824 for 8 days. Serum creatinine levels rose in both the 800- and 1000-mg/day PA-824 groups,
by an average of 0.18 mg/dL (19%) and 0.25 mg/dL (27%) in the two groups respectively by Day 8;
the largest individual increase was approximately 40% over baseline. In this study, although serum
creatinine levels rose, no meaningful effects were noted during the dosing period on GFR, ERPF,
BUN, uric acid or FF. As expected, creatinine clearance was reduced concomitantly with maximally
elevated serum creatinine levels relative to baseline. Taken together, these results indicate that
PA-824 does not negatively affect renal function. Instead, the drug can be assumed to cause its
effects on serum creatinine by inhibiting tubular creatinine secretion; such an effect has been
reported with other approved drugs (e.g. cimetidine) and is not considered clinically significant.
Across all studies, the great majority (>~95%) of AEs resolved without sequelae. In Study PA-824-
CL-003, one subject concluded the study with blurry/double vision that may have been related to
childhood strabismus, while another subject had ongoing swollen fingers on both hands at the end
of the study. In Study PA-824-CL-007, two patients exited the study with an ongoing AE (anemia
[mild, unrelated] and rash [mild, related]) and the outcomes for three AEs in one patient each
(anemia [mild, possibly related], elevated LFT [moderate, unrelated], and Wolff-Parkinson-White
syndrome [mild, unrelated] were not known due to loss to follow-up. In Study PA-824-CL-005, one
subject treated with 800 mg PA-824 was discharged with three ongoing AEs (proteinuria [nephrotic
range during the study, but non-nephrotic range in follow-up], hypoalbuminemia, and iron
deficiency). The proteinuria and hypoalbuminemia were moderate in severity and the iron
deficiency was mild. This subject has substantially improved, and the subject is being seen

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 43 of 142
periodically by a nephrologist and by trial site staff. A renal biopsy performed 20 months post-
study revealed focal segmental glomerulosclerosis likely secondary type, although the subject
remains fundamentally healthy with normal renal function indices and no signs of peripheral edema
or hypertension. A complete review of her screening and check-in lab values suggests in the
opinion of the Sponsor that she might have had a preexisting undiagnosed clinical condition
including atypical lipid profile, BUN below the lower limit of the normal range, and ALT and AST
above the upper limit of normal range. Furthermore, her eosinophil count was above-normal at
Screening at 6.7% and progressively rose during the study to 8.9% by Day 15 and she reported a
personal and family history of allergies and rhinorrea. The PI considered this individual normal and
meeting the protocol entry criteria, and enrolled this subject.
Among the healthy volunteers tested, any concomitant therapy for AEs was generally palliative in
nature. Occasionally, pains such as headaches, sore throat, and muscle ache were treated with
analgesics. One instance of iron deficiency (deemed not related to study drug) was treated with an
iron supplement. In Study PA-824-CL-007, multiple AEs, most of which were associated with
ongoing TB disease, were treated with medications or other interventions including hospitalization
(e.g., to treat hemoptysis).
In most of the completed Phase I studies, no subjects discontinued from the study as a result of
AEs. In Study PA-824-CL-002, dosing for all subjects in the 1000 mg dose group was discontinued
on Day 5 in response to rising serum creatinine levels. In Study PA-824-CL-005, one subject was
discontinued for safety reasons in relation to a severe rash that developed approximately 32 hours
after the 8th and last dose of PA-824 (1000 mg). The rash symptoms were treated with
diphenhydramine, prednisone, and hydroxyzine at various points during the ensuing approximately
9 days until the symptoms completely resolved. In Study PA-824-CL-007, two patients (one in the
200 mg/day PA-824 group and one in the control arm) were discontinued as a result of disease-
related hemoptysis. Each of these events was classified as an SAE, both resolved with treatment in
hospital and neither was considered possibly related to the study drugs.
Post-study follow-up ophthalmic examinations have been performed on subjects and patients
enrolled in two studies where participants received the highest doses of PA-824 for the longest
duration among the clinical studies conducted to date. Male and female healthy volunteers were

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 44 of 142
treated at doses up to 1000 mg/day for 8 days in Study PA-824-CL-005, and male and female TB
patients were treated at doses up to 1200 mg/day for 14 days in Study PA-824-CL-007.
For Study PA-824-CL-005, 30 of the randomized 47 subjects have been examined with no cataracts
being detected in any subject. For Study PA-824-CL-007, 46 of the 69 randomized patients received
post-study ophthalmic examinations and two were found with single, asymptomatic, unilateral
cataracts. One of these was among the 12 patients from the 1200 mg group who returned for the
eye examination; the other was from among the 5 patients from the control TB regimen (Rifafour®)
group who returned for the eye examination. For the PA-824 patient, the cataract was judged by
an external, independent ophthalmology review board (ORB) to be most consistent with an age-
related cataract based on the patient’s age (52 years at time of ophthalmologic examination) and
the unilateral nature of the cataract. Furthermore, the ORB has determined that there is no
evidence of an association between PA-824 exposure and cataracts in humans when PA-824 is
administered at the doses used and in the populations studied in the clinical studies completed to
date. The ORB recommended that slit-lamp eye exams (including at baseline) be incorporated into
future studies of PA-824. An additional expert clinical ophthalmologist also recommended for
future studies that ocular examinations be performed at baseline, and 3 to 6 months after dosing
for studies less than 12 weeks in duration. For studies of 12 or more weeks in duration, this
evaluation should be conducted at baseline, end of dosing and 3 to 6 months after dosing. The TB
Alliance intends to follow these guidelines in this study.
2.3.3. Moxifloxacin
Moxifloxacin has been approved in South Africa and most other countries around the world for the
treatment of acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis,
community-acquired pneumonia, skin and soft tissue infections. An extensive clinical program
supports the safety and efficacy of 400 mg moxifloxacin given once daily for 5 to 21 days in adults
for the above specified indications. For more detailed information, please refer to the IB for
Moxifloxacin (34).
Moxifloxacin is commonly used as second line therapy for TB in patients (e.g., MDR- TB), and has
also been evaluated in several clinical studies of patients with TB, including four 8-week treatment
period phase IIb studies (see Table 3 below).

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 45 of 142
Table 3 Summary of Phase IIb studies in which moxifloxacin was administered as part of a four-drug regimen during the intensive phase of treatment (total treatment duration: 2 months)
Study Sponsor Design Objectivea Patients
N Countries Status
Study 27 Tuberculosis Trials
Consortium (TBTC)
Randomized, double-blind,
controlled
To compare the sputum culture-conversion rate at the end of the 4-drug (intensive) phase of therapy using the standard 4-drug regimen
HRZE with a standard regimen with M replacing E.
336 USA, Canada, Uganda,
South Africa
Completed
OFLOTUB Study
South African Medical
Research Council
Randomized, Open label,
Controlled
To compare the bactericidal activities of regimens where gatifloxacin, M, and ofloxacin
were substituted for E in the 2-month initial phase of standard
TB treatment with HRZE.
217 South Africa
Completed
Johns Hopkins (FDA orphan drug) Study
Johns Hopkins
University & FDA orphan
drugs program
Randomized, double-blind,
Controlled
To compare the sputum culture conversion rate at the end of the 4-drug (intensive)
phase of therapy using a standard 4-drug regimen
(HRZE) with a regimen with M replacing E (HRZM).
170 Brazil Completed
Study 28 Tuberculosis Trials
Consortium (TBTC) /CDC
Randomized, double-blind,
Controlled
To compare the culture-conversion rate at the end of
the 4-drug (intensive) phase of therapy using a standard 4-drug regimen (HRZE 5 days
per week) with a regimen with M replacing H (MRZE 5 days
per week).
433 USA, Canada, Brazil, Spain,
Uganda, South Africa
Completed
aH = isoniazid; R = rifampin; E = ethambutol; Z = pyrazinamide; M = moxifloxacin
In Study 27 (23), the moxifloxacin-containing TB regimens (HRZM) were shown to be safe and well
tolerated. There was no difference in SAEs between the HRZM and HRZE treatment arms, and most
SAEs were hospitalizations thought to be unrelated to the study treatment. Patients treated with
moxifloxacin-containing regimen were more likely to report nausea (22% vs. 9%, p = 0.002), but this
was generally mild and did not lead to treatment discontinuation as similar proportions of patients
in both groups completed study drug treatment (88% HRZM-treated vs. 89% HRZE-treated). The
one death during the first 2 months of treatment was thought to be caused by pulmonary
embolism, unrelated to tuberculosis therapy.
In the OFLOTUB study (24), AEs and SAEs occurred with equal frequency in the four treatment arms.
The most frequent AEs were raised amylase (in 41% of patients due to HIV infection), raised
transaminase (10%), arthralgia (9%), anemia (7%), hypokalaemia (6%) and vomiting (5%). No
patient was withdrawn from the study due to an adverse event. There were 4 deaths in the study:
2 in the control group, 1 each in the moxifloxacin and ofloxacin arms. Importantly, patients in this

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 46 of 142
study who received moxifloxacin and pyrazinamide in addition to isoniazid and rifampin had more
rapid sputum clearance than patients on the four standard first line drugs (isoniazid, rifampin,
pyrazinamide and ethambutol). This difference was evident 14 days after initiation of treatment.
In the JHU Study (25), AEs did not differ by treatment group. There were 16 SAEs (8 in each group) in
12 patients. Only 1 event was judged related to study drug (grade 3 cutaneous reaction in the
ethambutol group). Eight patients died during the study, including 1 in each group still receiving
study phase treatment. No death was attributed to study treatment. Only 5 patients discontinued
treatment because of toxic effects; 2 patients in the moxifloxacin group stopped because of grade 2
nausea and vomiting and 1 because of grade 2 paraesthesias and ataxia. Two patients in the E
group stopped because of grade 2 rash and pruritis and 1 because of grade 3 peripheral
neuropathy. There was no change in the QTc interval on serial electrocardiograms taken during the
trial.
In Study 28 (26), the proportions of patients with SAEs during intensive phase treatment were similar
between arms (isoniazid 3.9% vs. moxifloxacin 4.2%; P = 0.88). Three SAEs attributed to study
treatment during the first 2 months occurred among the moxifloxacin group and two in the
isoniazid group. Seven patients died during the study including 3 patients receiving the
moxifloxacin treatment regimen and 4 patients receiving the isoniazid treatment regimen. All 3
moxifloxacin patients died during intensive phase TB treatment. Two patients died from advanced
pulmonary TB judged not related to study treatment and 1 subject (who developed diabetic
ketoacidosis considered possibly related to study treatment) died from possible acute pulmonary
embolus unrelated to study treatment. The 4 isoniazid deaths occurred during continuation phase,
and all 4 were considered unrelated to study treatment. Nausea was more common among
patients in the moxifloxacin arm than in the isoniazid arm (19.6% vs. 11.7%, respectively; P = 0.03)
although similar proportions reported vomiting. However, the proportions of patients with
hepatitis, defined as serum AST 3 times or greater than the upper limit of normal, were similar
between treatment arms during intensive phase (isoniazid 3.4% vs. moxifloxacin 3.3%; P = 0.93).
For additional information on moxifloxacin, refer to the IB(34) and the manufacturer’s package insert
(Appendix 7).

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 47 of 142
2.4. Known and Potential Risks and Benefits of the Investigational Medicinal Product (IMP) used
in the Study
2.4.1. TMC207
In the clinical studies conducted to date, a total of 173 healthy volunteers and 68 TB patients have
taken TMC207. Multiple dosing has extended for as long as 8 weeks. The most common side
effects or AEs observed across these studies are described above (Section 2.3.1), and most of them
were considered to be not or doubtfully related to TMC207.
2.4.2. PA-824
In the nine clinical studies completed to date, a total of 149 healthy volunteers and 61 TB
participants have taken PA-824. Multiple dosing has extended for as long as 14 days. As described
above (section 2.3.2), the most common side effects or AEs associated with PA-824 exposure
include:
Headache
Benign, isolated and reversible elevations of serum creatinine
Stomach discomfort (nausea, vomiting, flatulence, and/or diarrhea)
Post-study follow-up ophthalmic examinations have been performed on subjects and patients
treated at doses up to 1000 mg/day for 8 days in Study PA-824-CL-005, and male and female TB
participants treated at doses up to 1200 mg/day for 14 days in Study PA-824-CL-007. An
independent Ophthalmology Review Board concluded there was no evidence from these two
studies that PA-824 caused cataracts in humans. In addition, because exposures at the dose
planned for this study (200 mg/day) are anticipated to be lower than those associated with cataract
formation in rats (and substantially lower than exposures associated with cataracts in one monkey
study - a finding that could not be repeated in a more carefully conducted repeat monkey
toxicology study), the TB Alliance believes that the dose selected for the present study (200 mg/day
PA-824) does not represent a significant risk of cataract formation for participants enrolled into the
present study. For additional information, please refer to pages 40-47 and 133-134 in the PA-824 IB
(13).
2.4.3. Moxifloxacin
In the usual daily doses of 400 mg/day, moxifloxacin is well tolerated. As stated in the IB (34) (Table
5-41, pp 136-142), the most frequent individual drug related AEs observed in clinical studies with

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 48 of 142
400 mg oral moxifloxacin therapy were nausea and vomiting (8.2%) and diarrhea (4.9%). In
addition, moxifloxacin has been shown to prolong the QT interval of the electrocardiogram in some
patients. It should be used with caution in patients being treated concomitantly with other drugs
where an additive effect on the QT interval cannot be excluded.
2.4.4. Pyrazinamide
The doses of pyrazinamide that will be used in this study are those approved for the treatment of
TB *manufacturer’s package insert (Appendix 6)+. Pyrazinamide is mostly metabolized by the liver
and 3 to 4% of the administered dose is excreted unchanged by the kidneys.
The most serious side-effect is hepatotoxicity and its frequency appears to be dose-related.
Hyperuricemia commonly occurs, occasionally accompanied by arthralgia and may lead to attacks
of gout. Photosensitivity and skin rash have been reported less frequently. Other side-effects that
have been reported are anorexia, nausea and vomiting, malaise, fever, sideroblastic anemia and
dysuria. Pyrazinamide may decrease the efficacy of gout therapy (e.g. allopurinol, colchicine,
probenecid or sulphinpyrazone) and dosage adjustments of these medications may be necessary.
For additional information on pyrazinamide, refer to the manufacturer’s package insert (Appendix
6).
2.4.5. Standard, first-line TB treatment
Patients in the control group (control for Mycobacteriology laboratory methodology) will receive
standard, intensive phase pulmonary TB treatment as recommended in the SA National TB
Treatment Guidelines, which is RHZE 150/75/400/275 mg oral daily (Rifafour e-275) (R=rifampicin:
H=isoniazid: Z=pyrazinamide: E=ethambutol).
Please see the Rifafour e-275 Package Insert for its known and potential risks and benefits
(Appendix 2).
3. TRIAL RATIONALE AND OBJECTIVES
3.1. Trial Rationale
TMC207 and PA-824, new chemical entities with significant promise for shortening treatment of
active TB, have been under development for several years, and entered clinical trials in 2005. This
study will be the third study of each of these compounds in drug sensitive TB patients. As described
above, several studies of TMC207 and PA-824 have also been conducted previously in healthy
volunteers. Collectively, these studies in patients and healthy subjects have defined the efficacy,

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 49 of 142
safety and pharmacokinetic features of the drugs. Overall, TMC207 and PA-824 have
mycobactericidal activity in participants and are well tolerated and bioavailable after oral dosing.
Moxifloxacin is an approved drug for several indications and while not approved for TB, it is
regularly used in second line TB treatment. In addition, it is currently in late stage clinical
development for first-line TB treatment in combination with other TB drugs, including
pyrazinamide.
As previously stated, data from preclinical studies suggest that a regimen consisting of TMC207 and
pyrazinamide, TMC207 and PA-824, PA-824 and pyrazinamide or PA-824, pyrazinamide and
moxifloxacin may not only be appropriate for treating both drug-sensitive and MDR-TB but may
also shorten duration of therapy. The main purpose of this study is to determine whether these
combinations are as bactericidal in man as has been demonstrated in the mouse model of TB
(12,28,29). The dose of TMC207 for the current EBA study was selected to provide an average target
plasma concentration of ~2000 ng/mL to maximize TMC207’s mycobactericidal activity; the dose of
PA-824 selected for this study is based on data from two dose-ranging EBA studies; the currently
approved dose of pyrazinamide will be used, as will the approved dose of moxifloxacin (for other
indications; 400 mg per day), which is also the dose being evaluated in the ongoing Phase III clinical
TB trial.
EBA studies measure the time- and dose-dependent decline of viable bacteria in the patient's
expectorated sputum. Measuring the extended EBA of a new agent or regimen in TB patients is an
accepted method of determining mycobactericidal activity. In addition, these studies allow the
opportunity to characterize safety and tolerability profile in patients prior to evaluation in longer-
term studies.
Participants will not be exposed to undue risk in the proposed study. TMC207 has been used in TB
patients for up to 14 days at the 400-mg dose. (33) The proposed dosing regimen for this study is
being used in an ongoing 14-day EBA study in South Africa and preliminary results indicate that it is
well tolerated and produced significant mycobactericidal activity with no TMC-related SAEs. PA-
824 has been used in TB patients for up to 14 days at doses (up to 1200 mg) much higher than what
is being proposed (200 mg); 400 mg moxifloxacin per day has been used in TB patients for up to
four months, while pyrazinamide is approved for TB treatment and is used for 8 or more weeks
depending on whether drug-sensitive or drug-resistant TB patients are being treated. Moxifloxacin
and pyrazinamide are being co-administered to TB patients for 4 months in the phase III TB
moxifloxacin trial and are often co-administered to MDR-TB patients in clinical practice for even

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 50 of 142
longer periods. This will be the first clinical study where PA-824 will be used in combination with
moxifloxacin or TMC207. Although preliminary results from the preclinical safety pharmacology
study conducted with PA-824 plus moxifloxacin in monkeys and PA-824 plus TMC207 in dogs do not
suggest the potential for a marked QT prolongation in humans (see pages 24-26), there will be daily
ECG safety monitoring of all study participants timed to approximate the Cmax for moxifloxacin,
TMC207 and PA-824 (Flow chart, page 18). In addition, study participants on PA-824 containing
treatment arms (PA-824 + Z and PA-824 + Z + M) will have intensive ECG monitoring on Day 1 (first
day of dosing) and Day 8 (midway through the study ensuring attainment of steady state for PA-
824) as well as time matched baseline ECGs on Day -1. Finally, specific discontinuation criteria
based on QT prolongation are listed in section 9.7 (page 96).
All participants will remain under constant medical attention and will be housed and monitored in
hospital from check-in through the duration of the treatment period; this will allow a continuous
monitoring of the health conditions of each participant, any of whom can be withdrawn at any
stage of the trial and removed from study treatment should his/her condition suggest to the
Investigator that this would be in his/her best interest. Upon discharge, the participants will be
given initial doses of standard TB treatment and immediately referred to the national TB treatment
program local TB clinic. The Investigators’ primary responsibility is to ensure participant safety.
Information gained in this study will provide critical insight into the efficacy, safety and
pharmacokinetic profile of the indicated drug combinations, as well as their suitability for further
evaluation in longer-term clinical trials.
3.2. Trial Objectives
To evaluate the early bactericidal activity (EBA), safety, tolerability and pharmacokinetics of
TMC207 alone, TMC207 plus pyrazinamide, TMC207 plus PA-824, PA-824 plus pyrazinamide and
PA-824 plus pyrazinamide and moxifloxacin, administered orally as once daily doses for 14
consecutive days, in adult patients with newly diagnosed, smear positive pulmonary tuberculosis, in
order to help select appropriate combination therapies for later stage clinical development.
3.3. Trial Endpoints
3.3.1. Primary Endpoint

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 51 of 142
The EBA (EBA0-14) as determined by the rate of change in logCFU per ml sputum over the period
Day 0 to Day 14 which may be described with linear, bi-linear or non-linear regression of logCFU on
time.
3.3.2. Secondary Endpoints
3.3.2.1. Efficacy
S econdary Efficacy Endpoints:
The EBA (EBA0-2, EBA2-14 and EBA7-14) as determined by the rate of change of logCFU in
sputum over the period Day 0 to Day 2, Day 2 to Day 14 and Day 7 to Day 14, in participants,
which may be described with linear, bi-linear or non-linear regression of logCFU on time.
The time to sputum culture positivity (TTP) (TTP0-2, TTP0-14, TTP2-14, and TTP7-14) in the
Mycobacterial Growth Indicator Tube (Bactec MGIT960) system as determined by the rate of
change in TTP in sputum over the periods Day 0 to Day 2, Day 0 to Day 14, Day 2 to Day 14 and
Day 7 to Day 14 in participants, which may be described with linear, bi-linear or non-linear
regression of TTP on time.
3.3.2.2. Safety and Tolerability
The proportion of participants with adverse events and proportion of participants who discontinue
due to an adverse event in each experimental arm. These data will be presented as descriptive
analyses, and no inferential tests will be carried out.
3.3.2.3. Pharmacokinetics
For participants in the TMC207 (excluding TMC207 plus PA-824 containing treatment
arm)containing treatment arms:
The following PK parameters will be estimated for TMC207, TMC207 metabolite M2 and
pyrazinamide on Day 14 only: the maximum observed plasma concentration (Cmax), the minimum
observed plasma concentration 24 hours following the last dose (Cmin), time to reach Cmax (Tmax),
area under the plasma concentration time (t) curve from zero to 24 hours (AUC(0-24). These data
will be presented as descriptive analyses, and no inferential tests will be carried out.
For participants in the PA-824 (excluding TMC207 plus PA-824 containing treatment arm)
containing treatment arms:

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 52 of 142
The following PK parameters will be estimated for PA-824, pyrazinamide and moxifloxacin on Days
1, 8 and 14: the maximum observed plasma concentration (Cmax), the minimum observed plasma
concentration 24 hours following the last dose (Cmin), time to reach Cmax (Tmax), area under the
plasma concentration time (t) curve from zero to 24 hours (AUC(0-24)). These data will be
presented as descriptive analyses, and no inferential tests will be carried out.
For participants in the TMC207 plus PA-824 containing treatment arms:
The following PK parameters will be estimated for PA-824, TMC207 and TMC207 metabolite M2 on
Day 14 only: the maximum observed plasma concentration (Cmax), time to reach Cmax (Tmax), the
minimum observed plasma concentration (Cmin) 24 hours following the last dose, area under the
plasma concentration time (t) curve from zero to 24 hours (AUC(0-24)). These data will be
presented as descriptive analyses, and no inferential tests will be carried out.
No pharmacokinetic sampling is planned on participants receiving Rifafour e275®.
3.3.2.4. Pharmacokinetics-Pharmacodynamics (PK-PD)
For TMC207 and PA-824 containing treatment arms, the following will be estimated for TMC207,
PA-824, pyrazinamide and moxifloxacin, as appropriate per the above PK analyses performed:
The EBAs vs. the following PK variables (Day 14 only) will be presented:
Cmax;
AUC(0-24);
Time over Minimum inhibitory concentrations (MIC) (for TMC207, PA-824 and moxifloxacin).
These data will be presented as descriptive analyses, and no inferential tests will be carried out.
3.3.2.5. Extent of QT Prolongation
For TMC207 and PA-824 containing treatment arms, the potential correlations between the plasma
concentration of IMP and the change from baseline of QT interval corrected by Fridericia’s method
(QTcF) and change from baseline of QT interval corrected by Bazett’s method (QTcB) with respect
to time for the different treatment groups will be explored. These data will be presented as
descriptive analyses, and no inferential tests will be carried out.
3.4. Mycobacteriological Characterization

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 53 of 142
In order to characterize the participants’ infecting mycobacterium, with respect to species and TB
drug-sensitivity, and to determine the in vitro potency of each infecting strain with respect to each
treatment and Rifafour e-275® once daily, for 14 days, the overnight sputum collections from
Day -2 and Day 14 will be assessed as follows:
MIC of TMC207, PA-824 and moxifloxacin;
Drug susceptibility testing of the M. tuberculosis isolates with the MGIT system for sensitivity to
isoniazid, rifampicin, ethambutol and pyrazinamide;
Speciation of the infecting organism by polymerase chain reaction (PCR).
3.5. Participant Inclusion/Exclusion Criteria
3.5.1. Inclusion Criteria
Participants are required to meet all of the following inclusion criteria in order to be randomized.
1. Provide written, informed consent prior to all trial-related procedures including HIV testing.
2. Male or female, aged between 18 and 65 years inclusive.
3. Body weight (in light clothing and with no shoes) between 40 and 90 kg, inclusive.
4. Newly diagnosed, previously untreated, sputum smear-positive pulmonary TB.
5. A chest X-ray picture which in the opinion of the Investigator is compatible with TB.
6. Sputum positive on direct microscopy for acid-fast bacilli (at least 1+ on the IUATLD/WHO
scale (Appendix 3)).
7. Ability to produce an adequate volume of sputum as estimated from a spot assessment
(estimated 10 ml or more overnight production).
8. Females may participate if they are: 1) of non-childbearing potential (have had a bilateral
oophorectomy and/or hysterectomy or have been postmenopausal for at least 12
consecutive months), 2) if they are using effective birth control methods and are willing to
continue practicing birth control methods throughout treatment or 3) be non-
heterosexually active, practice sexual abstinence or have a vasectomized partner (confirmed
sterile). Therefore to be eligible for this study women of childbearing potential should
either: 1) use a double barrier method to prevent pregnancy (i.e. use a condom with either
diaphragm or cervical cap) or 2) use hormonal based contraceptives in combination with a
barrier contraceptive, or 3) use an intrauterine device in combination with a barrier
contraceptive. They must also be willing to continue these contraceptive measures until 6

