有機ホウ素化合物の 新しい合成方法と 発光性メカノクロミズム材料 北海道大学大学院工学研究院 伊藤 肇 住友化学・健康・農業関連事業研究所 講演会 2014年12月15日

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

有機ホウ素化合物の新しい合成方法と発光性メカノクロミズム材料
北海道大学大学院工学研究院 伊藤 肇
住友化学・健康・農業関連事業研究所 講演会 2014年12月15日

本日の講演内容
1. 銅触媒によるホウ素化反応:光学活性アルキルホウ素化合物の合成方法
3. メカノ応答性をもつ発光性金錯体
2. BBSホウ素化反応:遷移金属フリーでかつ官能基共存性のある芳香族ホウ素化合物の合成方法

有機ホウ素化合物の基本的な性質
有機ホウ素反応剤は高い安定性(保存性)官能基許容性をもつ
■ ホウ素は他の金属に比べて電気陰性度が大きい
■ 有機ホウ素化合物は、安定性(保存性)と官能基許容性をもつ
C Bδ+δ–
2.6 2.0C Mgδ+δ–
2.5 1.3C Liδ+δ–
2.5 1.0

有機ホウ素化合物の基本的な性質
LUMO of BMe3
R BR
R
✔ 弱い求核性と同時に空の2p軌道に 由来したルイス酸性をもつ
✔ R3Bはラジカル的に酸素と反応する。 空気下で不安定。
空の2p軌道
■トリアルキルボラン:R3B
R B
O
O
■アルキルボロン酸エステル:RB(OR)2
酸素のlone pairからの空p軌道への電子供与 →ルイス酸性の低下
✔ ルイス酸性の低下と共有結合性の 増加 → 化合物の安定性向上
HOMO-2 of MeB(OR)2
酸素へのσ電子流れ込み→共有結合性の増加
BO
OR