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 54 of 142
months after the last dose of study medication or 6 months after discontinuation from study
medication in case of premature discontinuation. (Note: Hormone-based contraception
alone may not be reliable when taking IMP; therefore, hormone-based contraceptives alone
cannot be used by female participants to prevent pregnancy).
9. Male participants who are having heterosexual intercourse with females of child-bearing
potential are required to use one of the following birth control methods during their
participation in the trial and for 12 weeks after their last dose of study medication to
prevent pregnancy:
- a double barrier method which can include a male condom, diaphragm, cervical cap,
or female condom; or
-a barrier method combined with hormone-based contraceptives or an intra-uterine
device for the female partner.
The use of the above mentioned birth control method does not apply if the male participant
has been vasectomised or has had a bilateral orchidectomy minimally one month prior to
screening, or is not heterosexually active, or practice sexual abstinence or if the female
sexual partner has had a bilateral oophorectomy and/or hysterectomy or has been
postmenopausal for at least 12 consecutive months.
3.5.2. Exclusion Criteria
Participants will be excluded from participation if they meet any of the following criteria.
Medical History
1. Evidence of clinically significant (as judged by the investigator), metabolic, gastrointestinal,
cardiovascular, musculoskeletal, ophthalmological, pulmonary, neurological, psychiatric or
endocrine diseases, malignancy, or other abnormalities (other than the indication being
studied).
2. Poor general condition where any delay in treatment cannot be tolerated per discretion of
the Investigator.
3. A history of previous TB.
4. Clinically significant evidence of extrathoracic TB (miliary TB, abdominal TB, urogenital TB,
osteoarthritic TB, TB meningitis), as judged by the Investigator.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 55 of 142
5. History of allergy to the IMP or related substances, including a known allergy to any
fluoroquinolone antibiotic, history of tendinopathy associated with quinolones or suspected
hypersensitivity to any rifamycin antibiotics.
6. Isoniazid-resistant and/or Rifampicin-resistant bacteria detected with a sputum specimen
collected within the pre-treatment period and tested at the study laboratory.
7. Known or suspected, current or history of within the past 2 years, alcohol or drug abuse,
that is, in the opinion of the Investigator, sufficient to compromise the safety or cooperation
of the participant.
8. HIV infected participants:
a. having a CD4+ count <300 cells/µL;
b. or having received antiretroviral therapy medication within the last 90 days;
c. or having received oral or intravenous antifungal medication within the last 90 days;
d. or with an AIDS-defining opportunistic infection or malignancies (except pulmonary TB).
9. Having participated in other clinical studies with investigational agents within 8 weeks prior
to trial start.
10. Significant cardiac arrhythmia requiring medication.
11. Participants with the following at screening. Where applicable to ECGs, the central
cardiologists reading must be used:
a. Marked prolongation of QT/QTc interval, e.g., confirmed demonstration of QTcF (Fridericia
correction) or QTcB (Bazett correction) interval >450 ms at screening;
b. History of additional risk factors for Torsade de Pointes, e.g., heart failure, hypokalemia,
family history of Long QT Syndrome;
c. Use of concomitant medications that prolong the QT/QTc interval (see exclusion criterion
22 as well as list of disallowed medication in Section 4.7.2);
d. Pathological Q waves (defined as >40ms or depth >0.4-0.5mV);
e. ECG evidence of ventricular pre-excitation;
f. ECG evidence of complete or incomplete left bundle branch block or right bundle branch
block;
g. ECG evidence of second or third degree heart block;
h. Intraventricular conduction delay with QRS duration >120ms;
i. Bradycardia as defined by sinus rate <50bpm.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 56 of 142
12. Females who are pregnant, breast-feeding, or planning to conceive a child within 6 months
of cessation of treatment.
13. Males planning to conceive a child within twelve weeks of cessation of treatment.
14. History and/or presence (or evidence) of neuropathy or epilepsy.
15. Diabetes Mellitus requiring insulin.
16. History of lens opacity or evidence of lens opacity on slit lamp ophthalmologic examination.
17. For males, any evidence or history of a clinically significant abnormality in the reproductive
system, including but not limited to the following: serum testosterone, luteinizing hormone
(LH), and/or follicle-stimulating hormone (FSH) levels outside the laboratory reference
range. An evaluation resulting in an isolated abnormal value (i.e., only 1 of the 3 hormones
is abnormal) may be repeated using a morning (ideally, 8am) serum specimen. If the
laboratory value on the repeat specimen is outside the laboratory reference range, unless
the result is deemed not clinically significant by the Investigator in consultation with the
Sponsor Medical Monitor, the participant should be excluded.
Specific Treatments
18. Previously received treatment with TMC207 or PA-824 as part of a clinical trial.
19. Treatment received with any drug active against MTB within the 3 months prior to Visit 1
(e.g. isoniazid, ethambutol, amikacin, cycloserine, rifabutin, rifampicin, streptomycin,
kanamycin, para-aminosalicylic acid, rifapentine, pyrazinamide, thioacetazone,
capreomycin, fluoroquinolones, thioamides, metronidazole).
20. Any diseases or conditions in which the use of the standard TB drugs or any of their
components is contra-indicated, including but not limited to allergy to any TB drug, their
component or to the IMP.
21. Any disease or conditions in which any of the medicinal products listed in the section
pertaining to prohibited medications is used.
22. Concomitant use of any drug known to prolong QTc interval (including amiodarone, bepridil,
chloroquine, chlorpromazine, cisapride, clarithromycin, disopyramide dofetilide,
domperidone, droperidol, erythromycin, halofantrine, haloperidol, ibutilide, levomethadyl,
mesoridazine, methadone, pentamidine, pimozide, procainamide, quinidine, sotalol,
sparfloxacin, thioridazine). The exception is moxifloxacin which is one of the drugs being
evaluated in this study, with extensive ECG monitoring to help ensure patient safety.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 57 of 142
23. Use of any drugs or substances within 30 days prior to dosing known to be strong inhibitors
or inducers of cytochrome P450 enzymes (such as quinidine, tyramine, ketoconazole,
testosterone, quinine, gestodene, metyrapone, phenelzine, doxorubicin, troleandomycin,
cyclobenzaprine, erythromycin, cocaine, furafylline, cimetidine, dextromethorphan).
Exceptions may be made for participants that have received 3 days or less of one of these
drugs or substances, if there has been a wash-out period before administration of IMP
equivalent to at least 5 half-lives of that drug or substance.
24. Use of any therapeutic agents known to alter any major organ function (e.g., barbiturates,
opiates, phenothiazines, cimetidine) within 30 days prior to dosing.
25. Use of systemic glucocorticoids within one year prior to dosing.
Based on Laboratory Abnormalities
26. Participants with the following toxicities at screening as defined by the enhanced Division of
Microbiology and Infectious Disease (DMID) adult toxicity table (November 2007) (Appendix
4):
a. creatinine grade 2 or greater (>1.5 times upper limit of normal [ULN]);
b. lipase grade 3 or greater (>2.0 x ULN);
c. hemoglobin grade 4 (<6.5 g/dL);
d. platelets grade 2 or greater (under 50x109 cells/L);
e. serum potassium grade 2 or greater (<3.5 mEq/L);
f. aspartate aminotransferase (AST) grade 3 (≥3.0 x ULN) to be excluded;
g. alanine aminotransferase (ALT) grade 3 (≥3.0 x ULN) to be excluded;
h. alkaline phosphatase (ALP) grade 4 (>8.0 x ULN) to be excluded, grade 3 (≥3.0 x ULN)
must be discussed with the sponsor Medical Monitor;
i. total bilirubin grade 3 or greater (>2.0 x ULN, or >1.50 x ULN when accompanied by any
increase in other liver function test) to be excluded, grade 2 (>1.50 x ULN, or >1.25 x ULN
when accompanied by any increase in other liver function test) must be discussed with
the sponsor Medical Monitor.
All inclusion and no exclusion criteria must be met. If no single variable/value is outside of the
ranges of acceptability, but when multiple values are close to the limits and/or whenever the
Investigator has reason to suspect that there might be a health problem (other than TB), enrolment
should only be considered after discussing the case with the sponsor medical monitor.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 58 of 142
4. STUDY DESIGN AND DURATION
4.1. Summary of Study Design
This is a Phase II, two-center, partially double-blinded, randomized clinical trial conducted in six
parallel groups. Participants, study investigators and staff, including laboratory staff, will be
partially blinded. All will know whether participants are on a TMC207, PA-824 or Rifafour e-275
containing treatment arm. However, the following blinding will occur:
TMC207 alone treatment arm blinded against TMC207 plus pyrazinamide treatment arm;
PA-824 plus pyrazinamide treatment arm blinded against PA-824 plus pyrazinamide plus
moxifloxacintreatment arm;
TMC207 plus PA-824 and Rifafour e-275 treatment arms will be open-label.
The trial will be performed at two centers located in Cape Town, South Africa. A total of 85 eligible
adult participants (including males and females) who meet all of the inclusion criteria and none of
the exclusion criteria, aged between 18 and 65 years (inclusive), with newly diagnosed, smear-
positive pulmonary TB will be randomized to one of 6 treatment groups:
1. TMC207 700 mg Day 1; 500 mg Day 2; 400 mg Days 3-14; + pyrazinamide placebo (dosed by
weight) Days 1-14;
2. TMC207 700 mg Day 1; 500 mg Day 2; 400 mg Days 3-14 + pyrazinamide (dosed by weight)
Days 1-14;
3. TMC207 700 mg Day 1; 500 mg Day 2; 400 mg Days 3-14 + PA-824 200 mg Days 1-14
4. PA-824 200 mg + pyrazinamide (dosed by weight) + moxifloxacin 400 mg placebo Days 1-14;
5. PA-824 200 mg + pyrazinamide (dosed by weight) + moxifloxacin 400 mg Days 1-14;
6. Rifafour e-275 which is the standard, first-line TB treatment group (isoniazid, rifampicin,
pyrazinamide and ethambutol per South African National TB Control Programme
requirements) Days 1-14.
Each of the TMC207 and PA-824 containing treatment groups will comprise 15 male or female adult
participants and the Rifafour e-275 treatment group will comprise 10 male or female adult
participants with newly diagnosed, smear-positive pulmonary TB. The Rifafour e-275 treatment
group is included as a control for the EBA quantitative mycobacteriology. With the exception of the

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 59 of 142
PK variables, the same variables as with the investigational medicinal product groups will be
measured for the Rifafour e-275 group.
4.2. Treatment Plan
See the Study Flow Chart (Section 1.2) for the timing of all procedures and laboratory samples to
done at each visit. For details regarding specific laboratory tests or procedures see Sections 7.2.
The trial will comprise three periods:
Pre-Treatment: Visits 1 to 4 (Days -9 to -1);
Treatment: Visits 5 to 19 (Days 1 to 15);
Follow-Up:
TMC207 containing treatment arms (excluding TMC207 plus PA-824 containing treatment
arm), Visit 20 (Day 28 ± 1 day or 14 days after last dose of IMP) and Visit 21 (Day 42 ± 3 days
or 28 days after last dose of IMP);
PA-824 containing treatment arms (excluding TMC207 plus PA-824 containing treatment
arm), Visit 20 (Day 28 ± 1 day or 14 days after last dose of IMP) and Visit 22 (Day 104 ±14
days or 90 days after last dose of IMP);
TMC207 plus PA-824 containing treatment arm and Rifafour e275® treatment arm, Visit 20
(Day 28 ± 1 day or 14 days after last dose of IMP), Visit 21 (Day 42 ± 3 days or 28 days after
last dose of IMP) and Visit 22 (Day 104 ±14 days or 90 days after last dose of IMP).
4.2.1. Pre-treatment Visits 1 to 4 (Days -9 to -1)
During the pre-treatment period, participants are not receiving treatment for TB whilst results are
being obtained and baseline sputum is collected. This time period will be kept to a minimum to the
greatest extent possible and will be up to a maximum of 9 days, consisting of up to a maximum of 6
days screening followed by 3 days of baseline sputum collection. If the investigator considers it
advisable for the participant to remain at the study site during the entire pre-treatment period, the
participant will be hospitalized. Hospital admission will occur at the latest at Visit 2 (Day -3). Should
a site’s ethics committee request that this period be shortened, the agreed upon maximum pre-
treatment period will be implemented for that site, and documented in the sponsor and site trial
files. This does not affect the data collected, as all sites may shorten this time period to meet their
specific sites’ needs, as long as baseline sputum collection starts on day -3.
Visit 1 (Days -9 to -4):

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 60 of 142
Prior to Visit 1, participants must have a positive TB sputum smear microscopy result from their TB
clinic or site of initial diagnosis.
The following information will be collected and procedures performed:
o Written informed consent obtained from the participant.
o Hospital admission (on advice of investigator).
o Demographic data: Date of birth, race, and gender.
o Inclusion and exclusion criteria will be verified.
o Spot sputum for confirmation of TB diagnosis and adequacy of bacterial count.
o Clinically significant medical and treatment history.
o Chest X-ray for confirmation of radiological picture compatible with TB.
o Physical examination including weight, height and history of menstrual cycle (e.g. possible
irregularities, missed cycles) for women.
o Ophthalmologic examination with visual acuity, and slit lamp examination of the lens.
o Fundoscopy.
o Vital signs.
o 12-lead ECG (single).
o Clinical laboratory tests: See Section 7.2 for list of tests.
o Urine drug screen (should be repeated at Visit 2 (Day -3) if participant not hospitalized at
screening visit).
o Urine test for isoniazid (should be repeated at Visit 2 (Day -3) if participant not hospitalized
at screening visit).
o Serum pregnancy test (women of child bearing potential only, whether they are sexually
active or not).
o Serum endocrinology (males only).
o HIV test and CD4 count.
o Isoniazid and Rifampicin -resistance test: sputum-based rapid test.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 61 of 142
o Concomitant medication/s.
Participants may proceed with the Visit 2 (Day -3) assessments as soon as their Visit 1 (Days -9
to -4) assessments have been completed. Visits 1 and 2 may occur on the same day if the
screening results are available in time for randomization. Overnight sputum collection will start
not later than Day -3, however may be collected for a number of more days if the screening
results are delayed, or the mycobacterial testing on the first spot sputum shows an
indeterminate result in which case the test may be repeated on a freshly collected spot sputum
or an overnight sputum and that result used.
Visit 2 (Day -3):
The following information will be collected and procedures performed:
o Eligibility assessment.
o Hospital admission (if not already admitted at Visit 1(Days -9 to -4)).
o Physical examination including weight.
o Vital signs.
o Overnight sputum collection (sputum sample will be used to ensure adequate volume and is
not required for efficacy variable tests, retention sample collection or mycobacterial
characterization testing).
o Concomitant medication/s.
o Adverse events.
Visit 3 (Day -2):
The following information will be collected and procedures performed:
o Physical examination including weight.
o Vital signs.
o Overnight sputum (includes sputum required for efficacy variable tests, retention sample
collection and mycobacterial characterization testing).

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 62 of 142
o Randomization by the pharmacist/registered dispenser may occur once all the screening
results are available and the investigator has determined that the participant is eligible for
the trial. Randomization must occur prior to the Visit 4 (Day -1) ECGs.
o Concomitant medication/s.
o Adverse events.
Visit 4 (Day-1)
The following information will be collected and procedures performed:
o Physical examination including weight.
o Vital signs.
o Overnight sputum (includes sputum required for efficacy variable tests and retention
sample collection).
o 12-lead ECG: For PA-824 (excluding TMC207 plus PA-824) containing treatment arms only.
To be performed in triplicate at 0, 1, 2, 3, 4, 6, 8, 12 and 24 hours. The hour 0 ECG should be
performed at the time it is anticipated that dosing will occur on Visit 5 (Day 1). The 24 hour
ECGs on Visit 4 (Day -1), will serve as the pre-dose (hour 0) ECGs for Visit 5 (Day 1).
o Concomitant medication/s.
o Adverse events.
4.2.2. Treatment Period (Visits 5 to 19 [Days 1 to 15])
Visit 5 (Day 1):
The following information will be collected and procedures performed:
o Pre-dosing (for TMC207 plus PA-824 treatment arm pre-PA-824 dose):
Eligibility assessment.
Physical examination including weight.
Vital signs.
Clinical laboratory tests: See Section 7.2 for list of tests.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 63 of 142
Retention sample collection: plasma in parallel with the pre-dose blood draw sample for
all participants; urine in parallel with the pre-dose urinalysis sample for all participants;
serum (males only) in parallel with the pre-dose blood draw sample for PA-824
(including TMC207 plus PA-824) and Rifafour e275® containing treatment arms.
o 12 lead ECG:
TMC207 (excluding TMC207 plus PA-824) and Rifafour e275® containing treatment
arms: (single) performed pre-dose (up to approximately 16 hours before the dose) and 5
hours post-dose.
PA-824 (excluding TMC207 plus PA-824) containing treatment arms: to be performed in
triplicate pre-dose and 1, 2, 3, 4, 6, 8, 12 and 24 hours post-dose. The 24 hour Visit 4
(Day -1) ECG will serve as the Visit 5 (Day 1) pre-dose ECG. The 24 hour Visit 5 (Day 1)
ECG will serve as the Visit 6 (Day 2) pre-dose ECG and will therefore be performed in
triplicate.
TMC207 plus PA-824 treatment arm to be performed in single pre-PA-824 dose, 4 hours
post-PA-824/pre-TMC207 dose and 9 hours post-PA-824/5-hours post TMC207 dose.
o Overnight sputum collection (includes sputum required for efficacy variable tests and
retention sample collection).
o PK sampling:
TMC207 containing treatment arms: performed pre-dose and 5 hours post-TMC207
dose.
PA-824 (excluding TMC207 plus PA-824) containing treatment arms: performed pre-
dose and 1, 2, 3, 4, 6, 8, 12 and 24 hours post-dose. The 24 hour sample is to be
collected pre-dose on Visit 6 (Day 2). For TMC207 plus PA-824 containing treatment
arm: performed pre-PA-824 dose and 4 and 8 hours post-PA-824 dose
o -IMP administration.
o Compliance check (‘hand-and-mouth’).
o Concomitant medication/s.
o Adverse events.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 64 of 142
Visits 6 to 17 (Days 2 to 13)
The following information will be collected and procedures performed:
o Daily:
Physical examination including weight.
Vital signs.
Overnight sputum collection (includes sputum required for efficacy variable tests and at
Visit 6 (Day 2), Visit 7 (Day 3), Visit 8 (Day 4), Visit 10 (Day 6), Visit 12 (Day 8), Visit 14
(Day 10) and Visit 16 (Day 12) includes retention sample collection).
IMP administration.
Compliance check (‘hand-and-mouth’).
Concomitant medication/s.
Adverse events.
o On Day 2:
PA-824 (excluding TMC207 plus PA-824) containing treatment arms:
12-lead ECG (triplicate) performed pre-dose as the Visit 5 (Day 1) 24 hour ECG will be
considered the pre-dose ECG for Visit 6 (Day 2).
12 lead ECG (single) performed 2 and 5 hours post-dose.
o On Day 3:
12 lead ECG (single):
TMC207 (excluding TMC207 plus PA-824) and Rifafour e275® containing treatment
arms; performed pre-dose and 5 hours post-dose.
PA-824 (excluding TMC207 plus PA-824) containing treatment arms: performed pre-
dose, 2 and 5 hours post dose.
TMC207 plus PA-824 treatment arm to be performed pre-PA-824 dose, 4 hours post-
PA-824/pre-TMC207 dose and 9 hours post-PA-824/5-hours post TMC207 dose.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 65 of 142
PK sampling:
TMC207 containing treatment arms; performed pre-dose and 5 hours post-TMC207 dose.
o On Days 4 to 7:
12 lead ECG (single): PA-824 (excluding TMC207 plus PA-824) containing treatment
arms; performed pre-dose, 2 and 5 hours post-dose.
o On Day 8:
12 lead ECG:
TMC207 (excluding TMC207 plus PA-824) and Rifafour e275® containing treatment
arms; (single) performed pre-dose and 5 hours post-dose.
PA-824 (excluding TMC207 plus PA-824) containing treatment arms; (triplicate)
performed pre-dose and 1, 2, 3, 4, 6, 8, 12 and 24 hours post-dose. The 24 hour Visit
12 (Day 8) ECG will serve as the Visit 13 (Day 9) pre-dose ECG and will therefore be
performed in triplicate.
TMC207 plus PA-824 treatment arm to be performed in single pre-PA-824 dose, 4
hours post-PA-824/pre-TMC207 dose and 9 hours post-PA-824/5-hours post TMC207
dose.
PK sampling:
TMC207 containing treatment arms: performed pre-dose and 5 hours post-TMC207
dose.
PA-824 (excluding TMC207 plus PA-824) containing treatment arms: performed pre-
dose, 1, 2, 3, 4, 6, 8, 12 and 24 hours post-dose Sampling. The 24 hour sample is to
be collected pre-dose on Visit 13 (Day 9). For TMC207 plus PA-824 containing
treatment arm:
TMC207 plus PA-824 treatment arm: performed pre-PA-824 dose and 4 and 8 hours
post-PA-824 dose.
Retention sample collection: plasma in parallel with the Hour 0 PK blood draw sample
for all participants; urine in parallel with the pre-dose urinalysis sample for all
participants; serum (males only) in parallel with the Hour 0 PK blood draw sample for

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 66 of 142
PA-824 (including TMC207 plus PA-824) and Rifafour e275® containing treatment arms.
For the TMC207 plus PA-824 treatment arm these are to be collected pre the PA-824
dose.
Clinical laboratory tests (pre-dose, for TMC207 plus PA-824 treatment arm pre-PA-824
dose): See Section 7.2 for list of tests.
o On Day 9:
PA-824 (excluding TMC207 plus PA-824) containing treatment arms:
12-lead ECG (triplicate) performed pre-dose as the Visit 12 (Day 8) 24 hour ECG will
be considered the pre-dose ECG for Visit 13 (Day 9).
12 lead ECG (single) performed 2 and 5 hours post-dose.
o On Days 10 to 13:
12 lead ECG (single) – PA-824 (excluding TMC207 plus PA-824) containing treatment
arms; performed pre-dose, 2 and 5 hours post-dose.
Visit 18 (Day 14):
The following information will be collected and procedures performed:
o Physical examination including weight.
o Vital signs.
o PK sampling:
TMC207 and PA-824 (excluding TMC207 plus PA-824) containing treatment arms
performed pre-dose and 1, 2, 5 and 8 hours post-dose.
TMC207 plus PA-824 treatment arm:
For TMC207 PK analyses - performed pre-TMC207 dose and 1, 2, 5 and 8 hours
post-TMC207 dose.
For PA-824 PK analyses - performed pre-PA-824 dose and 1, 2, 5 and 8 hours
post-PA-824 dose.
o 12 lead ECG –

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 67 of 142
TMC207 (excluding TMC207 plus PA-824) and Rifafour e275® containing treatment
arms; (single) performed pre-dose (prior to PK sampling) and 5 hours post-dose.
PA-824 (excluding TMC207 plus PA-824) containing treatment arms; (single) performed
pre-dose (prior to PK sampling) and at 2 and 5 hours post-dose.
TMC207 plus PA-824 treatment arm; performed in single pre-PA-824 dose, 4 hours post-
PA-824/pre-TMC207 dose and 9 hours post-PA-824/5-hours post TMC207 dose.
o Overnight sputum collection (includes sputum required for efficacy variable tests, retention
sample collection and mycobacterial characterization testing. If the participant was treated
with study drug for less than 9 days, mycobacteriology testing will be performed on the 32
(Day-2) isolate only for these participants).
o IMP administration.
o Compliance check (‘hand-and-mouth’).
o Concomitant medication/s.
o Adverse events.
Visit 19 (Day 15):
The following information will be collected and procedures performed:
o Physical examination including weight.
o Vital signs.
o Fundoscopy (TMC207 (including TMC207 plus PA-824) and Rifafour e275® containing
treatment arms).
o PK sampling (TMC207 and PA-824 containing treatment arms (including TMC207 plus PA-
824 arm, for whom two samples will be collected, one for TMC207 and one for PA-824
analyses) ) performed 24 hours after Day 14 dosing.
o Clinical laboratory tests: See Section 7.2 for list of tests.
o Retention sample collection: plasma in parallel with the Hour 0 PK blood draw sample for all
participants; urine in parallel with the pre-dose urinalysis sample for all participants; serum
(males only) in parallel with the Hour 0 PK blood draw sample for PA-824 (including TMC207
plus PA-824) and Rifafour e275® containing treatment arms.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 68 of 142
o Discharge from hospital with referral letter to National TB treatment programme. The
participants on the investigational medicinal product arms will receive the first dose (s) of
standard TB therapy per SA national TB treatment guidelines. The participants in the group
receiving standard TB treatment will receive an additional dose(s) to continue on this
treatment. All participants will be referred to the local community TB clinics for a full course
of standard antituberculosis chemotherapy according to National Guidelines. The
participants will be provided with a referral letter to take with them to the TB Clinic. A
follow-up call will be made by the study site staff to the clinic to determine if the participant
attended the clinic on the date as arranged. Participants will be provided with sufficient TB
medication (Rifafour e-275®) to cover the period from attending their last visit at the study
clinic until their scheduled visit at the TB clinic.
o Concomitant medication/s.
o Adverse events.
4.2.3. Follow-Up Period
Visit 20 (Day 28 [± 1 day window period][14 day ± 1 days after the last dose of IMP]):
All participants.
A safety follow-up assessment will be conducted at the hospital 14 days ± 1 day after the last IMP
dose. The following information will be collected and procedures performed:
o Physical examination including weight.
o Vital signs.
o 12 lead ECG (single)
o Clinical laboratory tests: See Section 7.2 for list of tests.
o Concomitant medication.
o Adverse events.
o Document in the Case Report Form (CRF) and in the source documents whether the
participant is attending the local TB clinic and included into the National TB Programme.
Visit 21 (Day 42 [± 3 days window period][28 days ± 3 days after the last dose of IMP]):

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 69 of 142
Only for participants in the TMC207 (including TMC207 plus PA-824) and Rifafour e275®
containing treatment arms.
A safety follow-up assessment will be conducted at the hospital 28 days ± 3 days after the last IMP
dose. The following information will be collected and procedures performed:
o Physical examination including weight.
o Fundoscopy.
o Vital signs.
o PK sampling (TMC207 (including TMC207 plus PA-824) containing treatment arms).
o 12-lead ECG (single).
o Retention sample collection: plasma in parallel with PK blood draw sample (TMC207
(including TMC207 plus PA-824) and Rifafour e275® containing treatment arms).
o Concomitant medications.
o Adverse events.
Visit 22 (Day 104 [± 14 day window period][90 days ± 14 day after the last dose of IMP])
Only for participants in PA-824 (including TMC207 plus PA-824) and Rifafour e275® containing
treatment arms.
A safety follow-up assessment will be conducted at the hospital 90 days ± 14 day after the last IMP
dose. The following information will be collected and procedures performed:
o Ophthalmologic examination with visual acuity and slit lamp examination of the lens.
o Concomitant medications.
o Adverse events - For the period from Visit 20 (Day 28) to Visit 22 (Day 104) for participants
on the PA-824 (excluding TMC207 plus PA-824) treatment arms and Visit 21 (Day 42) to Visit
22 (Day 104) for participants on the TMC207 plus PA-824 and Rifafour e275® treatment arm,
only ophthalmologic related adverse events, and all serious adverse events, will be
collected.
4.2.4. Early Withdrawal

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 70 of 142
In case of early withdrawal during the treatment period of the study, all efforts shall be made to
complete the Early Withdrawal assessments. At the early withdrawal visit the following
information will be collected and procedures performed:
o Physical examination including weight.
o Vital signs.
o Fundoscopy
o PK sampling (TMC207 and PA-824 containing treatment arms).
o Retention sample collection: plasma in parallel with the PK blood draw sample for all
participants; urine in parallel with the pre-dose urinalysis sample for all participants; serum
(males only) in parallel with the Hour 0 PK blood draw sample for PA-824 (including
TMC207 plus PA-824) and Rifafour e275® containing treatment arms.
o 12-lead ECG (single)
o Clinical laboratory tests: See Section 7.2 for list of tests.
o Concomitant medication.
o Adverse events.
o Discharge from hospital with referral letter to National TB treatment programme. The
participants on the investigational medicinal product arms will receive the first dose(s) of
standard TB therapy per SA national TB treatment guidelines. The participants in the group
receiving standard TB treatment will receive an additional dose to continue on this
treatment. All participants will be referred to the local community TB clinics for a full course
of standard antituberculosis chemotherapy according to National Guidelines. The
participants will be provided with a referral letter to take with them to the TB Clinic. A
follow-up call will be made by the study site staff to the clinic to determine if the participant
attended the clinic on the date as arranged. Participants will be provided with sufficient TB
medication (Rifafour e-275®) to cover the period from attending their last visit at the study
clinic until their scheduled visit at the TB clinic.
All participants who received at least one dose of IMP will be requested to return for the safety
follow up visits, Visit 20 (14 days ± 1 day after their early withdrawal visit), Visit 21 (28 days ± 3
days after their early withdrawal visit for all TMC207 (including TMC207 plus PA-824) and

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 71 of 142
Rifafour e275®participants) and Visit 22 (90 days ± 14 days after their early withdrawal visit for
all PA-824 (including TMC207 plus PA-824) and Rifafour e275® participants).
4.3. Participant Withdrawal Criteria
A participant can be prematurely withdrawn from the trial as a result of the following:
At their own request or at the request of their legally acceptable representative.
If, in the investigator’s opinion, continuation in the trial would be detrimental to the well-being
of the participant.
At the specific request of the sponsor.
QTc prolongation as described in section 9.7.
A serious adverse event occurs.
Fails to comply with the protocol.
Participants who withdraw from the trial after having received IMP will not be replaced. The
exception is those participants who are discontinued due to QT prolongation on Day 1, who will be
replaced. Any participant who received at least one dose of IMP and is withdrawn or withdraws
early from the study during the treatment phase will undergo an early withdrawal visit and will be
requested to return for the applicable follow up visits.
4.4. Trial Treatment Discontinuation
Trial treatment must be immediately discontinued for the following reasons:
Withdrawal of informed consent.
Investigator considers it for safety reasons in the best interest of the participant that he/she be
withdrawn.
QTc prolongation as described in section 9.7.
Participant experiences specific toxicities as outlined in Section 0.
Pregnancy.
Termination of the study by the sponsor.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 72 of 142
Participants who withdraw from the trial after having received IMP will not be replaced. The
exception is those participants who are discontinued due to QT prolongation on Day 1, who will be
replaced.
4.5. Stopping Rules
There are no stopping rules in this trial.
4.6. Participant Progress Definitions
4.6.1. Screening Failure
A screen failure participant is one from whom informed consent is obtained and is documented in
writing (that is, participant signs an informed consent form), but who’s not randomized.
4.6.2. Completed Treatment
Participants who complete their treatment will be defined as completed treatment.
4.6.3. Early Withdrawal
Participants who withdraw from the study prior to the completion of their treatment will be
defined as early withdrawal.
4.6.4. Completed Trial
Participants who complete the treatment period and attend the follow-up visit: Visit 21 (Day 42) for
TMC207 (excluding TMC207 plus PA-824) containing treatment arms and Visit 22 (Day 104) for PA-
824 (including TMC207 plus PA-824) and Rifafour e275® containing treatment arms, will be defined
as completed trial.
4.6.5. Lost to Follow-up
Participants who cannot be contacted on or before Visit 21 (Day 42) for TMC207 (excluding
TMC207 plus PA-824) participants or Visit 22 (Day 104) for PA-824 (including TMC207 plus PA-824)
and Rifafour e275® treatment participants and who do not have a known reason for
discontinuation (for example, withdrew consent or adverse event) will be defined as lost to follow-
up.
4.7. Restrictions
4.7.1. Foods and Beverages

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 73 of 142
Consumption of foods and beverages containing the following substances will be prohibited as
indicated:
Methyl-xanthines/caffeine: From 48 hours prior to Visit 5 (Day 1) until after Visit 19 (Day 15) or
early withdrawal visit i.e. until discharge from the study clinic. Decaffeinated drinks are allowed.
Poppy seeds: 7 days prior to Visit 1 (Days -9 to -4) until after Visit 19 (Day 15) or early
withdrawal visit i.e. until discharge from the study clinic.
Alcohol: 72 hours prior to Visit 5 (Day 1) until after Visit 19 (Day 15) or early withdrawal visit i.e.
until discharge from the study clinic.
Grapefruit or grapefruit juice: 10 days prior to prior to Visit 5 (Day 1) until after Visit 19 (Day 15)
or early withdrawal visit i.e. until discharge from the study clinic.
4.7.2. Prior and Concomitant Medications
Any medication taken within 30 days prior to IMP administration or during the trial until visit 21
(Day 42) for TMC207 (excluding TMC207 plus PA-824) participants and until Visit 22 (Day 104) for
PA-824 (including TMC207 plus PA-824) and Rifafour e275® participants is defined as concomitant
medication and is to be reported on the Concomitant Therapy page of the CRF. Reported
information will include a description of the type of the drug, treatment period, dosing regimen,
route of administration, and its indication. Any changes in the dosage of concomitant medication
must also be reported on the Concomitant Therapy page of the CRF. Data on concomitant
medication will be collected up to the last follow-up visit, even after early withdrawal of a subject.
Concomitant treatments should be kept to a minimum during the trial. However, if concomitant
treatments are considered to be necessary for the participant’s welfare and are unlikely to interfere
with the investigational medication, they must be given at the discretion of the Investigator. For
any concomitant therapy given as a treatment for a new condition or a worsening of an existing
condition occurring after signing of the informed consent form (ICF), the condition must be
documented on the Adverse Event pages of the CRF.
The prescribing information for all concomitant medication should be consulted and reviewed
carefully. The determinations listed in the respective contraindicated, warning, and precaution
sections must be respected in order to prevent any potentially serious and/or life-threatening drug
interactions.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 74 of 142
The following medications are disallowed during administration of IMP and up to 1 month after the
last dose of IMP:
the systemic use of CYP3A4 inhibitors (e.g., azole antifungals: ketoconazole, voriconazole,
itraconazole, fluconazole; ketolides such as telithromycin; and macrolide antibiotics other
than azithromycin) for more than 3 consecutive days;
the systemic use of CYP3A4 inducers (e.g., phenytoin, carbamazepine, phenobarbital, St.
John’s wort);
The following medicinal products are prohibited from Visit 1 (Days -9 to -4) until after Visit 19 (Day
15) or early withdrawal visit i.e. until discharge from the study clinic (unless part of study
treatment):
Medications of the statin class of compounds;
Tricylic antidepressants, including amtriptyline, doxepin, desipramine, imipramine,
clomipramine;
Nonsedating antihistamines astemizole and terfenadine
The neuroleptics-phenothiazines thioridazine, haloperidol, chlorpromazine, trifluoperazine,
percyline, prochlorperazine, fluphenazine, sertindole and pimozide;
The prokinetic cisapride;
Quinoline antimalarials (e.g., chloroquine and quinacrine);
Diuretics that deplete potassium;
Systemic glucocorticoids;
Medicinal products used to treat pulmonary TB: moxifloxacin, gatifloxacin, isoniazid,
ethambutol, amikacin, cycloserine, rifabutin, rifampicin, streptomycin, kanamycin, para-
aminosalicylic acid, rifapentine, thioacetazone, capreomycin, quinolones, thioamides, and
metronidazole.
4.7.3. Activity
Participants will be requested not to undertake vigorous exercise during the period from Visit 1
(Days -9 to -4) until after the final laboratory safety tests at follow up Visit 20 (Day 28 or 14 days + 1
day post last dose of IMP).
Participants will not engage in strenuous activity at any time during the in-hospitalization period.
5. INVESTIGATIONAL MEDICINAL PRODUCT

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 75 of 142
5.1. Trial Treatments
Participants will receive oral once-daily dosing. They will be randomized to one of the following
arms:
1. TMC207 700 mg Day 1; 500 mg Day 2; 400 mg Days 3-14; + pyrazinamide placebo (dosed by
weight) Days 1-14;
2. TMC207 700 mg Day 1; 500 mg Day 2; 400 mg Days 3-14 + pyrazinamide (dosed by weight)
Days 1-14;
3. TMC207 700 mg Day 1; 500 mg Day 2; 400 mg Days 3-14 +PA-824 200 mg Days 1-14;
4. PA-824 200 mg + pyrazinamide (dosed by weight) + moxifloxacin 400 mg placebo Days 1-14;
5. PA-824 200 mg + pyrazinamide (dosed by weight) + moxifloxacin 400 mg Days 1-14;
6. Rifafour e-275 which is the standard, first-line TB treatment group (isoniazid, rifampicin,
pyrazinamide and ethambutol per South African National TB Control Programme
requirements) Days 1-14.
5.2. Methods of Assigning Participants to Treatment Groups
Eligible participants who have given written, informed consent will be enrolled onto the trial during
Visit 1 (Days -9 to -4) and will be identified by a study generated participant identification code for
anonymity (subject number). Participants who meet all of the inclusion criteria and none of the
exclusion criteria will be randomized and assigned a treatment number.
The Investigational Medicinal Product will be centrally randomized by persons not directly involved
with the trial. Two separate randomisations will occur:
Randomization (1):
TMC207 and PA-824 (excluding the TMC207 plus PA-824) containing treatment arms plus 8 Rifafour
e275 participants;
Randomization (2):
TMC207 plus PA-824 containing treatment arm plus 2 of the Rifafour 2e75 participants.
The randomization (2) participants will be enrolled after full enrollment for the other 5 treatment
arms is complete. Participants, study investigators and staff, site pharmacists/dispensers, including
laboratory staff, sponsor staff and applicable CROs will be partially blinded. All will know whether

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 76 of 142
the participant is on a TMC207, PA-824 or Rifafour e-275 containing treatment arm. However,
the following blinding will occur:
TMC207 alone treatment arm blinded against TMC207 plus pyrazinamide treatment arm;
PA-824 plus pyrazinamide treatment arm blinded against PA-824 plus pyrazinamide plus
moxifloxacin treatment arm;
TMC207 plus PA-824 and Rifafour e-275 treatment arms will be open-label.
A treatment pack will be available for each participant which will be identified by a treatment
number.
In order to ensure there is no bias, the study medication will be retained by the site
pharmacist/registered dispenser. The site pharmacist/registered dispenser will not have any
interaction with the participants or participant care responsibilities. On randomization, the site will
request the site pharmacist/registered dispenser to assign an IMP treatment pack to the
participant. The site pharmacist/registered dispenser will assign to the participant the next
available treatment pack, in a sequential basis starting from the lowest unused treatment number.
This process will be fully documented. Randomization by the pharmacist/registered dispenser may
occur once all the screening results are available and the investigator has determined that the
participant is eligible for the trial. Randomization must occur prior to the Visit 4 (Day -1) ECGs.
Participants who withdraw from the trial after having received IMP will not be replaced. The
exception to this is those participants who are discontinued due to QT prolongation on Day 1, who
will be replaced. For these participants the site staff will inform the Randomization Monitor that
the participant was withdrawn due to QT prolongation on Day 1 and the treatment number of that
participant. The randomization monitor who will determine and inform the applicable site
pharmacist/registered dispenser of the next available sequential treatment number for the next
patient to be randomized. Replacement participant will be assigned to the treatment group of the
participant who was withdrawn but may be assigned to any site. The randomization monitor will
be responsible for working with the site pharmacists/registered dispensers to ensure that the
above process is managed so that the required number of participants per treatment arm are
randomized onto the study.
5.3. IMP Administration
5.3.1. TMC207 and PA-824 Treatment Groups

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 77 of 142
The TMC207 containing treatment arms will be administered in the morning (within 30 minutes
after breakfast) with 240 ml water.
The PA-824 containing treatment arms will be administered under fasting conditions in the morning
(at least 4 hours before breakfast) with 240 ml water. The minimum duration of the fast is to be
eight hours prior to dosing and 4 hours post-dosing.
The TMC207 plus PA-824 containing treatment arm will be administered as follows:
The PA-824 and TMC207 open-label tablets will be administered at different times, per their
dosing requirements, that is:
o The PA-824 open-label tablets will be administered under fasting conditions in the
morning (at least 4 hours before breakfast) with 240 ml water. The minimum
duration of the fast is to be eight hours prior to dosing and 4 hours post-dosing.
o The TMC207 open-label tablets will be administered in the morning (within 30
minutes after breakfast) with 240 ml water.
5.3.2. First-line standard TB treatment per SA national guidelines (Rifafour e-275 tablets)
Treatment Group
The Rifafour e-275 will be administered with a full glass of water 1 hour before, or 2 hours after a
meal (as per the package insert (Appendix 2)).
5.3.3. Participant Compliance
The IMP will be administered by the investigator/designated site personnel. The date, time and
number of tablets administered will be recorded in the participant’s CRF and in the source
documents. All IMP will be administered with water. Participants will be checked for IMP
compliance by the Investigators or trial personnel via the hand-and-mouth procedure. Both the
hand and mouth of the participant will be checked to ensure that the participant has swallowed the
IMP.
5.4. Blinding and Procedures for Breaking the Blind
Individual treatment code break envelopes for the blinded treatment arms will be supplied to:
the sponsor’s medical monitor;
the site;

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 78 of 142
the monitoring CRO.
The treatment code must not be broken except in the case of a medical emergency, where
treatment of the participant is influenced by the knowledge of what dose and type of IMP the
participant is receiving. Prior to breaking the code, if practical, the Investigator is to discuss the
breaking of the blind with the sponsor’s medical monitor. The Investigator must document on the
code break envelope and in the participant’s source documentation that the blind was broken, plus
the reason for breaking it. The time and date of breaking the blind must be completed. The
sponsor must be informed that the blind has been broken within 24 hours of the event. The
sponsor reserves the right to break the blind in order to fulfill any regulatory requirements
regarding reporting of SAEs.
In the absence of any medical emergencies requiring a code break, the randomization code will
only be broken once all clinical data, as well as all outcome parameters have been captured and no
more data queries are pending. The statistical analysis plan will also have been finalized. The only
exception to this rule is that the blind may be broken prior to:
The availability of results of specific microbiological tests of isolated M. tuberculosis cultures
from sputum (MIC against PA-824, TMC207 and moxifloxacin, first-line drug-susceptibility
testing, strain speciation by PCR). Un-blinding will occur before these results are known and
therefore before definitive database closure. The rationale for this exception lies in the long
time needed for completion of these microbiological assessments (they will take at least 6
weeks and can be further delayed by technical difficulties with the subsequent repetition of
tests), and these tests are objective parameters not influenced by blinding/un-blinding.
Visit 20 (Day 28), by the biostatistician responsible for the EBA efficacy analyses only. All other
sponsor, CRO, laboratory, statistical and site personnel will remain blinded. The efficacy
biostatistician may perform the efficacy analyses and inform the sponsor of the high level EBA
efficacy results by treatment arm only, in order for this information to be utilised in the decision
making for future clinical trial design. No un-blinded individual subject results will be supplied
by the biostatistician until formal database closure has occurred. The EBA biostatistician has no
role in the remaining analyses.
5.5. IMP Packaging and Labeling
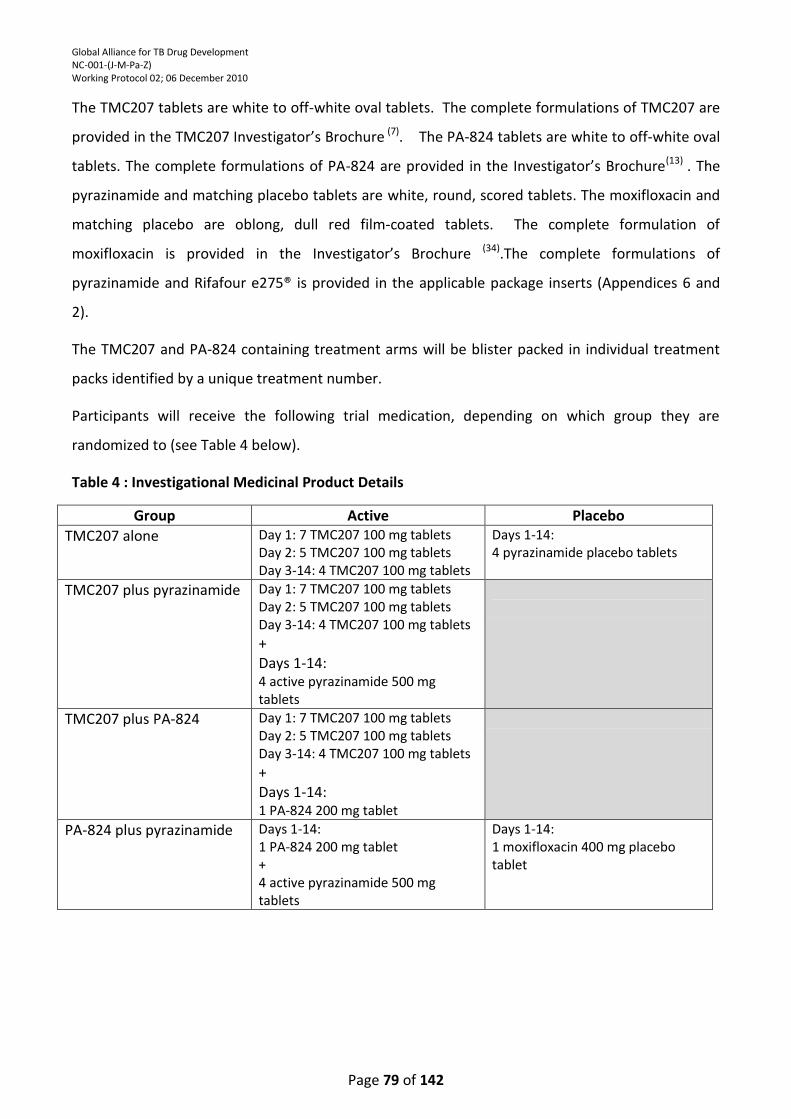
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 79 of 142
The TMC207 tablets are white to off-white oval tablets. The complete formulations of TMC207 are
provided in the TMC207 Investigator’s Brochure (7). The PA-824 tablets are white to off-white oval
tablets. The complete formulations of PA-824 are provided in the Investigator’s Brochure(13) . The
pyrazinamide and matching placebo tablets are white, round, scored tablets. The moxifloxacin and
matching placebo are oblong, dull red film-coated tablets. The complete formulation of
moxifloxacin is provided in the Investigator’s Brochure (34).The complete formulations of
pyrazinamide and Rifafour e275® is provided in the applicable package inserts (Appendices 6 and
2).
The TMC207 and PA-824 containing treatment arms will be blister packed in individual treatment
packs identified by a unique treatment number.
Participants will receive the following trial medication, depending on which group they are
randomized to (see Table 4 below).
Table 4 : Investigational Medicinal Product Details
Group Active Placebo
TMC207 alone Day 1: 7 TMC207 100 mg tablets Day 2: 5 TMC207 100 mg tablets Day 3-14: 4 TMC207 100 mg tablets
Days 1-14: 4 pyrazinamide placebo tablets
TMC207 plus pyrazinamide Day 1: 7 TMC207 100 mg tablets Day 2: 5 TMC207 100 mg tablets Day 3-14: 4 TMC207 100 mg tablets + Days 1-14: 4 active pyrazinamide 500 mg tablets
TMC207 plus PA-824 Day 1: 7 TMC207 100 mg tablets Day 2: 5 TMC207 100 mg tablets Day 3-14: 4 TMC207 100 mg tablets + Days 1-14: 1 PA-824 200 mg tablet
PA-824 plus pyrazinamide Days 1-14: 1 PA-824 200 mg tablet + 4 active pyrazinamide 500 mg tablets
Days 1-14: 1 moxifloxacin 400 mg placebo tablet
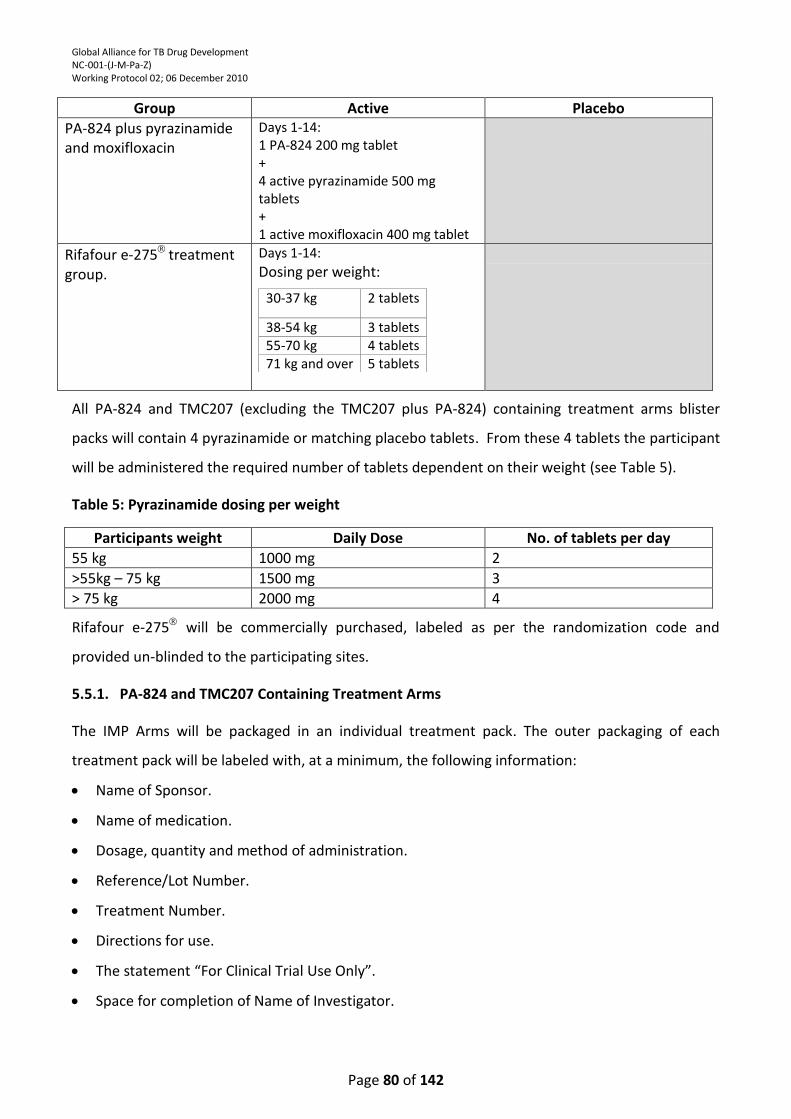
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 80 of 142
Group Active Placebo
PA-824 plus pyrazinamide and moxifloxacin
Days 1-14: 1 PA-824 200 mg tablet + 4 active pyrazinamide 500 mg tablets + 1 active moxifloxacin 400 mg tablet
Rifafour e-275 treatment group.
Days 1-14:
Dosing per weight:
30-37 kg 2 tablets
38-54 kg 3 tablets 55-70 kg 4 tablets 71 kg and over 5 tablets
All PA-824 and TMC207 (excluding the TMC207 plus PA-824) containing treatment arms blister
packs will contain 4 pyrazinamide or matching placebo tablets. From these 4 tablets the participant
will be administered the required number of tablets dependent on their weight (see Table 5).
Table 5: Pyrazinamide dosing per weight
Participants weight Daily Dose No. of tablets per day
55 kg 1000 mg 2
>55kg – 75 kg 1500 mg 3
> 75 kg 2000 mg 4
Rifafour e-275 will be commercially purchased, labeled as per the randomization code and
provided un-blinded to the participating sites.
5.5.1. PA-824 and TMC207 Containing Treatment Arms
The IMP Arms will be packaged in an individual treatment pack. The outer packaging of each
treatment pack will be labeled with, at a minimum, the following information:
Name of Sponsor.
Name of medication.
Dosage, quantity and method of administration.
Reference/Lot Number.
Treatment Number.
Directions for use.
The statement “For Clinical Trial Use Only”.
Space for completion of Name of Investigator.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 81 of 142
Storage conditions.
The statement “Keep out of reach of children”.
Expiry Date.
The inner packaging of each treatment pack will be labeled with, at a minimum, the following
information:
Name of Sponsor.
Name of medication.
Dosage, quantity and method of administration.
Reference Number.
Treatment Number.
Directions for use.
The statement “For Clinical Trial Use Only”.
5.5.2. Standard, first line TB treatment
Labels will be provided to be added to the commercial pack to denote the treatment code and that
it is for use in this clinical trial. These labels will conform to the requirements as described in
Section 5.5.1.
5.6. Storage
All study medication will be kept securely stored by the site pharmacist/registered dispenser in a
secured area with limited access to designated site personnel only.
Blinded treatment arms will be stored in the supplied containers between 15 to 25 degrees Celsius.
Rifafour e-275 will be stored at <25 degrees Celsius, in the tightly closed container that it is
supplied in, protected from light.
5.7. Dispensing and Accountability
The site pharmacist/registered dispenser will be responsible for dispensing the IMP. Accurate
accountability records will be kept by the site to assure that the IMP will not be dispensed to any
person who is not a participant under the terms and conditions set forth in this protocol i.e.
delivery to site, inventory at site, use by participant, destruction etc. The Investigator/designee will
immediately inform the sponsor of any quality issues arising with respect to the trial medication.
The sponsor will take whatever action is required should such a situation arise.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 82 of 142
The Investigator undertakes to use the trial medication only as indicated in this protocol.
5.8. Returns and Destruction
Upon completion or termination of the trial, all unused and/or partially used IMPs must be
returned to sponsor (or designated contractor). If no supplies remain, this fact will be indicated in
the drug accountability section of the final report.
6. TRIAL VARIABLES
6.1. Demographic and Background Variables
The following demographic and background variables will be collected:
Written informed consent.
Demographic data: Date of birth, race, gender.
Clinically significant medical and treatment history (including ophthalmological history and for
females menstrual history including any history of irregular or missed periods).
Inclusion and exclusion criteria.
Chest X-ray for confirmation of radiological picture compatible with TB.
Urine drug screen.
Urine test for isoniazid.
Serum pregnancy test (women of child-bearing potential only, whether they are sexually active
or not).
Serum endocrinology (males only): testosterone, luteinizing hormone (LH), follicle-stimulating
hormone (FSH).
Serology: HIV and CD4 count.
Spot sputum:
o rapid test for isoniazid and rifampicin resistance;
o confirmation of MTB prior to randomization;
o Adequate bacterial load;
o Estimate of adequate sputum production.
Concomitant Medications.
Height (meters (m)).

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 83 of 142
Method of Birth Control: Male and Female participants.
Restrictions: restricted food and beverages, restricted activity, prohibited mediation.
Post Trial Treatment TB Programme Attendance: attendance at the local TB clinic, inclusion
onto the National TB Programme.
Hospitalization.
6.2. Efficacy Variables
The following efficacy variables will be assessed for evaluation of the efficacy endpoints:
The change in number of sputum CFU of M. tuberculosis on solid media (logCFU) over time.
The change in TTP in liquid media (the MGIT system) over time.
6.3. Pharmacokinetic Variables
The following pharmacokinetic variables will be assessed for the evaluation of the pharmacokinetics
of PA-824, TMC207, TMC207 metabolite M2, pyrazinamide and moxifloxacin (except in the Rifafour
e275® treatment arms):
Maximum observed plasma concentration (Cmax).
Time at which Cmax is observed (Tmax).
Minimum observed plasma concentration 24 hours following the last dose (Cmin).
Area under the plasma concentration time (t) curve from zero to 24 hours AUC(0-24)
No PK sampling will be performed on the Rifafour e275® treatment arm.
6.4. Safety Variables
The following safety variables will be assessed for evaluation of the safety endpoints:
Laboratory Parameters:
o Hematology: hemoglobin, hematocrit, red blood cell count, white blood cell count with
differential, platelet count.
o Coagulation: activated partial thromboplastin time (APTT), prothrombin time (PT),
international normalized ratio (INR) for PT.
o Clinical Chemistry: Albumin, urea, creatinine, direct, indirect and total bilirubin, uric acid,
cholesterol (total), triglycerides, total protein, total amylase, lipase, alkaline phosphatase
(ALP), creatine phosphokinase (CPK), AST, ALT, gamma-glutamyl transferase (GGT), lactate

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 84 of 142
dehydrogenase (LDH), phosphate, sodium, potassium, calcium (corrected for albumin),
chloride, random/fasting glucose, bicarbonate/CO2.
o Urinalysis; pH, specific gravity, protein, glucose, micro-albumin, ketones, bilirubin,
creatinine, nitrite, sodium, urobilinogen, blood, leukocytes, microscopy.
12-lead ECG: Normal, Abnormal, Heart rate, PR interval, QT, QTc (QTcB and QTcF), QRS.
Physical Examination. Height is measured at screening only.
Ophthalmological Exam [to include visual acuity, fundoscopy and slit lamp examination.
Vital signs: supine systolic and diastolic blood pressure (SBP and DBP), heart rate, axillary body
temperature, weight, calculated body mass index (BMI).
Adverse Events.
Concomitant medication.
6.5. Mycobacterial Characterization
The following characteristics of the participants’ MTB strains will be assessed:
MIC of PA-824, TMC207 and moxifloxacin.
Drug susceptibility testing of the M. tuberculosis isolates with MGIT system for sensitivity to
isoniazid, rifampicin, ethambutol and pyrazinamide.
Speciation of the infecting organism by PCR.
Due to the complexities involved with the above mycobacteriological testing, which can result in
repeated testing being required in order to produce a result, the above tests will be performed
once. If a result is not available from this test it will be repeated once. Should this repeated test
also not produce a result the sample will be reported as “no result available”.
7. TRIAL PROCEDURES
7.1. Sputum Sampling
7.1.1. Spot Sputum
Purpose 1:
Determining participant eligibility: Direct microscopy for acid-fast bacilli. This serves as
confirmation of MTB infection prior to randomization and ensures adequate bacterial load (at
least 1+ on the IUATLD/WHO scale (Appendix 3).
Purpose 2:

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 85 of 142
Determining participant eligibility: Rapid test for isoniazid and rifampicin resistance.
Sample Collection Methodology:
The spot sputum sample collection methodology and requirements will be described in a
separate document, The Laboratory Manual, which will be provided to the site prior to trial
start.
Spot sputum samples are to be collected in the appropriate clean, 40 ml sterile containers
supplied. The screw caps must be fitted tightly to avoid leakage. The spot specimens should
have a volume of 3-5 ml. In defining a good sputum specimen, satisfactory quality implies the
presence of mucoid or mucopurulent particles. This is of greater significance than quantity.
The samples will be sent via courier to the designated trial mycobacteriology laboratory for
handling and assessment.
Testing performed by:
Trial appointed mycobacteriology laboratory.
7.1.2. Overnight Sputum
Purpose 1:
Calculating efficacy variables: The change of sputum colony forming units of M tuberculosis
(logCFU) over time and the change of time to culture positivity (TTP) in the MGIT system over
time.
Note: Should the spot sputum results for direct microscopy for acid-fast bacilli as described in
section 5.1.1 purpose 1 be negative or insufficient, but the results from the day -2 overnight
sample be positive as required and described in Section 5.1.1 the participant may be
randomized based on these results.
Purpose 2:
Determining mycobacteriologic characteristics of the participants’ MTB strains:
o MIC of PA-824, TMC207 and moxifloxacin.
o Drug susceptibility testing of the M. tuberculosis isolates with MGIT system for sensitivity to
isoniazid, rifampicin, ethambutol and pyrazinamide.
o Speciation of the infecting organism by PCR.
Sample Collection Methodology:

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 86 of 142
The overnight sputum sample collection methodology and requirements will be described in a
separate document, The Laboratory Manual, which will be provided to the site prior to trial
start.
Overnight sputum sampling will start on Visit 2 (Day -3) and will be collected from the afternoon
and for 16 hours overnight. The 16-hour sputum sampling for each of these sampling days must
be finished prior to the administration of each day’s IMP. Each participant’s overnight sputum
samples will be collected (pooled) in an appropriate clean, 120-180 ml container. The screw
caps must fit tightly to avoid leakage. Each overnight pooled specimen should consist of
approximately 20-30 ml with a minimum of 10 ml being required. In defining a good sputum
specimen, satisfactory quality implies the presence of mucoid or mucopurulent particles. This is
of greater significance than quantity. Pooled specimens must remain at the participant’s
bedside until the 16-hour collection is completed. The samples will be sent via courier to the
designated trial mycobacteriology laboratory for handling and assessment.
Testing performed by:
Trial appointed mycobacteriology laboratory.
7.2. Clinical Laboratory Tests
7.2.1. Safety Laboratory Tests
Test Details:
Hematology: hemoglobin, hematocrit, red blood cell count, white blood cell count with
differential, platelet count.
Coagulation: APTT, PT, INR for PT.
Clinical Chemistry: Albumin, urea, direct, indirect and total bilirubin, uric acid, cholesterol
(total), triglycerides, total protein, total amylase, lipase, alkaline phosphatase (ALP), creatinine,
creatine phosphokinase (CPK), AST, ALT, gamma-glutamyl transferase (GGT), lactate
dehydrogenase (LDH), phosphate, sodium, potassium, calcium (corrected for albumin), chloride,
random/fasting glucose, bicarbonate/CO2. If total amylase results are Grade 2 or higher, further
testing of pancreatic amylase and trypsin-like immunoreactivity should be considered after
consultation with the Sponsor Medical Monitor. Note: It is preferable for a fasting glucose to
be obtained at screening. However if the participant arrives at the screening visit not in a

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 87 of 142
fasting condition, a random glucose may be done. If the resulting glucose is significantly
elevated (>11.1 mmol/L) the following procedures must be followed during screening:
o The investigator is to question the participant about possible clinical symptoms and look for
possible clinical signs of diabetes.
o The urinalysis glucose results are also to be taken into consideration.
If based on these results, it is the opinion of the investigator that the participant may have a
significant endocrine abnormality the participant should not be enrolled (screening failure).
Urinalysis: pH, specific gravity, protein, glucose, microalbumin, ketones, bilirubin, creatinine,
nitrite, sodium, urobilinogen, blood, leukocytes, microscopy.
Purpose:
Safety Monitoring
Sample Collection Methodology:
The laboratory sample collection methodology and requirement will be described in a separate
document, The Laboratory Manual, which will be provided to the site prior to trial start.
Testing performed by:
Trial appointed safety laboratory.Other laboratory Tests
Test Details:
Pregnancy: Serum βHCG. Women of child-bearing potential only whether sexually active or not.
HIV and CD4 count. Prior to HIV testing and on receipt of the results, participants will be
counseled on HIV by trained counselors. Approval for this to be performed will be obtained
from participants in the written informed consent process.
Urine Drug Screen: cannabinoids, cocaine, amphetamines, opiates, benzodiazepines,
barbiturates.
Urine test for isoniazid.
Serum Endocrinology: testosterone, luteinizing hormone (LH), follicle-stimulating hormone
(FSH). These will be performed on male participants only. The serum endocrinology sampling (in
males, only) may be repeated in the morning (ideally 8 am) if the original results show an
isolated abnormal value (i.e., only 1 of the 3 hormones is abnormal). Only the item which was
abnormal in the original testing needs to be repeated.
Purpose:

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 88 of 142
Determining participant eligibility.
Sample Collection Methodology:
The laboratory sample collection methodology and requirement will be described in a separate
document, The Laboratory Manual, which will be provided to the site prior to trial start.
Testing performed by:
Trial appointed safety laboratory or mycobacteriology laboratory.
7.3. Plasma Pharmacokinetics (PK)
Purpose:
Determining pharmacokinetic variables.
Sample Collection Methodology:
4 ml blood sample will be collected in a vacutainer tube containing:
Lithium Heparin for TMC207 (excluding TMC207 plus PA-824) Containing Treatment Arms;
K2 EDTA for PA-824 (excluding TMC207 plus PA-824) Containing Treatment Arms;
The TMC207 plus PA-824 Containing Treatment Arm will be collected in vacutainers containing:
o Lithium Heparin for TMC207 identified time points (Flow Chart Section 1.3);
o K2 EDTA for PA-824 identified time points (Flow Chart Section 1.3).The sample will be
centrifuged, plasma separated, and stored in a freezer set at -80°C. The overall duration of
sample processing, from collection to freezing, should be less than 2 hours. The following
processing, storage, and shipping procedures should be followed:
1. After obtaining the 4 ml blood sample, mix collection tube thoroughly by slowly inverting the
collection tube several times.
2. Within 60 minutes of collection, process collection tubes in a centrifuge set at approximately
3000 RPM for 10 minutes at room temperature.
3. Transfer a 1.0 ml aliquot of plasma using standard laboratory technique into each of two
appropriately-labeled tubes (or the remaining plasma if 1.0 ml is not available). These aliquots
will be designated for the PK testing.
4. Tube labels will be provided by the designated laboratory. Labels should be secured to each
storage tube so they do not come off during storage at -80°C or shipping on dry ice. Labels will
contain the following information: Protocol number; site number, participant number; and time
point of sample collection (e.g., 5 hours post-dose).
5. Samples will be stored in an upright position at -80°C until shipped.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 89 of 142
6. Plasma samples for PK determinations will be couriered in batches to the designated laboratory
in two separate shipments packed in dry ice sufficient to last for 3 days. The first set of aliquots
from all participants will be sent to the designated laboratory first. Once the lab confirms the
first shipment has arrived in good condition, the second set of aliquots will be sent. Shipments
should be made by overnight courier.
Testing performed by:
Sponsor-appointed pharmacokinetics laboratories.
7.4. Electrocardiography (ECG)
Purpose:
Safety monitoring (please refer to PK-ECG flow charts in section 1.3) for immediate on-site
safety assessment and central evaluation at prospectively defined time points.
For the designated on-site safety assessment ECGs (section 1.3.), the site staff will immediately
check the ECG readings performed by the provided ECG machine for QT prolongation safety
margins, defined for this purpose as a QTc interval of 470msec or an increase of 40msec from
baseline (for PA-824 (excluding TMC207 plus PA-824) containing treatment arms, the comparison
will be to the time-matched pre-treatment ECG). If these safety margins are not met, the site staff
will immediately notify the designated appropriately trained physician who will manually read the
ECG tracing in question to determine if the QTc prolongation criteria for patient withdrawal (see
section 9.7) have been met.
In addition, prior to dosing on each treatment day, clinical assessment of the previous day’s safety
ECGs are to be performed for safety monitoring (section 9.7) by the designated appropriately
trained physician. When ECGs are obtained in triplicate, the first recorded ECG should be used for
the on-site QT safety evaluation. The investigator’s interpretation of each safety ECG will be
recorded in the CRF. For PA-824 containing treatment arms, the post-treatment ECG evaluations
will be compared to the time-matched pre-treatment ECGs. All clinically significant changes must
be accompanied by further specification in the CRF.
If the initial physician-conducted review of the post-treatment ECGs suggests that the corrected QT
interval (corrected QTcB and QTcF) has increased by more than 60 msec, additional ECGs will be
collected at 15 minute intervals until the corrected QT interval has returned to baseline or the
increase has been satisfactorily explained. However, because this assessment is based on an initial
(on-site) impression prior to centralized measurement by the centralized ECG laboratory, an

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 90 of 142
increase in corrected QT interval of more than 60 msec need not be reported as an AE unless it is
accompanied by signs or symptoms of myocardial ischemia or developing cardiac arrhythmia or
that the overall value is above 500 msec.
Methodology:
ECGs will be recorded for 10 seconds. Timing and registration technique for ECGs will be
standardized for all participants. Participants should be lying down (recumbent) for at least 5
minutes prior to each 12-lead ECG evaluation. ECGs will be performed as single (TMC207
(including TMC207 plus PA-824) and Rifafour e275® containing treatment arms) or in single or
triplicate (PA-824 (excluding TMC207 plus PA-824) containing treatment arms). Please refer to
Section 1.3 PK and ECG Chart. The inter-ECG interval being 30 to 180 seconds for triplicate
ECGs. For each participant, the ECGs should to every extent possible be collected at
approximately the same time of day and in the same fed/fast state (e.g. 4 hours after lunch).
7.5. Ophthalmology Examinations
Purpose:
Safety monitoring and participant eligibility.
Methodology:
The same ophthalmologist should perform the Visit 1 (Day -9 to -4) and Visit 22 (Day
104±14days) ophthalmology examination for a given participant, if at all possible. A limited
number of ophthalmologists will be involved in the study to reduce observer variability. The
exception to this is the fundoscopy which is performed by the Investigator at Visit 19 (Day 15)
and Visit 21 (Day 42) or at early withdrawal.
An ophthalmologic medical history will be obtained from the participant.
One or all of the following examinations will be performed per the timelines in section 1.3 at
the designated visit:
o Visual acuity test and slit lamp examination after dilatation.
o Fundoscopy (to be performed by the Ophthalmologist at Screening and by the
Investigator at subsequent visits).
The slit lamp examination results will be documented using the AREDS2 Clinical Lens Opacity
Classification and Grading System(35-37). With this system the lens will be evaluated and graded
by comparison to a series of standard photographs for each possible type of lens opacity, the

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 91 of 142
location and degree of lens opacity being determined. The possible type of lens opacity and its
grading will be documented. The possible lens opacity types are nuclear (9 possible grades),
cortical (9 possible grades) and posterior subcapsular (PSC) (9 possible grades). The
ophthalmologists will be trained on the AREDS2 clinical lens classification and grading system
prior to performing the study ophthalmologic examinations.
AREDS2 Lens Opacity Classification and Grading System Methodology:
The AREDS2 Clinical Lens Opacity Classification and Grading System methodology and
requirements will be described in a separate document which will be provided to the study
ophthalmologists prior to trial start.
7.6. Physical Examinations and Vital Signs
Purpose:
Safety monitoring.
Methodology:
The investigator will conduct a complete physical examination and any clinically significant
findings will be recorded. Complete physical exam includes a history of menstrual cycle (e.g.
possible irregularities, missed cycles) at the screening visit.
Systolic and diastolic blood pressure (mmHg) to be measured supine (after 5 minutes of rest)
using an appropriately sized cuff, and using the same type of sphygmomanometer, if possible by
the same observer, at each relevant visit.
Heart rate (bpm).
Axillary body temperature (ºC).
Weight (kg) (in light clothing and with no shoes).
Height (m) only at Visit 1 (Day -9 to -4)
To be performed within 2 hours before dosing on Days 1 through 14 and within 2 hours before the
time dosing would have occurred on Day 15. For the TMC207 plus PA-824 treatment arm these are
to be performed within 2 hours before the PA-824 dose.
7.7. Chest X-ray
Purpose:

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 92 of 142
Determining participant eligibility: Chest x-ray for confirmation of radiological picture
compatible with TB.
Methodology:
A chest x-ray picture will be obtained from the clinic appointed radiology department. The
investigator is responsible for its review and analysis.
7.8. Retention Sample Collection
Purpose:
Additional plasma, serum, urine and sputum collections will be made on certain days when
samples are collected for other protocol-specified activities. These collections will be stored for
possible future analysis (e.g., for TMC207 or PA-824 metabolism). These samples will not be
used for genetic testing. The collection of these samples will be described in the informed
consent document.
7.8.1. Plasma and Serum
Methodology:
The laboratory sample collection methodology and requirement will be described in a separate
document, The Laboratory Manual, which will be provided to the site prior to study start. The
plasma and serum samples will be collected, handled and couriered to the designated archive
laboratory.
The designated archive laboratory will be determined prior to study start and all contact and
shipping details provided to the sites.
7.8.2. Sputum
Methodology:
If sufficient overnight sputum is available, the microbiology laboratory will collect up to three
1-ml aliquots from the overnight sputum samples collected by the investigational site. The
investigational site will not be required to process these retention samples in any way. The
microbiology laboratory will sample, freeze and ship the aliquots to the designated archive
laboratory.
The designated archive laboratory will be determined prior to study start and all contact and
shipping details provided to the laboratory.
7.8.3. Urine

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 93 of 142
Methodology:
The urine sample collection methodology and requirement will be described in a separate
document, The Laboratory Manual, which will be provided to the site prior to study start.
The designated archive laboratory will be determined prior to study start and all contact and
shipping details provided to the sites.
8. ADVERSE EVENTS
The Investigators are responsible for eliciting adverse events by observing the participant and
recording all adverse events observed by him/her or reported by the participant during the trial.
8.1. Definitions
8.1.1. Adverse Event (AE)
Any untoward medical occurrence in a patient or clinical investigation subject administered a
pharmaceutical product and which does not necessarily have a causal relationship with this
treatment. An adverse event can therefore be any unfavorable and unintended sign (including an
abnormal laboratory finding), symptom, or disease temporally associated with the use of a
medicinal product, whether or not related to the medicinal product.
8.1.2. Serious Adverse Event (SAE)
Any untoward medical occurrence that at any dose:
results in death;
is life threatening (any event in which the subject was at risk of death at the time of the event; it
does not refer to an event, which hypothetically might have caused death if it were more
severe);
requires inpatient hospitalization or prolongation of existing hospitalization;
results in persistent or significant disability/incapacity;
is a congenital anomaly/birth defect; or
is a medically important event.
Note:
Medical and scientific judgment should be exercised in deciding which is a medically important
event that may not be immediately life-threatening or result in death or hospitalization, but may
jeopardize the participant or may require medical or surgical intervention to prevent one of the
outcomes listed above. Examples of such events are intensive treatment in an emergency room or

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 94 of 142
at home for allergic bronchospasm, blood dyscrasias or convulsions that do not result in
hospitalization, or the development of drug dependency or drug abuse. A “suspected transmission
of infectious agent by a medicinal product” is also considered a serious adverse event under the
SAE criterion “Other medically important condition”.
8.1.3. Unlisted (Unexpected) Adverse Event
An adverse reaction, the nature or severity of which is not consistent with the applicable product
information (e.g., Investigator’s Brochure for an unapproved investigational product or package
insert/summary of product characteristics for an approved product).
8.1.4. Life Threatening
Any event in which the subject was at risk of death at the time of the event; it does not refer to an
event, which hypothetically might have caused death if it were more severe.
8.1.5. Associated With the Use of the Drug
An adverse event is considered associated with the use of the drug if the attribution is possible,
probable or very likely.
8.2. Attribution Definitions
The following definitions for rating relatedness will be used:
Not Related: An adverse event, which is not related to the use of the drug.
Doubtful: An adverse event for which an alternative explanation is more likely, e.g.,
concomitant drug(s) or concomitant disease(s), and/or the relationship in time suggests that a
causal relationship is unlikely.
Possible: An adverse event, which might be due to the use of the drug. An alternative
explanation, e.g., concomitant drug(s) or concomitant disease(s), is inconclusive. The
relationship in time is reasonable; therefore the causal relationship cannot be excluded.
Probable: An adverse event, which might be due to the use of the drug. The relationship in time
is suggestive, e.g., confirmed by dechallenge. An alternative explanation is less likely, e.g.,
concomitant drug(s) or concomitant disease(s).
Very Likely: An adverse event, which is listed as a possible adverse reaction and cannot be
reasonably explained by an alternative explanation, e.g., concomitant drug(s) or concomitant
disease(s).
The definitions for rating severity will be based on the DMID Adult Toxicity Table (Appendix 4).

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 95 of 142
8.3. Reporting
8.3.1. Adverse Event Reporting
Adverse events will be collected by the Investigator from the time a participant signs the Informed
Consent Form through to:
for TMC207 (excluding TMC207 plus PA-824) containing treatment arms:
all AEs and SAEs - the end of Visit 21 (Day 42);
for PA-824 (excluding TMC207 plus PA-824) containing treatment arms:
all AEs and SAEs - the end of Visit 20 (Day 28);
only ophthalmologic related adverse events and all serious adverse events - from Visit 20
(Day 28) through to the end of Visit 22 (Day 104);
for TMC207 plus PA-824 and Rifafour e275® treatment arms:
all AEs and SAEs - the end of Visit 21 (Day 42);
only ophthalmologic related adverse events and all serious adverse events - from Visit 21
(Day 42) through to the end of Visit 22 (Day 104).
Any AE (serious or non-serious) observed by the Investigator or reported by the participant will be
recorded on the Adverse Event Case Report Form. Diagnosis of the AE, if possible, should be
recorded by the Investigator. In the case where an overall diagnosis cannot be made, each specific
sign and/or symptom should be recorded as individual AEs.
The Investigator will review each AE and assess its relationship to drug treatment based on all
available information at the time of the completion of the case report form. Each sign or symptom
reported will be graded on a 4-point severity scale using the definitions according to the DMID
Adult Toxicity Table November 2007 (Appendix 4). Additionally, the date and time of onset,
relationship to IMP, duration, action taken, and outcome (recovered with sequelae, recovered
without sequelae, persisting, fatal, or unknown (lost to follow-up)) of each event will be noted.
8.3.2. Serious Adverse Event Reporting
Any AE that occurs which is serious must be reported by the Investigator to the study monitor and
copied to the Sponsor Medical Monitor within 24 hours of the site first being aware of the SAE,
whether or not the serious event is deemed drug-related.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 96 of 142
In addition, the Investigator will provide a detailed, signed, written, and complete SAE report form
that addresses the Investigator’s estimates of the relationship to the study drug and seriousness of
the AE in question to the study monitor within 24 hours of becoming aware of the SAE.
The study monitor will confirm receipt of the SAE Form with the Investigator and review the initial
information on the SAE for diagnosis, consistency and completeness of data.
For submission of updated or additional information on a previously reported SAE, the Investigator
will provide the study monitor with a newly completed Serious Adverse Event Form, designated as
a follow-up report. This will be submitted to the study monitor within 24 hours of the Investigator
receiving the information.
The study monitor will query for additional information from the Investigator, if necessary, to
complete the profile of the SAE reported.
The Investigator/designee will inform Regulatory Authorities and/or IEC/IRB of all SAEs in
accordance with local requirements and ICH guidelines for GCP.
The sponsor/designee will forward Safety Notification letters to Investigator for submission to the
IEC/IRB.
8.3.3. Follow-up of Adverse Events
During and following a participant’s participation in a clinical trial, the Investigator must ensure that
adequate medical care is provided to the participant for all AEs, including significant laboratory
values related to the trial. The Investigator should inform the participant when medical care is
needed for any AEs he/she becomes aware of.
All AEs must be followed until satisfactory clinical resolution or stabilization or until the end of the
follow-up period, and until all queries on these AEs have been resolved. Certain long-term AEs
cannot be followed until resolution within the setting of this protocol; in these cases follow-up will
be the responsibility of the treating physician. However, this has to be agreed upon with the
sponsor.
8.3.4. Follow-up of Post Trial Adverse Events
Any new SAEs reported by the participant to the Investigator that occur after the last scheduled
contact, and are determined by the Investigator to be possible, probable or very likely related to

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 97 of 142
the use of the IMP, will be reported to the sponsor, IEC/IRB and regulatory authorities on an
expedited basis as required in accordance with local requirements and ICH guidelines for GCP.
8.3.5. Clinical Laboratory Adverse Events
Changes in the results of the Clinical Laboratory assessment results which the investigator feels are
clinically significant will be reported as adverse events. It is the investigators’ responsibility to
review the results of all laboratory tests as they become available. This review must be
documented by the investigators’ dated signature on the laboratory report. For each abnormal
laboratory test result, the investigator needs to ascertain and document if this is a clinically
significant change from baseline for that individual participant. This determination, however, does
not necessarily need to be made the first time an abnormal value is observed. The investigator may
repeat the laboratory test or request additional tests to verify the results of the original laboratory
tests. If this laboratory value is determined by the investigator to be a clinically significant change
from baseline for that participant, it is considered to be an adverse event.
8.3.6. Pregnancy
During the trial and for 1 week (PA-824 (excluding TMC207 plus PA-824) and Rifafour e275®
containing treatment arms) or 6 months (TMC207(including TMC207 plus PA-824) containing
treatment arms) after last dose of IMP, all women of childbearing potential will be instructed to
contact the Investigator immediately if they suspect they might be pregnant (for example, missed
or late menses). If pregnancy is suspected while the participant is receiving IMP, the IMP will be
withheld immediately until the result of the pregnancy test is known. If pregnancy is confirmed,
the IMP will be permanently discontinued in an appropriate manner and the participant withdrawn
from the trial.
The Investigator will immediately notify the sponsor of any pregnancy associated with IMP
exposure. The pregnancy will be recorded on the Pregnancy form and forwarded to the sponsor.
Protocol-required procedures for trial discontinuation and follow-up will be performed unless
contraindicated by the pregnancy. In addition, the Investigator will report to the sponsor follow-up
information regarding the outcome of the pregnancy, including perinatal and neonatal outcome.
Infants will be followed for 6 months. SAE reporting will also occur if the pregnancy outcome is a
congenital anomaly.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 98 of 142
Should the female partner of a male participant become pregnant during the study or in the 12
weeks after the completion of IMP and the Investigator becomes aware that this situation has
occurred, the Investigator will immediately notify the sponsor of any pregnancy associated with
IMP exposure. Consent will be requested from the female partner for collection of information on
her pregnancy history and for information on the current pregnancy and birth. If consent is
obtained, the pregnancy will be recorded on the Pregnancy forms and forwarded to the sponsor
and the Investigator will report to the sponsor information regarding the course of the pregnancy
including perinatal and neonatal outcome. Infants will be followed for 6 months. SAE reporting will
also occur if the pregnancy outcome is a congenital anomaly.
8.3.7. Disease Under Study
Symptoms of the disease under study (TB) experienced by the participant whilst on the study will
be assessed by the Investigator. If the symptom has worsened whilst the participant is in the study,
and the Investigator assesses it as clinically significant, it will be recorded as an adverse event. If
there is no change and the Investigator assesses the symptom as due to the participant’s TB and
not clinically significant, it will not be recorded as an AE and this will be noted in the participant’s
source documentation. If the Investigator is unsure as to whether the symptom is clinically
significant or not, it is to be classified as significant and reported as an AE.
All TB related symptoms that meet SAE criteria will be recorded and reported as a SAE.
9. MONITORING AND SAFETY FOR SPECIFIC TOXICITIES
AEs still ongoing at the end of treatment in the trial will be followed until satisfactory clinical
resolution or stabilization or until the end of the follow-up period, and until all queries on these AEs
have been resolved. The exception to this is all grade 3 and grade 4 laboratory abnormalities and
laboratory abnormalities resulting in an increase of 2 grades from baseline will be followed until
return to baseline or within one grade from baseline.
Note: For grade 3 or 4 laboratory toxicities, participants should have a confirmatory measurement
within 48 hours where possible. This management scheme is for confirmed lab abnormalities and
not for isolated events
Monitoring for specific toxicities is based upon target organs defined in preclinical toxicity studies
(see Investigator’s Brochures).
9.1. ALT and AST

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 99 of 142
Management will be at the discretion of the Investigator, according to generally accepted medical
practice standards.
Grade 1 (> 1.0 to < 2.0 x ULN), Grade 2 (≥ 2.0 to < 3.0 x ULN) AST or ALT elevation:
Patients may continue IMP. Patients should be followed until resolution (return to baseline) or
stabilization of AST/ALT elevation.
Grade 3 (≥ 3.0 to ≤ 8.0 x ULN) or Grade 4 (> 8.0 x ULN ) AST or ALT elevation:
Patients will permanently discontinue IMP and be withdrawn from the trial. It is recommended
that the investigator contacts the sponsor to discuss the case of AST or ALT elevation. Patients
should be followed until resolution (return to baseline) or stabilization of AST/ALT elevation.
9.2. Amylase elevation
Grade 1 (> 1.0 to ≤ 1.5 x ULN) or Grade 2 (> 1.5 to ≤ 2.0 x ULN):
Participants may continue IMP and should be carefully evaluated and followed closely.
Grade 3 (> 2.0 to ≤ 5.0 x ULN) or Grade 4 (> 5.1 x ULN):
Further testing of pancreatic amylase and trypsin-like immunoreactivity should be considered after
consultation with the Sponsor Medical Monitor.
9.3. Pancreatic amylase and/or lipase elevation
Management will be at the discretion of the Investigator, according to generally accepted medical
practice standards.
Grade 1 (> 1.0 to ≤ 1.5 x ULN) or Grade 2 (> 1.5 to ≤ 2.0 x ULN):
Participants may continue IMP and should be carefully evaluated and followed closely.
Grade 3 (> 2.0 to ≤ 5.0 x ULN) or Grade 4 (> 5.0 x ULN):
For asymptomatic grade 3 pancreatic amylase elevations with no history or concomitant disease of
pancreatitis, participants may continue IMP but should be carefully evaluated and followed closely.
For confirmed grade 4 elevations of pancreatic amylase and confirmed grade 3 or 4 elevations of
lipase, participants will permanently discontinue IMP and be withdrawn from the trial.
9.4. Trypsin-like immunoreactivity ≥ 1.5 times ULN

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 100 of 142
Participants should be followed closely. If there is clinical evidence of pancreatitis and elevation of
lipase and amylase of grade 3 or higher, participants will permanently discontinue IMP and be
withdrawn from the trial.
9.5. Musculo-skeletal System and Cardiac Muscle
Myalgia
Grade 1 (mild with no limitation of activity):
Participants may continue IMP and should be carefully evaluated and followed closely.
Grade 2 (muscle tenderness at site other than injection site or with moderate impairment of
activity) or Grade 3 (severe muscle tenderness with marked impairment of activity) or Grade
4 (frank myonecrosis):
Participants will permanently discontinue IMP and be withdrawn from the trial. CPK should be
fractionated for CPK-MB subunit.
Participants having grade 3 (3.1 to 6 x ULN) or grade 4 (> 6 x ULN) elevation in CPK-MB subunit will
permanently discontinue IMP and be withdrawn from the trial when grade 3 or 4 CPK –MB
elevation is accompanied by ECG and clinical evidence of ischemic heart disease.
9.6. LDH and LDH-isoenzymes
For participants with LDH elevation >2.5 x ULN, LDH isoenzymes will be assessed.
9.7. Cardiac Rhythm Disturbances
Grade 1 (asymptomatic) or Grade 2 (asymptomatic, transient rhythm abnormality not
requiring any treatment) cardiac rhythm disturbances:
Participants may continue IMP and should be carefully evaluated and followed closely.
Grade 3 (recurrent, persistent, symptomatic arrhythmia requiring treatment) or Grade 4
(unstable dysrhythmia requiring treatment) cardiac rhythm disturbances:
Participants will permanently discontinue IMP and be withdrawn from the trial.
QTc prolongation
A participant should be withdrawn if they have a QTcB and QTcF interval greater than 500 msec; or
an increase from baseline values greater than 60 msec, present on repeated ECGs and exceeding

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 101 of 142
the values 430 msec (for males) and 450 msec (for females) and accompanied with clinically
relevant T-wave morphology changes. For PA-824 containing treatment arms (excluding the
TMC207 plus PA-824 arm), the post-treatment ECG evaluations will be compared to the time-
matched pre-treatment ECGs.
If the initial review of the post-treatment ECGs suggests that the corrected QT interval (corrected
QTcB and QTcF) has increased by more than 60 msec, additional ECGs will be collected at 15 minute
intervals until the corrected QT interval has returned to baseline or the increase has been
satisfactorily explained. However, because this assessment is based on an initial (on-site)
impression prior to centralized measurement by the centralized ECG laboratory, an increase in
corrected QT interval of more than 60 msec need not be reported as an AE unless it is accompanied
by signs or symptoms of myocardial ischemia or developing cardiac arrhythmia or that the overall
value is above 500 msec.
9.8. Gastrointestinal System
Participants with Grade 4 elevation of gastrointestinal parameters will not be withdrawn from the
trial, however should be monitored closely.
9.9. Other toxicities
Grade 1 or 2
Participants who develop grade 1 or 2 AE or laboratory toxicity may continue intake of IMP.
Grade 3 or 4
Participants who develop grade 3 or 4 AE or laboratory toxicity (see Appendix 4 for specifics) will be
carefully evaluated by the Investigator. Participants may continue intake of IMP or be withdrawn
from the trial if, in the opinion of the Investigator, the AE or laboratory toxicity poses a significant
risk for the subject in case of continued participation in the trial. Participants should be followed as
appropriate until resolution of the AE or toxicity.
10. STATISTICAL ANALYSIS
There are no interim analyses planned for this study. All randomized participants will be included in
the analyses. Additional details of the analyses specified in this section will be provided in the
statistical analysis plan (SAP) and/or the clinical study report.
10.1. Sample Size

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 102 of 142
This is a Phase II trial to investigate the EBA and safety, tolerability and PK of TMC207 alone,
TMC207 plus pyrazinamide, TMC207 plus PA-824, PA-824 plus pyrazinamide and PA-824 plus
pyrazinamide and moxifloxacin. The planned sample size of 15 participants per treatment group is
in keeping with other Phase II trials of this type and accounts for the possibility of up to 3 drop-outs
per arm, which based on previous studies of this type conducted at these sites, represents a
conservative estimate of the expected drop-out rate. Previous EBA studies indicate that the
between participant standard deviation of logCFU can be approximately 0.2. Therefore, assuming
similar variability in the new trial the expected standard errors of group mean EBA and
corresponding width of 95% confidence intervals are in Table 6.
Table 6: Expected Standard Errors of Group mean log CFU and confidence intervals
Group Size Standard Error Width of 95%CI
15 0.052 0.101
10 0.063 0.124
10.2. Primary Efficacy Analysis
The primary efficacy analysis will be the rate of change in CFU over the period Day tx-ty, with tx
being Day 0 and ty being Day 14, which may be described with linear, bi-linear or non-linear
regression of logCFU over time.
The values of EBA(tx - ty) will be calculated using the following formula:
EBA(tx -ty)=[mean logCFU(Day tx)- logCFU(Day ty)]/ (ty- tx)
It is important to note that the regression may be non-linear, i.e. to accommodate the possibility of
varying rate of change. One form of non-linear regression to be considered is bi-linear in which the
trend in logCFU is linear with a certain slope over the period Day tx to Day , and then linear with a
different slope over Day to Day ty; note that is a parameter to be estimated. Some other form
of non-linear regression may be fitted. Whatever its form, bi-linear or otherwise, the essential
measure of efficacy is the rate of change described over the period Day x-y. Fitting of all regression
relations will be by the method of least squares. Regressions will be fitted to the data of individual
participants and then appropriately averaged to estimate population trends and to compare the
treatment groups. This is, in effect, a non-linear random effects model approach.
10.3. Secondary Efficacy Analysis

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 103 of 142
The secondary efficacy endpoints are:
The EBA (EBA0-2, EBA2-14 and EBA7-14) as determined by the rate of change of logCFU in
sputum over the period Day 0 to Day 2, Day 2 to Day 14 and Day 7 to Day 14, in participants,
which may be described with linear, bi-linear or non-linear regression of logCFU on time.
The time to sputum culture positivity (TTP) (TTP0-2, TTP0-14, TTP2-14, and TTP7-14) in the
Mycobacterium Growth Indicator Tube (Bactec MGIT960) system as determined by the rate
of change in TTP in sputum over the periods Day 0 to Day 2, Day 0 to Day 14, Day 2 to Day
14 and Day 7 to Day 14 in participants, which may be described with linear, bi-linear or non-
linear regression of TTP on time.
These EBAs will be based on the fitted regressions as described above, and their summary statistics,
for example means and confidence limits, will be obtained.
Time to positivity (TTP) in the MGIT system will be recorded for the same time points as the logCFU.
Serial TTP measurements are a relatively new and not fully validated method for the assessment of
the change of the viable bacterial load on sputum over time. Because of its technical simplicity it is
anticipated that TTP might replace logCFU in future trials so that collecting such data for this trial is
essential. In analogy to the primary outcome parameter described above, linear or some other
form of non-linear regression will be fitted by the method of least squares.
In addition, post hoc analyses of the EBA and TTP data over time periods other than those stated
above (0-2, 0-14, 2-14 and 7-14) may be performed.
10.4. Pharmacokinetic Analysis
The following PK parameters will be estimated for TMC207, TMC207 metabolite M2, PA-824,
pyrazinamide and moxifloxacin from participants’ individual plasma concentrations (except in the
Rifafour e275® treatment arm) by applying a noncompartmental approach using PK software such
as WinNonlin Professional. Concentration values reported as below the limit of quantitation will be
set to zero. Each parameter will be calculated separately using plasma concentrations of TMC207,
TMC207 metabolite M2, PA-824, pyrazinamide or moxifloxacin. PK parameters will be estimated for
TMC-207 treatment arms on Day 14 only and for PA-824 on Days 1, 8 and 14.
Cmax: Maximum observed plasma drug concentration.
Tmax: Time at which Cmax is observed (obtained without interpolation).
Cmin: Minimum observed plasma drug concentration 24 hours following the last dose.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 104 of 142
AUC(0-24):Area under the drug concentration-time curve calculated using linear trapezoidal
summation from time zero to time 24 hours.
Descriptive statistics including mean, standard deviation, coefficient of variation, median, minimum
and maximum, geometric mean and geometric mean CV% will be computed for each PK parameter
by group. Grouping will be by gender.
In addition, mean and median concentration-versus-time graphs will be provided (with error bars as
appropriate).
10.5. Pharmacokinetic and Pharmacodynamic Statistical Analysis
For the TMC207 and PA-824 containing treatment arms, the following PK-PD correlations will be
calculated and presented for TMC207, PA-824, pyrazinamide and moxifloxacin:
EBAs versus the following PK variables (Day 14 only) separately and by gender:
Cmax;
AUC(0-24) ;
Time over MIC (for PA-824, TMC207 and moxifloxacin).
For each of the above the following will be presented:
A plot of all the individual values for each PK-PD comparison on an X vs. Y graph, with r
(Pearson’s correlation coefficient) noted.
Tables including a simple r.
10.6. Safety and Tolerability Analyses
All adverse events will be coded using the Medical Dictionary for Regulatory Activities
(MedDRA) and will be presented by Preferred Term within each MedDRA System Organ Class
(SOC).
The incidence of the following events will be summarized by treatment group for further
medical analysis:
o Treatment-emergent adverse events (TEAEs) by severity.
o Potentially IMP-related TEAEs.
o TEAEs with a fatal outcome.
o Serious TEAEs.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 105 of 142
o Discontinuations due to TEAEs.
Other safety variables: Laboratory Parameters, Physical Examination, Ophthalmological Exam,
Vital signs (see Appendix 5), Concomitant medication. Descriptive summary statistics will be
presented.
Cardiovascular Safety (see Appendix 5). ECGs will be centrally read. QT intervals will be
adjusted using Fridericia’s correction and Bazett’s correction. QT/QTc values and changes from
pre-dose (average of Screening and Day 1 values) at each time point will be summarized using
descriptive statistics by group and time of collection. For TMC207 and PA-824 containing
treatment arms, the potential correlations between the plasma concentration of IMP and the
change from baseline of QT interval corrected by Fridericia’s method (QTcF) and change from
baseline of QT interval corrected by Bazett’s method (QTcB) with respect to time for the
different treatment groups will be explored. These will be presented as descriptive analyses,
and no inferential tests will be carried out.
o Post-baseline QT/QTc intervals will be classified into the following categories:
o QT/QTc < 450 msec
o 450 msec < QT/QTc < 480 msec
o 480 msec < QT/QTc < 500 msec
o QT/QTc > 500 msec
o QTc changes from baseline will be classified into the following categories:
o increase < 30 msec,
o > 30 msec and < 60 msec, and
o increase > 60 msec.
Frequency counts will be used to summarize the number of participants at each time point
according to the above categories.
o ECG results will be classified as normal or abnormal and summarized using frequency counts
by dose group and time of collection.
10.7. Mycobacteriology Characterization

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 106 of 142
Descriptive summary statistics of the mycobacterial characteristics of the participants’ MTB strains
assessed will be presented.
11. RECORDS MANAGEMENT
11.1. Data Collection
All CRF pages will be completed for each participant who receives any amount of IMP. For
screening failure participants a screening failure CRF will be completed. For participants who are
prematurely withdrawn, the visits up to withdrawal plus the withdrawal and follow-up visits need
to be completed.
11.2. Source Documents
Source data are defined as all information in original records and certified copies of original records
of clinical findings, observations, or other activities in a clinical trial necessary for the reconstruction
and evaluation of the trial. Source documents will include, but are not limited to, progress notes,
electronic data, screening logs, and recorded data from automated instruments.
All source documents pertaining to this trial will be maintained by the Investigators. The
Investigator has to permit trial-related monitoring, audits, Independent Ethics
Committee/Institutional Review Board (IEC/IRB) review and regulatory inspections providing
authorized persons direct access to source data/documents.
11.3. File Management at the Trial Center
It is the responsibility of the Investigators to ensure that the trial center files are maintained in
accordance with the Guidelines for Good Practice in the Conduct of Clinical Trials in Human Patients
in South Africa 2006 (SA GCP), International Conference on Harmonization Good Clinical Practice
(ICH GCP) and the ethical principles that have their origin in the Declaration of Helsinki.
11.4. Records Retention at the Trial Center
The Principal Investigator is obliged to retain records and data from the trial for safety reasons and
for audit and inspection subsequent to trial completion. The essential documents should be
retained for not less than 5 years after the last approval of a marketing application and until there
are no pending or contemplated marketing applications or at least 5 years have elapsed since the
formal discontinuation of clinical development of the IMP.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 107 of 142
The sponsor will make financial provisions for the Investigator to deposit the documents at an
external site for safekeeping for as long as required by regulations and the sponsor.
12. QUALITY CONTROL AND QUALITY ASSURANCE
12.1. Site Procedures
The Investigator undertakes to perform the clinical trial in accordance with this protocol, SA GCP,
ICH GCP and the ethical principles that have their origin in the Declaration of Helsinki.
The Investigator undertakes to complete the CRFs according to the Sponsor’s requirements, in a
timely, accurate and legible manner. Print-outs of laboratory reports and ECG tracings will be
attached to the applicable page of the CRF. CRF entries will be verifiable to source documentation
other than the CRF.
Site Standard Operating Procedures will be adhered to for all clinical and bioanalytical activities
relevant to the quality of the study. Participant compliance will be monitored throughout the study
via procedures such as a Screening questionnaire to review inclusion and exclusion criteria, urine
drug and alcohol screens at Screening, mouth check following dosing, and confinement for conduct
of all procedures with clinical research staff on site at all times.
The Investigator will sign and date any analysis results (e.g. laboratory, ECG, etc.) to verify that the
results have been reviewed.
The Investigator may appoint other sub-investigators to assist with the study. However the
Investigator maintains responsibility for the study and will supervise the sub-investigators. Written
IEC/IRB approval will be obtained prior to involvement in the study.
The Investigator will ensure that all site personnel are adequately trained in SA GCP, ICH GCP, the
protocol, IB and all study procedures and requirements.
12.2. Monitoring
The Principal Investigator is responsible for the validity of all data collected at the clinical site and
must accept the various monitoring procedures employed by the Sponsor. The purpose of
monitoring is to verify that the rights and well being of human patients are protected; that trial
data are accurate, complete and verifiable with source data; and that the trial is conducted in
compliance with the protocol, SA GCP, ICH GCP and the ethical principles that have their origin in
the Declaration of Helsinki and the applicable regulatory requirements.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 108 of 142
Monitors assigned by the Sponsor will conduct regular site visits for the purpose of monitoring
various aspects of the study. Visits will take place usually within a predetermined interval, but this
may vary during the course of the study. The Investigator and site staff will allow the study monitor
and authorized representatives of the Sponsor to (1) inspect all CRFs, written informed consent
documents and corresponding source documents (e.g. original medical records), participant records
and laboratory raw data, and (2) access clinical supplies, dispensing and storage areas. The
Investigator and site staff should also (1) agree to assist with monitoring activities if requested and
(2) provide adequate time and space for monitoring visits.
The monitor will query any missing, confusing, spurious, or otherwise ambiguous data with the
Investigator. All queries should be resolved in a timely manner. A monitoring log will be
maintained recording each visit, the reason for the visit, the monitor’s signature and Investigator or
designee’s confirmation signature.
12.3. Auditing
For the purpose of compliance with SA GCP, ICH GCP and regulatory agency guidelines, it may be
necessary for Sponsor-authorized Quality Assurance personnel and/or authorized personnel from
an external regulatory agency to conduct an audit or inspection of the investigational site. The
purpose of an audit is to assess the quality of data with regard to accuracy, adequacy and
consistency, and to assure that studies are in accordance with the guidelines. Having the highest
quality data from studies is an essential aspect of drug development.
The Investigator and site staff will be given sufficient notice to prepare for such visits, which will
usually last between one and two days and may be conducted at any stage during the study. The
audit will involve the review of all study-related documentation required by GCP to be maintained
by each site; drug storage, dispensing and return; all study-related supplies; and source documents
against the CRFs to assure the adequacy and accuracy of the information which has been recorded,
including the verification of any AEs which have occurred.
In the event of the site being notified of a Regulatory Inspection, the Sponsor will help with
preparation. It is essential that the Sponsor be notified of the inspection as soon as possible.
13. ETHICS AND RESPONSIBILITY
13.1. Basic Principles

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 109 of 142
This research will be carried out in accordance with South African GCP guidelines Second Edition
2006, the principles enunciated in the Declaration of Helsinki (revised version of Seoul, October
2008; and the ICH harmonized tripartite guideline regarding Good Clinical Practice (E6 Consolidated
Guidance, 10 June 1996).
13.2. Independent Ethics Committee/Institutional Review Board (IEC/IRB) Review
The protocol and required study related documents will be reviewed by the sites respective
IEC/IRB. The study will not start until the IEC/IRB has approved the protocol, written informed
consent, any written information to be provided to the participant or any modification thereof, plus
any other study related documents required for review. The IEC/IRB shall be constituted and shall
operate in accordance with SA GCP guidelines Second Edition 2006, principles enunciated in the
Declaration of Helsinki (revised version of Seoul, October 2008); and the ICH GCP (E6 Consolidated
Guidance, 10 June 1996). The Investigator will maintain an accurate and complete record of all
submissions made to the IRB/IEC. The records should be filed in the Investigator’s Study File, and
copies will be sent to the Sponsor.
13.3. Regulatory Authorities
The Regulatory Authorities will receive the protocol, amendments, reports on SAEs, and the
Integrated Clinical Trial Report according to national regulations. Written approval will be obtained
from the Regulatory Authorities prior to commencement of the trial.
13.4. Informed Consent
Written informed consent will be obtained from all participants (or legally acceptable
representative) before any trial-related procedures (including any screening or pre-treatment
procedures) are performed. Investigators may discuss the availability of the trial and the
opportunity for entry with a potential participant without first obtaining consent. However,
informed consent must be obtained and documented prior to initiation of any procedures that are
performed solely for the purpose of determining eligibility for research, including withdrawal from
current medication(s). When this is done in anticipation of, or in preparation for, the research, it is
considered to be part of the research.
The Investigators have both ethical and legal responsibility to ensure that each participant being
considered for inclusion in this trial is given a full explanation of the protocol. This shall be
documented on a written informed consent form that shall be approved by the same IEC/IRB

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 110 of 142
responsible for approval of this protocol. Each informed consent form shall include the elements
required by the SA GCP and ICH GCP guidelines and must adhere to the ethical principles that have
their origin in the Declaration of Helsinki.
Once the appropriate essential information has been provided to the participant and fully explained
by the Investigators (or qualified designees) and it is felt that the participant understands the
implications of participating, the IEC/IRB approved written informed consent form will be signed
and dated by both the participant and the person obtaining consent (Investigators or designees),
and by any other parties required by the IEC/IRB.
In accordance with the SA GCP guidelines, the original signed informed consent form will be kept
with the trial records and a copy of signed informed consent form will be provided to the
participant. Another copy of the signed informed consent form and a source document identifying
the trial and recording the dates of participation will be placed in the participant’s medical record.
All of the above mentioned activities will be completed prior to the participant’s participation in the
trial.
The monitor will inspect the original completed consent form(s) for all participants.
13.5. Confidentiality
All site staff, the Sponsor, and any sponsor representatives will preserve the confidentiality of all
participants taking part in the study, in accordance with ICH GCP, applicable South African national
and local regulations and, to the extent applicable, the U.S. Health Insurance Portability and
Accountability Act of 1996 (“HIPAA”). Subject to the requirement for source data verification by
the study personnel by reference to the participant’s notes, confidentiality of all participant
identities will be maintained. Only participant study number and initials will be used on the CRF
and in all study correspondence, as permitted. No material bearing a participant’s name will be
kept on file by the Sponsor. The written informed consent will contain a clause granting permission
for review of the participants’ source data.
13.6. Publication Policy
The definition of publication for this purpose is any public presentation of the data emerging from
this study.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 111 of 142
All unpublished information given to the Investigator by the Sponsor shall not be published or
disclosed to a third party, other than to the responsible IEC/IRB, within the understanding of the
confidentiality of their nature, without the prior written consent of the Sponsor.
Results of this research will be submitted for publication as soon as feasible upon completion of the
study in the form of a joint publication(s) between Sponsor and Investigator(s), including site
clinical and laboratory investigators, as appropriate.
13.7. Protocol Amendment Policy
Any change to the protocol will be effected by means of a protocol amendment. Any changes
which affect participant safety or welfare will be submitted to the IEC/IRB and Regulatory
Authorities prior to implementation. The Investigator, IEC/IRB, and Sponsor must agree on all
amendments. No amendment will be implemented until approved by the relevant Authorities and
signed by all required parties. Exceptions to this are when the Investigator considers that the
participant’s safety is compromised.
Protocol amendments detailing minor administrative changes should be submitted by the
Investigator to the IEC/IRB and Regulatory Authorities for notification purposes as appropriate.
13.8. Financial Aspects, Insurance and Indemnity
The study sponsor is the Global Alliance for TB Drug Development (TB Alliance). The TB Alliance is a
not for profit, product development partnership accelerating the discovery and development of
new TB drugs that will shorten treatment, be effective against susceptible and resistant strains, be
compatible with antiretroviral therapies for those HIV-TB participants currently on such therapies,
and improve treatment of latent infection.
The TB Alliance works with public and private partners worldwide. It is committed to ensuring that
approved new regimens are affordable, adopted and available to those who need them.
The TB Alliance operates with funding mainly from the Bill & Melinda Gates Foundation, the
Netherlands Ministry of Foreign Affairs (DGIS), the United Kingdom Department for International
Development (DFID), and the United States Agency for International Development (USAID).
The participants will not receive any incentives for their involvement in the study. The sponsor has
made provision to reimburse the participants for out-of-pocket expenses such as travelling to and
from the study site and other miscellaneous costs as a result of their study participation.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 112 of 142
The sponsor certifies that it has liability insurance coverage for itself and will provide an associated
certificate upon request. The insurance does not relieve the Investigators of the obligation to
maintain their own liability insurance as required by applicable law. The sponsor does not assume
any obligation for the medical treatment of other injuries and illnesses.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 113 of 142
14. REFERENCES
1. Maggi N, Pasqualucci CR, Ballotta R, Sensai P. Rifampicin: A new orally active rifamycins.
Chemotherapy 1966; 11 (5): 285-92.
2. Jindani A, Aber VR, Edwards EA, Mitchison DA. The early bactericidal activity of drugs in
patients with pulmonary tuberculosis. Am Rev Respir Dis 1980; 121 (6): 939-49.
3. Dietze R, Teixeira L, Rocha LMC, et al. Safety and bactericidal activity of rifalazil in patients with
pulmonary tuberculosis. Antimicrob Agents Chemother 2001; 47 (7): 1972-6.
4. Donald PR, Sirgel FA, Venter A, et al. Early bactericidal activity of antituberculosis agents.
Expert Rev Anti-infect Ther 2003; 1 (1): 141-55.
5. Johnson JL, Hadad DJ, Boom WH, et al. Early and extended early bactericidal activity of
levofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. Int J Tuberc Lung Dis
2006; 10 (6): 1-8.
6. Rustomjee R, Diacon AH, Allen J, et al. Early bactericidal activity and pharmacokinetics of the
diarylquinoline TMC207 in treatment of pulmonary tuberculosis. Antimicrob Agents
Chemother 2008 Aug; 52(8): 2831-5. Epub 2008 May 27.
7. TMC207 Investigator’s Brochure, Tibotec Pharmaceuticals Ltd., Edition 5 June 2008, Addendum
May 2009.
8. Koul A, Dendouga N, Vergauwen K, et al. Diarylquinolines target subunit c of mycobacterial
ATP synthase. Nat Chem Biol 2007; 3: 323-4.
9. Andries K, Verhasselt P, Guillemont J, et al. A diarylquinoline drug active on the ATP synthase
of Mycobacterium turberculosis. Science 2005; 307: 223-7.
10. Huitric E, Verhasselt P, Andries K, Hoffner SE. In vitro antimycobacterial spectrum of a
diarylquinoline ATP synthase inhibitor. Antimicrob Agents Chemother 2007; 51: 4202-4.
11. Koul A, Vranckx L, Dendouga N, et al. Diarylquinolines are bactericidal for dormant
mycobacteria as a result of disturbed homeostasis. J Biol Chem 2008; 283: 25273-80.
12. Ibrahim M, Andries K, Lounis N, et al. Synergistic activity of R207910 combined with
pyrazinamide against murine tuberculosis. Antimicrob Agents Chemother 2007; 51: 1011-15.
13. Investigator’s Brochure, Global Alliance for TB Drug Development, PA-824, 11 June 2009.
14. Stover, C. K., P. Warrener, D. R. VanDevanter, et al. 2000. A small-molecule nitroimidazopyran
drug candidate for the treatment of tuberculosis. Nature 405:962–966.
15. Anne J. Lenaerts, Veronica Gruppo, Karen S. Marietta, Christine M. Johnson, Diane K. Driscoll,
Nicholas M. Tompkins, Jerry D. Rose, Robert C. Reynolds, and Ian M. Orme. Preclinical Testing
of the Nitroimidazopyran PA-824 for Activity against Mycobacterium tuberculosis in a Series of
In Vitro and In Vivo Models. Antimicrob Agents Chemother. 2005 June; 49(6): 2294–2301.
16. Sandeep Tyagi, E. Nuermberger, T. Yoshimatsu, K. Williams, I. Rosenthal, N. Lounis, W. Bishai,
and J. Grosset. Bactericidal Activity of the Nitroimidazopyran PA-824 in a Murine Model of
Tuberculosis. Antimicrob Agents Chemother. 2005 June; 49(6): 2289–2293.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 114 of 142
17. Baohong PI, Lounis N, Maslo C, Truffot-Pernot C, Bonnafous P, Grosset J. In vitro and in vivo
activities of moxifloxacin and clinafloxacin against Mycobacterium tuberculosis.
Antimicrob.Agents Chemother. 1998; 42(8):2066-9.
18. Miyazaki E, Miyazaki M, Chen JM, Chaisson RE, Bishai WR. Moxifloxacin (BAY12-8039), a new 8-
methoxyquinolone, is active in a mouse model of tuberculosis. Antimicrob. Agents Chemother.
1999; 43(1):85-9.
19. Nuermberger EL, Yoshimatsu T, Tyagi S, O'Brien RJ, Vernon AN, Chaisson RE et al. Moxifloxacin-
containing regimen greatly reduces time to culture conversion in murine tuberculosis.
Am.J.Respir.Crit.Care Med. 2003; 169:421-6.
20. O'Brien RJ. Scientific blueprint for tuberculosis drug development. Tuberculosis 2001;
81(S1):19-45.
21. Gosling RD, Uiso LO, Sam NE, Bongard E, Kanduma EG, Nyindo M et al. The bactericidal activity
of moxifloxacin in patients with pulmonary tuberculosis. Am J Respir Crit Care Med 2003; 168
(11):1342-5.
22. Pletz MWR, De Roux A, Roth A et al. Early Bactericidal Activity of Moxifloxacin in Treatment of
Pulmonary Tuberculosis: a Prospective, Randomized Study. Antimicrob. Agents Chemother
2004; 48 (3); 780-2.
23. Burman W, Goldberg S, Johnson JL, et al. Moxifloxacin versus ethambutol in the first 2 months
of treatment for pulmonary tuberculosis. Am J Respir Crit Care Med 2006; 174: 331-38.
24. Rustomjee R, Lienhardt C, Kanyok T et al. Gatifloxacin for TB (OFLOTUB) study team. A phase II
study of the sterilizing activities of ofloxacin, gatifloxacin and moxifloxacin in pulmonary
tuberculosis. Int J Tuberc Lung Dis 2008; 12:128-38.
25. Conde MB, Efron A, Loredo C et al. Moxifloxacin versus ethambutol in the initial treatment of
tuberculosis: a double-blind, randomized, controlled phase II trial. Lancet 2009; 373: 1183-89.
26. Dorman SE, Johnson JL, Goldberg S, et al. Substitution of Moxifloxacin for Isoniazid during
Intensive Phase Treatment of Pulmonary Tuberculosis. Am J Respir Crit Care Med 2009; 180:
273-80.
27. Zhang Y and Mitchison D. The curious characteristics of pyrazinamide: a review. Int J Tuberc
Lung Dis; 7 (1): 6-21.
28. Rokeya Tasneen, Sandeep Tyagi, Kathy Williams, Jacques Grosset, and Eric Nuermberger
Enhanced Bactericidal Activity of Rifampin and/or Pyrazinamide When Combined with PA-824
in a Murine Model of Tuberculosis. Antimicrob Agents Chemother 2008 Oct; 52(10): 3664–8
29. Nuermberger E, Tyagi S, Tasneen R, Williams K and Almeida D, Rosenthal I and Grosset J.
Powerful Bactericidal and Sterilizing Activity of a Regimen Containing PA-824, Moxifloxacin and
Pyrazinamide in a Murine Model of Tuberculosis. Antimicrob.Agents Chemother.
2008;52:1522-4

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 115 of 142
30. WORLD MEDICAL ASSOCIATION DECLARATION OF HELSINKI Ethical Principles for Medical
Research Involving Human Subjects Adopted by the 18th WMA General Assembly, Helsinki,
Finland, June 1964, and amended by the:
29th WMA General Assembly, Tokyo, Japan, October 1975
35th WMA General Assembly, Venice, Italy, October 1983
41st WMA General Assembly, Hong Kong, September 1989
48th WMA General Assembly, Somerset West, Republic of South Africa, October 1996
52nd WMA General Assembly, Edinburgh, Scotland, October 2000
53rd WMA General Assembly, Washington 2002 (Note of Clarification on paragraph 29 added)
55th WMA General Assembly, Tokyo 2004 (Note of Clarification on Paragraph 30 added)
59th WMA General Assembly, Seoul, October 2008.
31. INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR
REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE. ICH HARMONISED TRIPARTITE
GUIDELINE. GUIDELINE FOR GOOD CLINICAL PRACTICE, E6(R1). www.ich.org. 2002
32. Chaves AA, Keller WJ, O’Sullivan S, Williams MA, Fitzgerald LE, McPherson HE, et al.
Cardiovascular monkey telemetry: sensitivity to detect QT interval prolongation. J Pharmacol
Toxicol Methods. 2006 Sep-Oct;54(2):150-8
33. Diacon A, Pym A, Grobusch M, et al. The Diarylquinoline TMC207 for Multidrug-Resistant
Tuberculosis. 2009; 360 (23): 2397-405.
34. Moxifloxacin Investigator’s Brochure, Bayer Schering Pharma., V11,01 Feb 09.
35. The age-related eye disease study (AREDS) system for classifying cataracts from photographs:
AREDS report no. 4. American Journal of Ophthalmology, Volume 131, Issue 2, February 2001,
Pages 167-175. The Age-Related Eye Disease Study Research Group.
36. A Randomized, Placebo-Controlled, Clinical Trial of High-Dose Supplementation With Vitamins
C and E and Beta Carotene for Age-Related Cataract and Vision Loss: AREDS Report No. 9. Arch
Ophthalmol. 2001 October ; 119(10): 1439–1452. Age-Related Eye Disease Study Research
Group.
37. A Randomized, Double-Masked, Placebo-Controlled Clinical Trial of Multivitamin
Supplementation for Age-Related Lens Opacities Clinical Trial of Nutritional Supplements and
Age-Related Cataract Report No. 3. Ophthalmology 2008;115:599 – 607 © 2008 by the
American Academy of Ophthalmology. Clinical Trial of Nutritional Supplements and Age-
Related Cataract Study Group.
38. Mdluli, K and Nuermberger, E. Pre-Clinical Drug Combination Studies to Identify Novel
Regimens for TB. In: Recent Advances in TB Drug Development symposium, 40th Union World
Conference, International Union Against TB and Lung Disease, Cancun, Mexico, 2009.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 116 of 142
APPENDIX 1: PROTOCOL SIGNATURE PAGES
Not Applicable.
This document does not require formal approval as it is a working document containing a combination of:
Protocol 27 April 2010
Protocol Administrative Change Number 01; 09 September 2010
Protocol Amendment Number 01; 06 December 2010
which comprise the formal approved documents.

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 117 of 142
APPENDIX 2: RIFAFOUR E-275 PACKAGE INSERT
RIFAFOUR®e-275 TABLETS SCHEDULING STATUS: S4 PROPRIETARY NAME (and dosage form):
RIFAFOUR®e-275 TABLETS COMPOSITION: Each tablet contains: Rifampicin 150 mg Isoniazid 75 mg Pyrazinamide 400 mg Ethambutol 275 mg Contains sodium ascorbate as anti-oxidant. PHARMACOLOGICAL CLASSIFICATION: A 20.2.3 Tuberculostatic combinations PHARMACOLOGICAL ACTION: Rifafour e-275 tablets is a combination of four first line agents used in the treatment of tuberculosis. Rifampicin is a semi-synthetic, broad-spectrum bactericidal antibiotic. Isoniazid is a synthetic, antitubercular agent which is bacteriostatic against semi-dormant bacilli and bactericidal against actively dividing mycobacteria. Pyrazinamide may be bactericidal or bacteriostatic, depending on its concentration and the susceptibility of the organism. Ethambutol is a synthetic, bacteriostatic antitubercular agent. All agents are readily absorbed following oral administration, with wide distribution to most tissues and fluids including cerebrospinal fluid. INDICATIONS: Initial phase treatment of pulmonary and extrapulmonary tuberculosis in new adult patients and re-treatment of adult cases. CONTRA-INDICATIONS: Rifafour e-275 tablets are contra-indicated in: • patients with hypersensitivity to rifamycins, isoniazid, pyrazinamide, ethambutol or other chemically related
medication.
• the presence of jaundice or active hepatic disease.
• patients with optic neuritis. Safety in pregnancy has not been established. All agents of Rifafour e-275 tablets are excre
ted in breast milk. Safety during lactation has not beer established.
Rifafour e-275 should not be used in children under 13 years of age. WARNINGS: Liver function should be checked before and during treatment and special care should be exercised in alcoholic patients, the elderly or those with pre-existing liver disease. Caution should be observed with the use of Rifafour e-275 tablets in the following patients: • Impaired kidney function: dosage adjustment may be required according to the serum concentration of ethambutol
• Patients with visual defects: should visual disturbances occur during treatment these must be reported immediately and the medicine discontinued pending visual evaluation
• Patients at risk of neuropathy or pyridoxine deficiency, including those who are diabetic, alcoholic, malnourished, uraemic or pregnant: pyridoxine supplementation (in a 10 mg to 50 mg daily dose) is usually required in these instances
• Patients with a history of gout
• Patients with porphyria
• Patients with epilepsy, as convulsions may be precipitated
• Patients with a history of psychosis
• Patients with diabetes: pyrazinamide may cause interference with urine ketone determinations
• Rifampicin may decrease the effect of oral contraceptives and patients are advised to change to non-hormonal methods of birth control

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 118 of 142
• Treatment with Rifafour e-275 tablets may produce reddish colouration of urine, tears and saliva. Contact lenses may be irreversibly stained.
DOSAGE AND DIRECTIONS FOR USE: Take Rifafour e-275 tablets with a full glass of water 1 hour before, or 2 hours after a meal. However, if gastrointestinal irritation occurs, the tablets may be taken with food. If aluminium-containing antacids are taken, administer one hour after the tablet dose. The recommended treatment dosages, based on the patient's body weight, given daily for the 2 month initial-phase treatment in adults and children over 13 years of age are as follows:
30-37 kg 2 tablets
38-54 kg 3 tablets
55 - 70 kg 4 tablets
71 kg and over 5 tablets
SIDE-EFFECTS AND SPECIAL PRECAUTIONS: Side-effects associated with rifampicin: Some patients may experience a cutaneous syndrome which presents 2 to 3 hours after a daily or intermittent dose i.e. facial flushing, itching, rash, eye irritation. A 12 hour "flu" syndrome, usually occurring after 3 to 6 months of intermittent treatment and usually with doses of 20 mg/kg or more, may present as fever, chills, bone pain and malaise. Gastrointestinal effects include nausea, vomiting, anorexia, diarrhoea and epigastric distress, which may be alleviated by administration with food. There have been reports of pseudomembranous colitis. Hepatitis and the prodromal symptoms of hepatitis may occur (nausea, vomiting, unusual tiredness/fatigue). Rifampicin can cause thrombocytopenia and purpura usually with intermittent regimens. Other haematological adverse effects include eosinophilia, leucopenia and haemolytic anaemia. Nervous system effects include headache, drowsiness, dizziness, ataxia, numbness, visual disturbances and muscular weakness. Alterations in kidney function and renal failure have occurred. Rifampicin may cause orange-red discoloration of urine and other body fluids. Menstrual disturbances have been reported. Side-effects associated with isoniazid: Elevated liver enzymes associated with clinical signs of hepatitis such as nausea, vomiting or fatigue may indicate hepatic damage. The incidence of liver damage is highest in patients over 35 years of age, those who are slow acetylators and those who consume alcohol on a daily basis. Gastrointestinal effects (nausea, vomiting, pellagra) and hypersensitivity reactions (skin eruptions including erythema multiforme, fever, lymphadenopathy, vasculitis) may occur. Haematological effects have been reported (sideroblastic anaemia, agranulocytosis, haemolytic anaemia, thrombocytopenia, neutropenia, eosinophiliaand less frequently, aplastic anaemia). Neurological effects include psychotic reactions and convulsions. Other effects: hyperglycaemia, metabolic acidosis, lupus-like syndrome, rheumatoid syndrome, urinary retention and gynaecomastia. Optic neuritis has also been reported. Peripheral neuropathy has also been associated with isoniazid administration. Pyridoxine supplementation prevents the development of peripheral neuritis, as well as most other nervous system dysfunctions. Side-effects associated with pyrazinamide: The most serious side-effect is hepatotoxicity and its frequency appears to be dose-related. Hyperuricaemia commonly occurs, occasionally accompanied by arthralgia and may lead to attacks of gout. Photosensitivity and skin rash have been reported less frequently. Other side-effects that have been reported are anorexia, nausea and vomiting, malaise, fever, sideroblastic anaemia and dysuria. Side-effects associated with ethambutol: Retrobulbar neuritis with a reduction in visual acuity, constriction of visual field, central or peripheral scotoma, and green-red colour blindness may occur, affecting one or both eyes. The degree of visual impairment appears to depend on the dose and duration of therapy. Retinal haemorrhage has occurred less frequently. Renal clearance of urate may be reduced and acute gout has been precipitated. Hypersensitivity reactions include skin rash, pruritis, leucopenia, fever and joint pains. Gastrointestinal disturbances include metallic taste, nausea, vomiting, anorexia and abdominal

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 119 of 142
pain. Other adverse effects: Confusion, disorientation, hallucinations, headache, dizziness, malaise, jaundice or transient liver dysfunction, peripheral neuritis. SPECIAL PRECAUTIONS: In the following cases, treatment with Rifafour e-275 tablets should be stopped immediately and the patient evaluated: jaundice, rash and fever, elevated liver enzymes associated with the clinical signs of hepatitis, visual impairment. If liver damage is confirmed, the medicine should not be recommenced. Treatment should be discontinued permanently should thrombocytopenia, purpura, shock or renal failure occur. Periodic eye examinations during treatment is suggested. INTERACTIONS: Rifampicin: Concurrent use of alcohol, acetaminophen, isoniazid and other hepatotoxic medication may increase the incidence of rifampicin-induced hepatotoxicity. The effectiveness of oestrogen-containing oral preparations is reduced. Rifampicin accelerates the metabolism of certain medicines by inducing microsomal enzymes. Medicines that are affected include: atorvaquone, azathioprine, chloramphenicol, cimetidine, clofibrate, corticosteroids, coumarin anticoagulants, cyclosporin, dapsone, diazepam and other benzodiazepines, doxycycline, azole antifungals (ketoconazole, itraconazole, fluconazole), haloperidol, hexobarbitone, methadone, oral hypoglycaemic agents, phenytoin, quinine, sulphasalazine, thyroxine, theophylline, zidovudine, beta-blockers, digitoxin, digoxin, antiarrhythmic agents (e.g. disopyramide, verapamil) and calcium channel blockers. Isoniazid: Chronic use of isoniazid may decrease the plasma clearance and prolong the duration of action of alfentanil, coumarin anticoagulants, benzodiazepines, carbamazepine, phenytoin, ethosuximide, chlorzoxazone, and theophylline. Appropriate adjustment of the anticonvulsant dose may be required. Concurrent use of paracetamol, alcohol, rifampicin and other hepatotoxic medication, may increase the potential for isoniazid-induced hepatotoxicity. Aluminium-containing antacids may delay absorption and decrease serum concentrations of isoniazid. Ingestion of certain types of cheese e.g. Swiss or Cheshire, or fish e.g. tuna, may result in itching of the skin, rapid or pounding heart, chills or headache. Glucocorticoid corticosteroids may increase hepatic metabolism and/or excretion of isoniazid. Concurrent use of cycloserine, disulfiram and other neurotoxic medicines may increase the potential for CNS toxicity. Isoniazid may increase the formation of potentially nephrotoxic inorganic fluoride metabolites when used concurrently with enflurane. Interactions with ketoconazole and miconazole have been reported. False positive reactions with copper sulphate urine glucose tests may occur. Pyrazinamide: Pyrazinamide may decrease the efficacy of gout therapy (e.g. allopurinol, colchicine, probenecid or sulphinpyrazone) and dosage adjustments of this medication may be necessary. Ethambutol: Concurrent administration of neurotoxic medication with ethambutol may potentiate neurotoxic effects such as optic and peripheral neuritis. KNOWN SYMPTOMS OF OVERDOSAGE AND PARTICULARS OF ITS TREATMENT: For symptoms of overdosage, refer to "Side-Effects". Treatment of overdosage consists of gastric lavage, symptomatic and supportive therapy. IDENTIFICATION: Purple, round, film coated tablets. PRESENTATION: Securitainers of 40, 60, 80, 100 and 500 tablets. STORAGE INSTRUCTIONS: Store in a cool place, below 25°C in well-closed containers, protected from light. KEEP OUT OF REACH OF CHILDREN. REGISTRATION NUMBER: 34/20.2.3/0187 NAME AND BUSINESS ADDRESS OF THE APPLICANT: Aventis Pharma (Pty) Ltd 2 Bond Street, Midrand, 1685 DATE OF PUBLICATION OF THIS PACKAGE INSERT: 11 February 2002 Aventis Pharma (Pty) Ltd 2 Bond Street Midrand 1685

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 120 of 142
APPENDIX 3: THE IUATLD SCALE
The IUATLD scale proposes five groups for reporting the results of reading smears for acid fast bacilli. They should be recorded as follows:
FINDING RECORDING
No acid-fast bacilli found in at least 100 fields
negative
1 to 9 acid-fast bacilli per 100 fields exact figure/100/scanty positive
10 to 99 acid-fast bacilli per 100 fields +
1 to 10 acid-fast bacilli per field in at least 50 fields
++
More than 10 acid-fast bacilli per field in at least 20 fields
+++
Reference: The Public Health Service National Tuberculosis Reference Laboratory and the National Laboratory Network. Minimum Requirements, Role and Operation in a Low-Income Country. International Union Against Tuberculosis and Lung Disease 1998

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 121 of 142
APPENDIX 4: DIVISION OF MICROBIOLOGY AND INFECTIOUS DISEASES (DMID) ADULT TOXICITY TABLE ABBREVIATIONS: Abbreviations utilized in the Table:
ULN = Upper Limit of Normal LLN = Lower Limit of Normal Rx = Therapy Req = Required Mod = Moderate IV = Intravenous ADL = Activities of Daily Living Dec = Decreased
ESTIMATING SEVERITY GRADE For abnormalities NOT found elsewhere in the Toxicity Tables use the scale below to estimate grade of severity: GRADE 1 Mild Transient or mild discomfort (< 48 hours); no medical
intervention/therapy required GRADE 2 Moderate Mild to moderate limitation in activity - some
assistance may be needed; no or minimal medical intervention/therapy required
GRADE 3 Severe Marked limitation in activity, some assistance usually required; medical intervention/therapy required, hospitalizations possible
GRADE 4 Potentially Life-threatening Extreme limitation in activity, significant assistance required; significant medical intervention/therapy required, hospitalization or hospice care probable
SERIOUS OR LIFE-THREATENING AEs ANY clinical event deemed by the clinician to be serious or life-threatening should be considered a grade 4 event. Clinical events considered to be serious or life-threatening include, but are not limited to: seizures, coma, tetany, diabetic ketoacidosis, disseminated intravascular coagulation, diffuse petechiae, paralysis, acute psychosis, severe depression. COMMENTS REGARDING THE USE OF THESE TABLES • Standardized and commonly used toxicity tables (Division of AIDS, NCI’s Common Toxicity
Criteria (CTC), and World Health Organization (WHO)) have been adapted for use by the Division of Microbiology and Infectious Diseases (DMID) and modified to better meet the needs of patients in DMID trials.
• For parameters not included in the following Toxicity Tables, sites should refer to the “Guide
For Estimating Severity Grade” located above. • Criteria are generally grouped by body system.
• Some protocols may have additional protocol specific grading criteria, which will supercede the use of these tables for specified criteria.
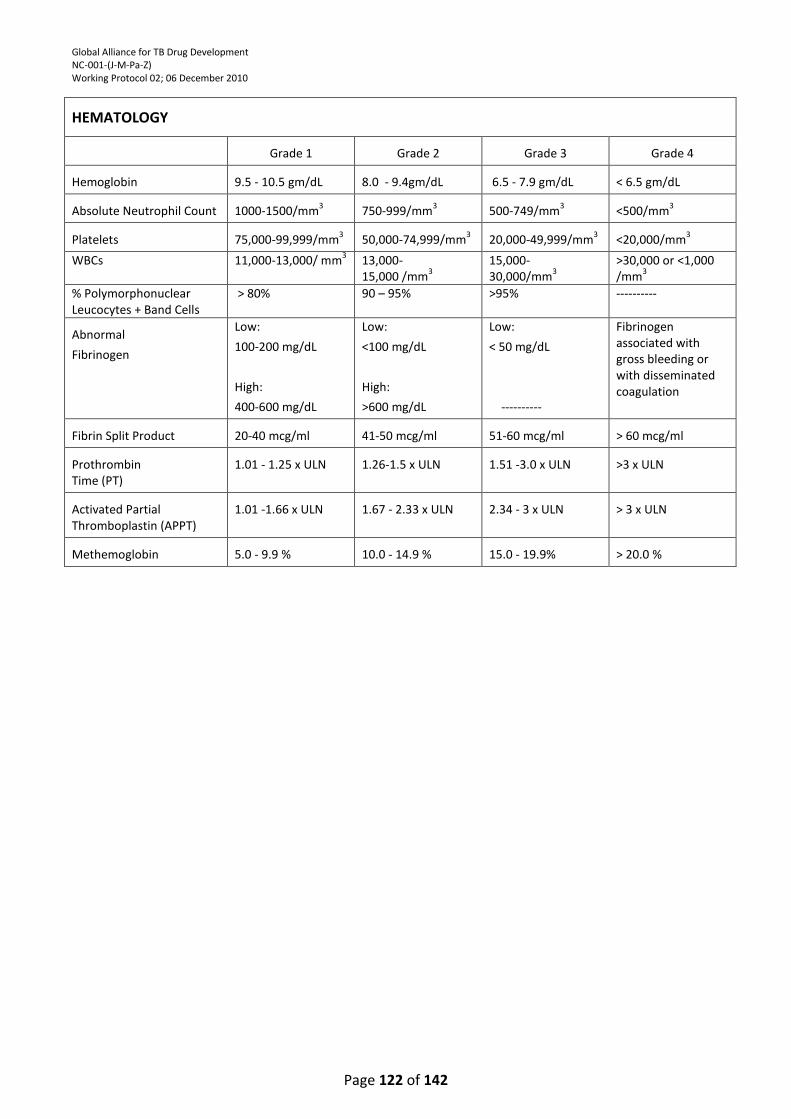
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 122 of 142
HEMATOLOGY
Grade 1
Grade 2
Grade 3
Grade 4
Hemoglobin
9.5 - 10.5 gm/dL
8.0 - 9.4gm/dL
6.5 - 7.9 gm/dL
< 6.5 gm/dL
Absolute Neutrophil Count
1000-1500/mm
3
750-999/mm
3
500-749/mm
3
<500/mm
3
Platelets
75,000-99,999/mm
3
50,000-74,999/mm
3
20,000-49,999/mm
3
<20,000/mm
3
WBCs 11,000-13,000/ mm3 13,000-
15,000 /mm3
15,000- 30,000/mm
3
>30,000 or <1,000 /mm
3
% Polymorphonuclear Leucocytes + Band Cells
> 80% 90 – 95% >95% ----------
Abnormal
Fibrinogen
Low:
100-200 mg/dL
High:
400-600 mg/dL
Low:
<100 mg/dL
High:
>600 mg/dL
Low:
< 50 mg/dL
----------
Fibrinogen associated with gross bleeding or with disseminated coagulation
Fibrin Split Product
20-40 mcg/ml
41-50 mcg/ml
51-60 mcg/ml
> 60 mcg/ml
Prothrombin Time (PT)
1.01 - 1.25 x ULN
1.26-1.5 x ULN
1.51 -3.0 x ULN
>3 x ULN
Activated Partial Thromboplastin (APPT)
1.01 -1.66 x ULN
1.67 - 2.33 x ULN
2.34 - 3 x ULN
> 3 x ULN
Methemoglobin
5.0 - 9.9 %
10.0 - 14.9 %
15.0 - 19.9%
> 20.0 %
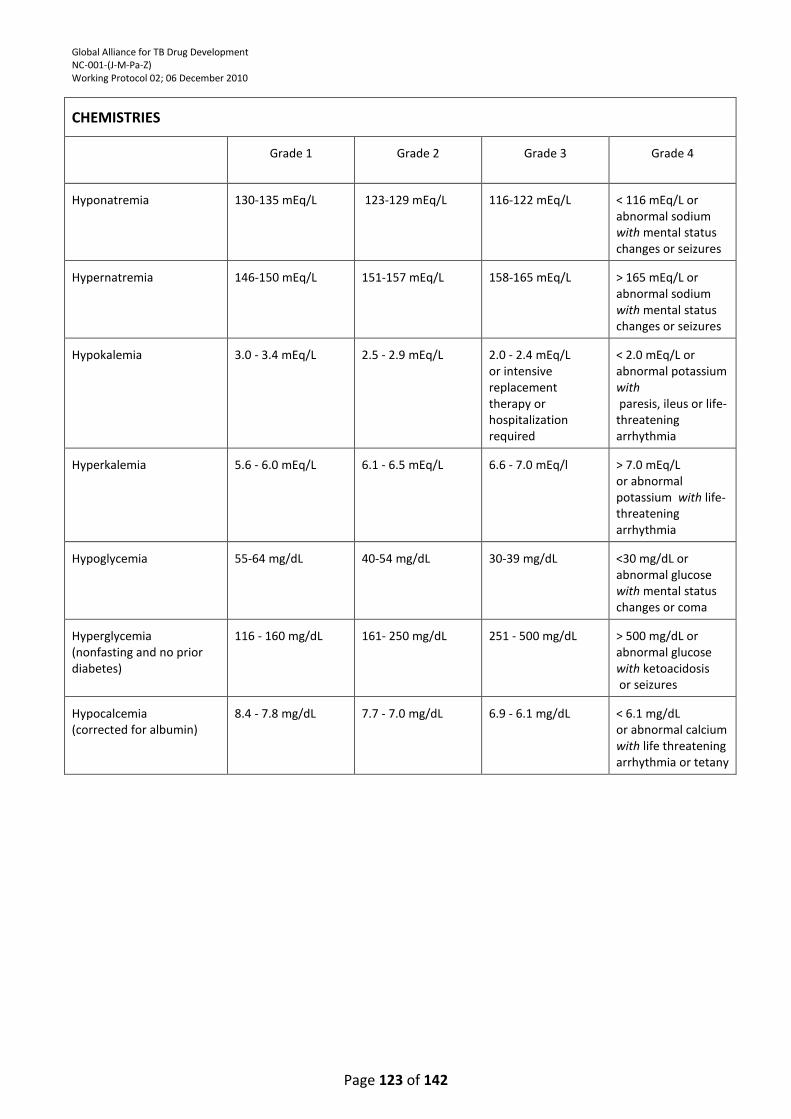
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 123 of 142
CHEMISTRIES
Grade 1
Grade 2
Grade 3
Grade 4
Hyponatremia
130-135 mEq/L
123-129 mEq/L
116-122 mEq/L
< 116 mEq/L or abnormal sodium with mental status changes or seizures
Hypernatremia
146-150 mEq/L
151-157 mEq/L
158-165 mEq/L
> 165 mEq/L or abnormal sodium with mental status changes or seizures
Hypokalemia
3.0 - 3.4 mEq/L
2.5 - 2.9 mEq/L
2.0 - 2.4 mEq/L or intensive replacement therapy or hospitalization required
< 2.0 mEq/L or abnormal potassium with paresis, ileus or life-threatening arrhythmia
Hyperkalemia
5.6 - 6.0 mEq/L
6.1 - 6.5 mEq/L
6.6 - 7.0 mEq/l
> 7.0 mEq/L or abnormal potassium with life-threatening arrhythmia
Hypoglycemia
55-64 mg/dL
40-54 mg/dL
30-39 mg/dL
<30 mg/dL or abnormal glucose with mental status changes or coma
Hyperglycemia (nonfasting and no prior diabetes)
116 - 160 mg/dL
161- 250 mg/dL
251 - 500 mg/dL
> 500 mg/dL or abnormal glucose with ketoacidosis or seizures
Hypocalcemia (corrected for albumin)
8.4 - 7.8 mg/dL
7.7 - 7.0 mg/dL
6.9 - 6.1 mg/dL
< 6.1 mg/dL or abnormal calcium with life threatening arrhythmia or tetany
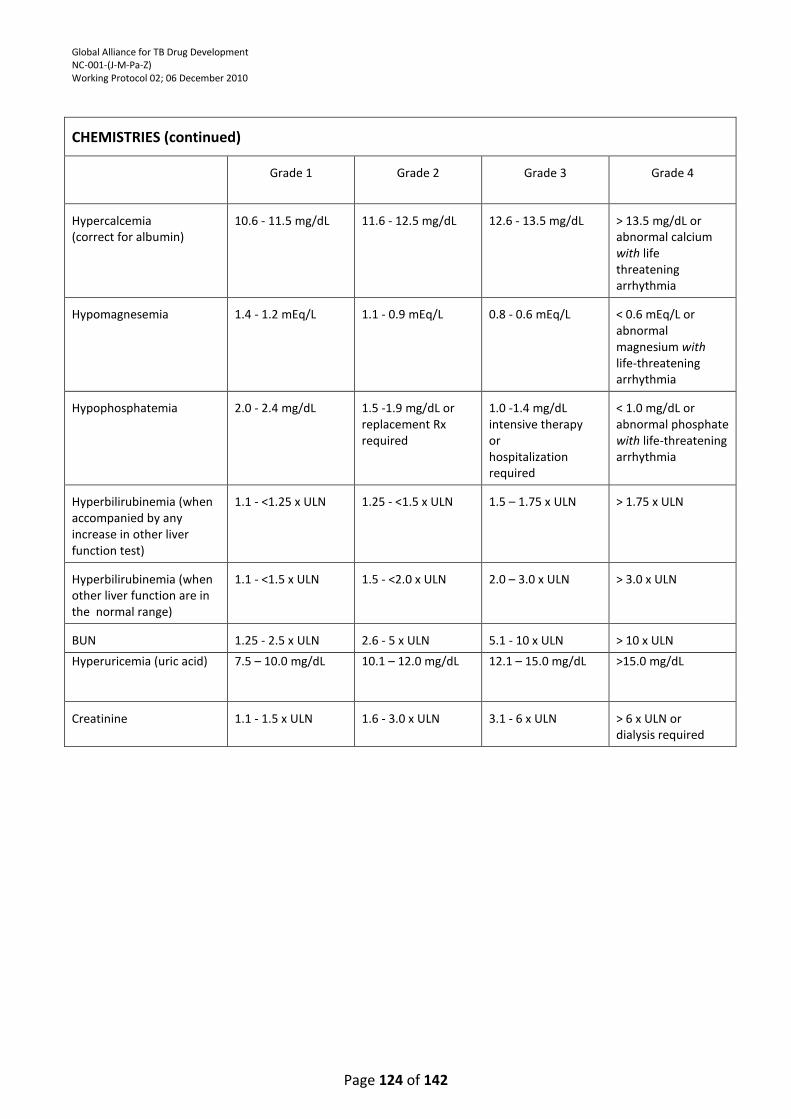
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 124 of 142
CHEMISTRIES (continued)
Grade 1
Grade 2
Grade 3
Grade 4
Hypercalcemia (correct for albumin)
10.6 - 11.5 mg/dL
11.6 - 12.5 mg/dL
12.6 - 13.5 mg/dL
> 13.5 mg/dL or abnormal calcium with life threatening arrhythmia
Hypomagnesemia
1.4 - 1.2 mEq/L
1.1 - 0.9 mEq/L
0.8 - 0.6 mEq/L
< 0.6 mEq/L or abnormal magnesium with life-threatening arrhythmia
Hypophosphatemia
2.0 - 2.4 mg/dL
1.5 -1.9 mg/dL or replacement Rx required
1.0 -1.4 mg/dL intensive therapy or hospitalization required
< 1.0 mg/dL or abnormal phosphate with life-threatening arrhythmia
Hyperbilirubinemia (when accompanied by any increase in other liver function test)
1.1 - <1.25 x ULN
1.25 - <1.5 x ULN
1.5 – 1.75 x ULN
> 1.75 x ULN
Hyperbilirubinemia (when other liver function are in the normal range)
1.1 - <1.5 x ULN
1.5 - <2.0 x ULN
2.0 – 3.0 x ULN
> 3.0 x ULN
BUN
1.25 - 2.5 x ULN
2.6 - 5 x ULN
5.1 - 10 x ULN
> 10 x ULN
Hyperuricemia (uric acid) 7.5 – 10.0 mg/dL 10.1 – 12.0 mg/dL 12.1 – 15.0 mg/dL >15.0 mg/dL
Creatinine
1.1 - 1.5 x ULN
1.6 - 3.0 x ULN
3.1 - 6 x ULN
> 6 x ULN or dialysis required
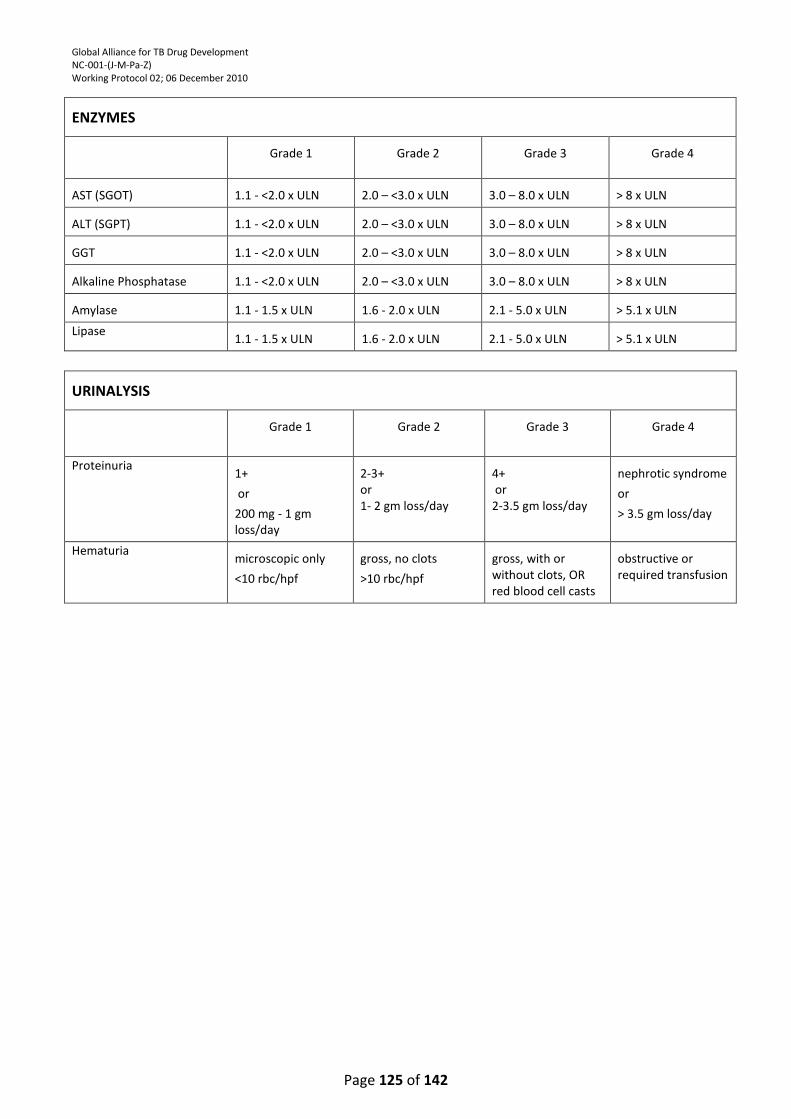
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 125 of 142
ENZYMES
Grade 1
Grade 2
Grade 3
Grade 4
AST (SGOT)
1.1 - <2.0 x ULN
2.0 – <3.0 x ULN
3.0 – 8.0 x ULN
> 8 x ULN
ALT (SGPT)
1.1 - <2.0 x ULN
2.0 – <3.0 x ULN
3.0 – 8.0 x ULN
> 8 x ULN
GGT
1.1 - <2.0 x ULN
2.0 – <3.0 x ULN
3.0 – 8.0 x ULN
> 8 x ULN
Alkaline Phosphatase
1.1 - <2.0 x ULN
2.0 – <3.0 x ULN
3.0 – 8.0 x ULN
> 8 x ULN
Amylase
1.1 - 1.5 x ULN
1.6 - 2.0 x ULN
2.1 - 5.0 x ULN
> 5.1 x ULN
Lipase 1.1 - 1.5 x ULN
1.6 - 2.0 x ULN
2.1 - 5.0 x ULN
> 5.1 x ULN
URINALYSIS
Grade 1
Grade 2
Grade 3
Grade 4
Proteinuria 1+
or
200 mg - 1 gm loss/day
2-3+ or 1- 2 gm loss/day
4+ or 2-3.5 gm loss/day
nephrotic syndrome
or
> 3.5 gm loss/day
Hematuria microscopic only
<10 rbc/hpf
gross, no clots
>10 rbc/hpf
gross, with or without clots, OR red blood cell casts
obstructive or required transfusion
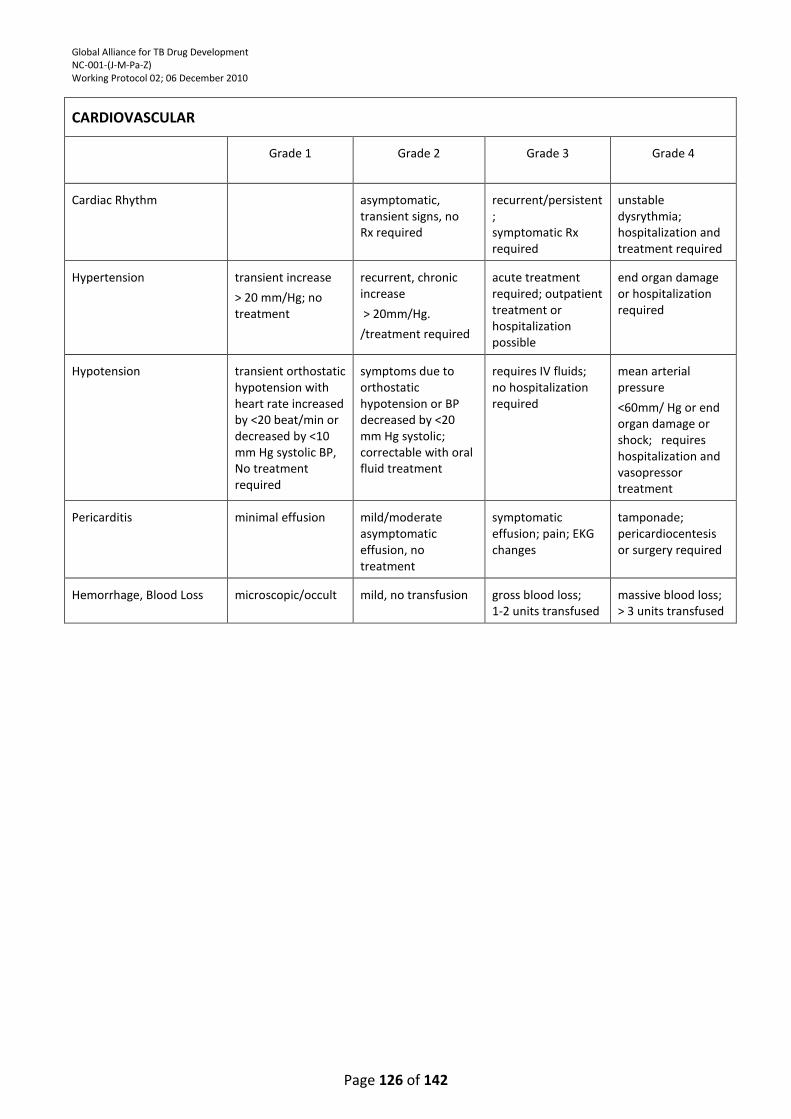
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 126 of 142
CARDIOVASCULAR
Grade 1
Grade 2
Grade 3
Grade 4
Cardiac Rhythm
asymptomatic, transient signs, no Rx required
recurrent/persistent; symptomatic Rx required
unstable dysrythmia; hospitalization and treatment required
Hypertension
transient increase
> 20 mm/Hg; no treatment
recurrent, chronic increase
> 20mm/Hg.
/treatment required
acute treatment required; outpatient treatment or hospitalization possible
end organ damage or hospitalization required
Hypotension
transient orthostatic hypotension with heart rate increased by <20 beat/min or decreased by <10 mm Hg systolic BP, No treatment required
symptoms due to orthostatic hypotension or BP decreased by <20 mm Hg systolic; correctable with oral fluid treatment
requires IV fluids; no hospitalization required
mean arterial pressure
<60mm/ Hg or end organ damage or shock; requires hospitalization and vasopressor treatment
Pericarditis
minimal effusion
mild/moderate asymptomatic effusion, no treatment
symptomatic effusion; pain; EKG changes
tamponade; pericardiocentesis or surgery required
Hemorrhage, Blood Loss
microscopic/occult
mild, no transfusion
gross blood loss; 1-2 units transfused
massive blood loss; > 3 units transfused
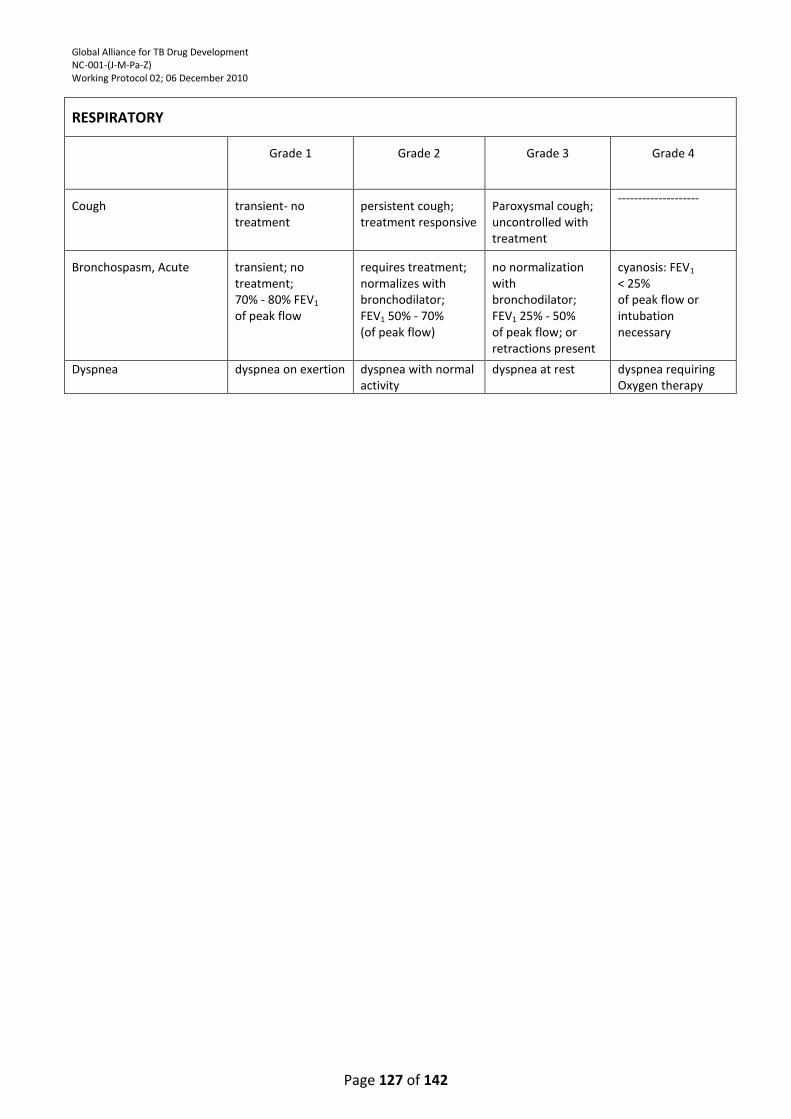
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 127 of 142
RESPIRATORY
Grade 1
Grade 2
Grade 3
Grade 4
Cough
transient- no treatment
persistent cough; treatment responsive
Paroxysmal cough; uncontrolled with treatment
--------------------
Bronchospasm, Acute
transient; no treatment; 70% - 80% FEV1
of peak flow
requires treatment; normalizes with bronchodilator; FEV1 50% - 70% (of peak flow)
no normalization with bronchodilator; FEV1 25% - 50% of peak flow; or retractions present
cyanosis: FEV1 < 25% of peak flow or intubation necessary
Dyspnea
dyspnea on exertion dyspnea with normal activity
dyspnea at rest dyspnea requiring Oxygen therapy
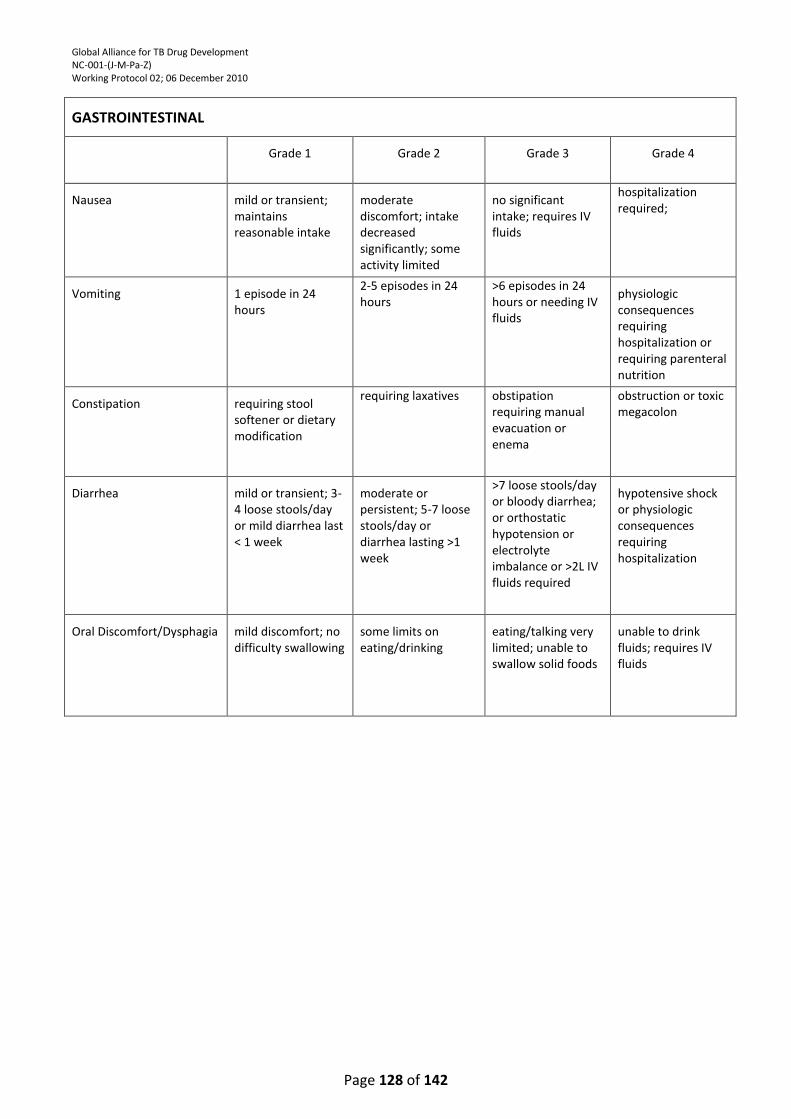
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 128 of 142
GASTROINTESTINAL
Grade 1
Grade 2
Grade 3
Grade 4
Nausea
mild or transient; maintains reasonable intake
moderate discomfort; intake decreased significantly; some activity limited
no significant intake; requires IV fluids
hospitalization required;
Vomiting
1 episode in 24 hours
2-5 episodes in 24 hours
>6 episodes in 24 hours or needing IV fluids
physiologic consequences requiring hospitalization or requiring parenteral nutrition
Constipation
requiring stool softener or dietary modification
requiring laxatives obstipation requiring manual evacuation or enema
obstruction or toxic megacolon
Diarrhea
mild or transient; 3-4 loose stools/day or mild diarrhea last < 1 week
moderate or persistent; 5-7 loose stools/day or diarrhea lasting >1 week
>7 loose stools/day or bloody diarrhea; or orthostatic hypotension or electrolyte imbalance or >2L IV fluids required
hypotensive shock or physiologic consequences requiring hospitalization
Oral Discomfort/Dysphagia
mild discomfort; no difficulty swallowing
some limits on eating/drinking
eating/talking very limited; unable to swallow solid foods
unable to drink fluids; requires IV fluids
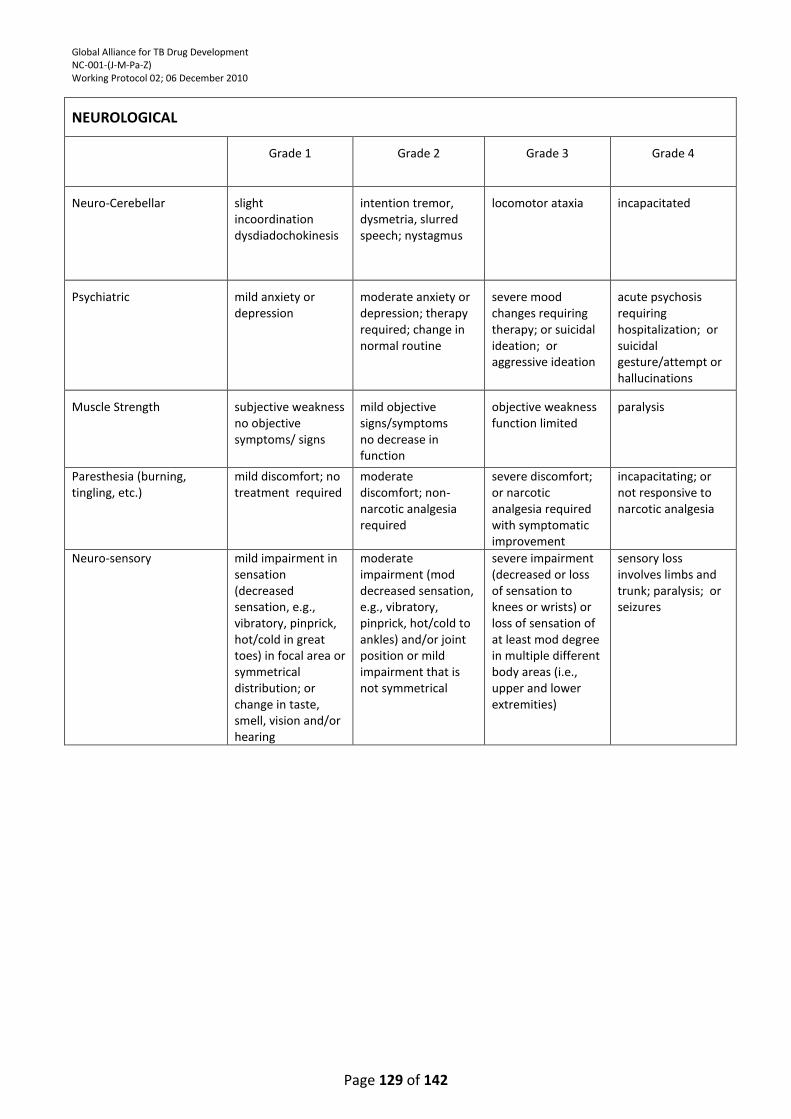
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 129 of 142
NEUROLOGICAL
Grade 1
Grade 2
Grade 3
Grade 4
Neuro-Cerebellar
slight incoordination dysdiadochokinesis
intention tremor, dysmetria, slurred speech; nystagmus
locomotor ataxia
incapacitated
Psychiatric
mild anxiety or depression
moderate anxiety or depression; therapy required; change in normal routine
severe mood changes requiring therapy; or suicidal ideation; or aggressive ideation
acute psychosis requiring hospitalization; or suicidal gesture/attempt or hallucinations
Muscle Strength
subjective weakness no objective symptoms/ signs
mild objective signs/symptoms no decrease in function
objective weakness function limited
paralysis
Paresthesia (burning, tingling, etc.)
mild discomfort; no treatment required
moderate discomfort; non-narcotic analgesia required
severe discomfort; or narcotic analgesia required with symptomatic improvement
incapacitating; or not responsive to narcotic analgesia
Neuro-sensory mild impairment in sensation (decreased sensation, e.g., vibratory, pinprick, hot/cold in great toes) in focal area or symmetrical distribution; or change in taste, smell, vision and/or hearing
moderate impairment (mod decreased sensation, e.g., vibratory, pinprick, hot/cold to ankles) and/or joint position or mild impairment that is not symmetrical
severe impairment (decreased or loss of sensation to knees or wrists) or loss of sensation of at least mod degree in multiple different body areas (i.e., upper and lower extremities)
sensory loss involves limbs and trunk; paralysis; or seizures
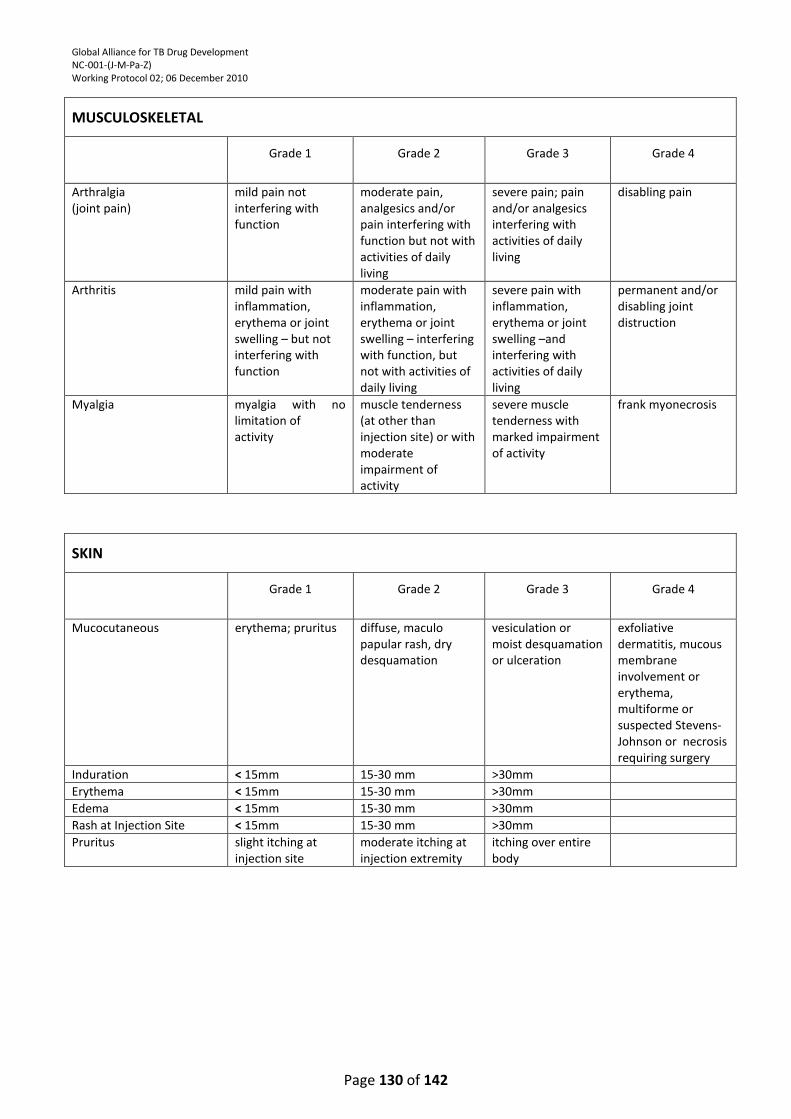
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 130 of 142
MUSCULOSKELETAL
Grade 1
Grade 2
Grade 3
Grade 4
Arthralgia (joint pain)
mild pain not interfering with function
moderate pain, analgesics and/or pain interfering with function but not with activities of daily living
severe pain; pain and/or analgesics interfering with activities of daily living
disabling pain
Arthritis mild pain with inflammation, erythema or joint swelling – but not interfering with function
moderate pain with inflammation, erythema or joint swelling – interfering with function, but not with activities of daily living
severe pain with inflammation, erythema or joint swelling –and interfering with activities of daily living
permanent and/or disabling joint distruction
Myalgia myalgia with no limitation of activity
muscle tenderness (at other than injection site) or with moderate impairment of activity
severe muscle tenderness with marked impairment of activity
frank myonecrosis
SKIN
Grade 1
Grade 2
Grade 3
Grade 4
Mucocutaneous erythema; pruritus diffuse, maculo papular rash, dry desquamation
vesiculation or moist desquamation or ulceration
exfoliative dermatitis, mucous membrane involvement or erythema, multiforme or suspected Stevens-Johnson or necrosis requiring surgery
Induration < 15mm 15-30 mm >30mm
Erythema < 15mm 15-30 mm >30mm
Edema < 15mm 15-30 mm >30mm
Rash at Injection Site < 15mm 15-30 mm >30mm
Pruritus slight itching at injection site
moderate itching at injection extremity
itching over entire body
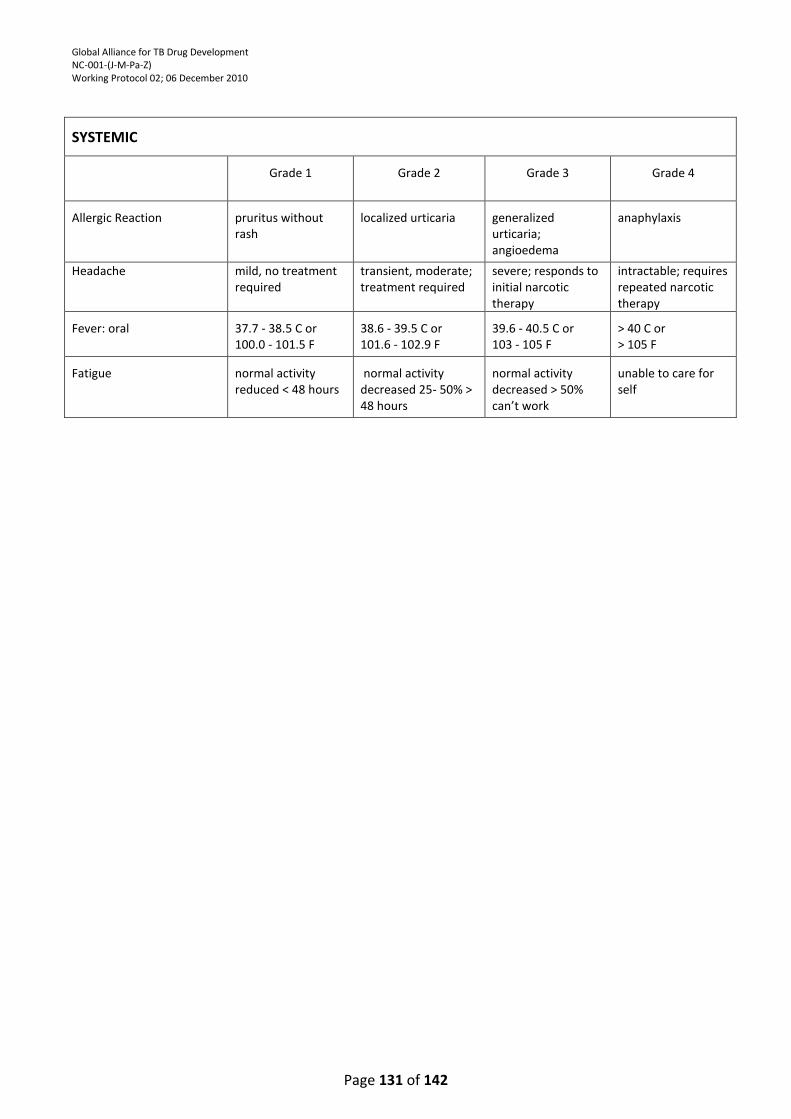
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 131 of 142
SYSTEMIC
Grade 1
Grade 2
Grade 3
Grade 4
Allergic Reaction
pruritus without rash
localized urticaria
generalized urticaria; angioedema
anaphylaxis
Headache mild, no treatment required
transient, moderate; treatment required
severe; responds to initial narcotic therapy
intractable; requires repeated narcotic therapy
Fever: oral
37.7 - 38.5 C or 100.0 - 101.5 F
38.6 - 39.5 C or 101.6 - 102.9 F
39.6 - 40.5 C or 103 - 105 F
> 40 C or > 105 F
Fatigue
normal activity reduced < 48 hours
normal activity decreased 25- 50% > 48 hours
normal activity decreased > 50% can’t work
unable to care for self
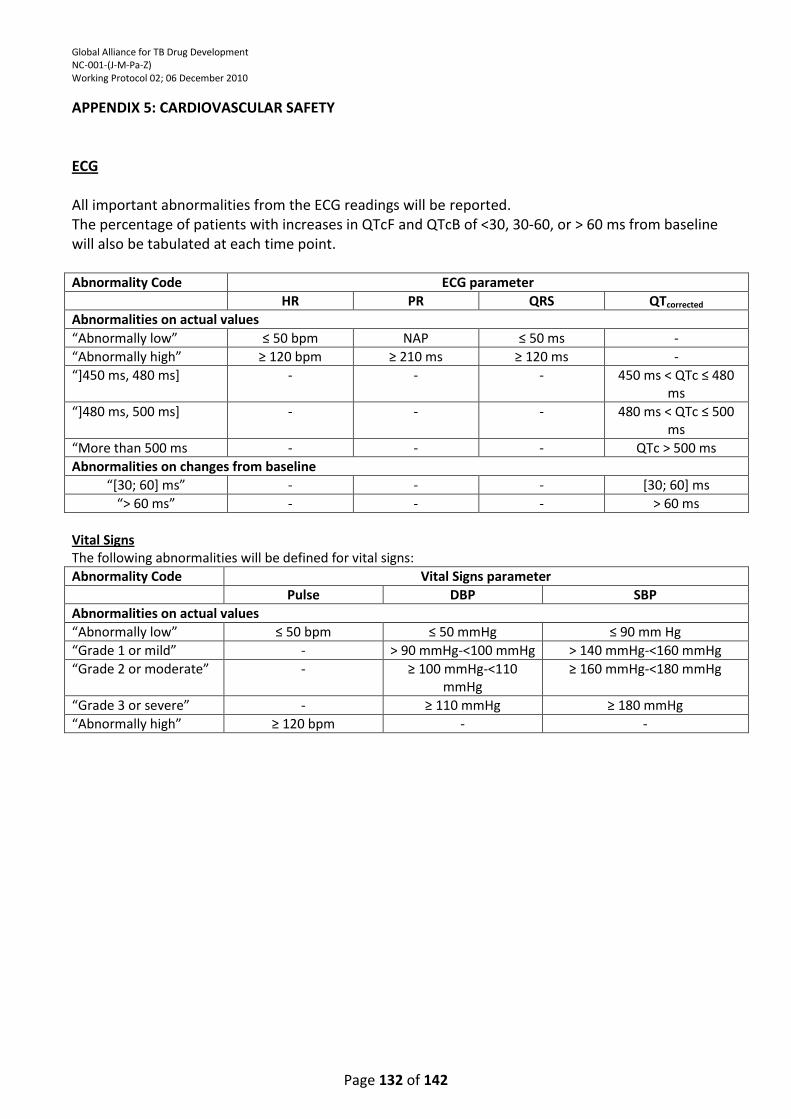
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 132 of 142
APPENDIX 5: CARDIOVASCULAR SAFETY ECG All important abnormalities from the ECG readings will be reported. The percentage of patients with increases in QTcF and QTcB of <30, 30-60, or > 60 ms from baseline will also be tabulated at each time point. Abnormality Code ECG parameter
HR PR QRS QTcorrected
Abnormalities on actual values
“Abnormally low” ≤ 50 bpm NAP ≤ 50 ms -
“Abnormally high” ≥ 120 bpm ≥ 210 ms ≥ 120 ms -
“+450 ms, 480 ms+ - - - 450 ms < QTc ≤ 480 ms
“+480 ms, 500 ms+ - - - 480 ms < QTc ≤ 500 ms
“More than 500 ms - - - QTc > 500 ms
Abnormalities on changes from baseline
“*30; 60+ ms” - - - [30; 60] ms
“> 60 ms” - - - > 60 ms
Vital Signs The following abnormalities will be defined for vital signs:
Abnormality Code Vital Signs parameter
Pulse DBP SBP
Abnormalities on actual values
“Abnormally low” ≤ 50 bpm ≤ 50 mmHg ≤ 90 mm Hg
“Grade 1 or mild” - > 90 mmHg-<100 mmHg > 140 mmHg-<160 mmHg
“Grade 2 or moderate” - ≥ 100 mmHg-<110 mmHg
≥ 160 mmHg-<180 mmHg
“Grade 3 or severe” - ≥ 110 mmHg ≥ 180 mmHg
“Abnormally high” ≥ 120 bpm - -

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 133 of 142
APPENDIX 6: PYRAZINAMIDE PACKAGE INSERT

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 134 of 142
APPENDIX 7: MOXIFLOXACIN PACKAGE INSERT

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 135 of 142

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 136 of 142

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 137 of 142

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 138 of 142
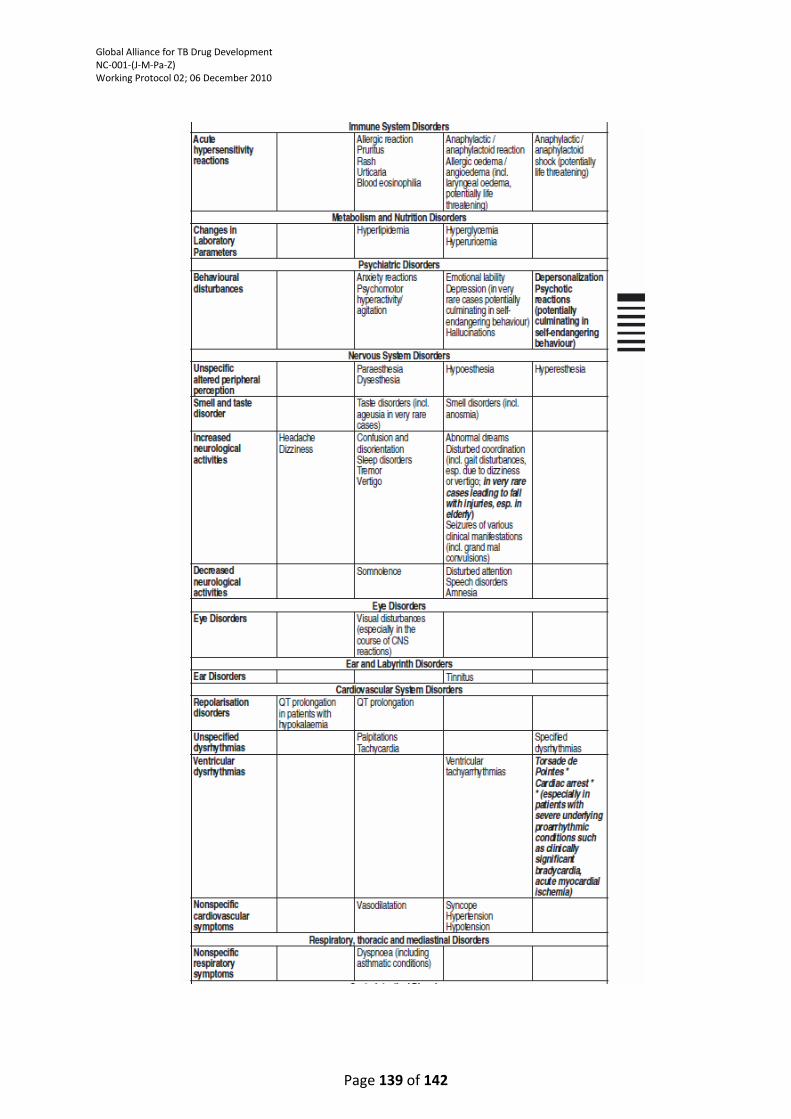
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 139 of 142
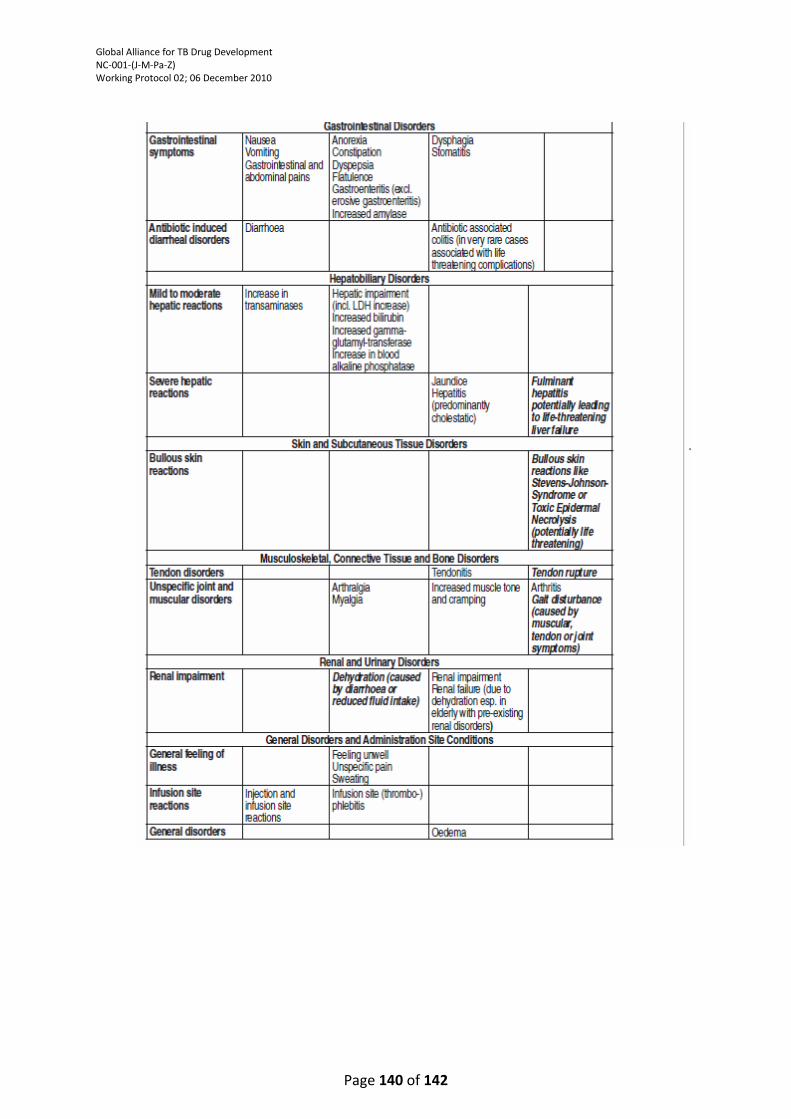
Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 140 of 142

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 141 of 142

Global Alliance for TB Drug Development NC-001-(J-M-Pa-Z) Working Protocol 02; 06 December 2010
Page 142 of 142
Related Documents