有機ホウ素化合物の基本的な性質
■ RB(OR)2 は塩基の添加により活性化される。
BOR
ORR
ORORB
OR
ORR B
OR
ORR OR
■ 様々なボロン酸エステル
O
OBR
O
OBR
O
OBR
O
OBR
R
R
R B(pin)R Bpin
OH
OHBR
OBOBO
B
R R
R
boroxine
–H2O
■ ボロン酸は縮合に注意
■ ボロン酸の保護基
R BF3-K+
B NO
O
O
O
MeRN
BN
R
H
H
Burke, 2008 Suginome, 2007R B(dan)MIDA boronate
BO
OOR
M+
Miyaura, 2008
BAr
■ その他の安定化
the anthracene moieties for the cyclization to occur selec-tively at the 1,8-positions and to prevent the strong aggrega-tion of the resulting PAH p skeleton. This anthryl group wasoriginally reported by Anderson and coworkers, and is widelyused for the synthesis of expanded p skeletons.[13]
The precursor 3 was prepared in 54% yield by thelithiation of 9-bromobis(mesityloxy)anthracene 4[13] withnBuLi, followed by treatment with dibromodiborapentacene5.[14] Compound 3 showed high stability to water and oxygenas a result of the steric protection of the boron atoms by thebulky anthryl groups. The cyclodehydrogenation of 3 with anexcess of FeCl3 proceeded successfully to form 1a in 51%yield as a deep purple solid. As expected, the doubly B-dopednanographene 1a is stable enough to handle in air and wasisolated by column chromatography on silica gel without anyspecial precautions. Compound 1a is sufficiently soluble incommon organic solvents, such as chlorobenzene(4.8 mgmL!1) and ortho-dichlorobenzene (10.8 mg mL!1),thus demonstrating its processability in solution.
The structure of 1 was unambiguously characterized bymass spectrometry, NMR spectroscopy (Figure 1), and finallyX-ray crystallography (Figure 2). These analyses revealed 1ato be a single compound. High-resolution atmospheric
pressure chemical ionization time-of-flight (APCI-TOF) MSshowed a parent ion signal for 1a at m/z 1157.4942 (calcd forC84H63O4B2 [M+H]+, m/z 1157.4931; see the SupportingInformation). Two sets of coupled signals (Ha and Hb, andHc and Hd in Figure 1) were observed at 6.96 and 9.12 ppm,and 7.76 and 9.02 ppm, respectively in the 1H NMR spectrum
of 1a in [D2]tetrachloroethane at 353 K (Figure 1). Therelatively downfield chemical shifts of the Hb and Hd signalsare attributed to the deshielding effect by the ring current ofthe neighboring benzene rings in the cove region. The otherdeshielded singlet signal at 10.85 ppm corresponds to the He
atom, which reflects the close contact with the oxygen atoms(see the Supporting Information).[13] Variable-temperature1H NMR measurements from 193 to 353 K did not show anysignificant change. This temperature independency indicatesa large energy gap between the singlet closed-shell groundstate and a triplet excited state. The gap was calculatedtheoretically to be 34.9 kcalmol!1 for 1a at the B3LYP/6-31G* level, which is far larger than that of the parent undopednanographene 2 (1.5 kcalmol!1) with an open-shell groundstate (see the Supporting Information). The broad 11B NMRsignal of 1a at 58.0 ppm is typical of tricoordinated boroncompounds.
The single crystals were obtained by slow diffusion ofheptane into a solution of 1a in chlorobenzene. The X-raycrystallographic analysis revealed a contorted polycyclicskeleton of the B-doped nanographene 1a composed of 48sp2-hybridized C atoms and two tricoordinated B atoms(Figure 2).[15] Fifteen six-membered rings are fused to formthe nanographene sheet with four cove regions and two zigzagedges. As a consequence of steric overcrowding of the Hb andHd atoms in the cove regions, the p-conjugated core skeletonis distorted away from planarity. Although the distancebetween the most deviated C atoms and the C48B2 meanplane is 1.02 !, the dihedral angles between the mostcontorted benzene rings and the central C4B2 ring is 19.78.The doping positions of the two B atoms were determinedunambiguously, as the B!C bond lengths of 1.507(2), 1.531(2),and 1.535(2) ! are significantly longer than those of the otherC!C bonds (1.37–1.48 !). Notably, these B!C bonds aremuch shorter than those of the nonfused triphenylborane(1.57–1.59 !).[16] This structural characteristic has alreadybeen observed in the other planarized triarylboranes pre-
Scheme 1. Stepwise boron doping of an extended polyaromatic hydro-carbon.
Scheme 2. Synthesis of B-doped nanographene 1a. Reagents andconditions: a) 4, nBuLi, Et2O, from 0 8C to 25 8C, then 5, toluene, from0 8C to 25 8C; b) FeCl3, CH3NO2 and CH2Cl2.
Figure 1. 1H NMR spectrum of 1a in [D2]tetrachloroethane at 353 K.
AngewandteChemie
12207Angew. Chem. Int. Ed. 2012, 51, 12206 –12210 ! 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org
平面化: Yamaguchi, 2012
立体障害導入

合成反応剤としての有機ホウ素化合物
■ Roush アリル化 (1985~)
William R. Roush photo: Wikipedia
B R1
R2
O
O
iPrO2C
iPrO2CR H
O
R
OH
R1 R2
O
B
RR1
R2
+
■ H. C. Brown: 有機ホウ素化合物が優れた合成反応剤であることを見いだした。
H. C. Brown (1912-2004) photo: Purdue Univ.H. C. Brown (1961)
BH2 +
H B(ipc)2 H OHoxidation
99% ee
■ 有機ホウ素化合物の活用により有機合成化学が大きく発展

NO
HO
OnBu
NNN
HN
Suzuki-Miyaura Coupling
■ Suzuki-Miyaura クロスカップリング (1979~)
Akira Suzuki
RB
B
XR
Pd catalyst
鈴木カップリングの登場
NNHN
NNH2
OCl
Cl F
Crizotinib, 肺がん治療薬Valsartan, 高血圧治療薬
■ Suzuki-Miyaura クロスカップリングは実験室~工業生産レベルまで 幅広く使われている。

the anthracene moieties for the cyclization to occur selec-tively at the 1,8-positions and to prevent the strong aggrega-tion of the resulting PAH p skeleton. This anthryl group wasoriginally reported by Anderson and coworkers, and is widelyused for the synthesis of expanded p skeletons.[13]
The precursor 3 was prepared in 54% yield by thelithiation of 9-bromobis(mesityloxy)anthracene 4[13] withnBuLi, followed by treatment with dibromodiborapentacene5.[14] Compound 3 showed high stability to water and oxygenas a result of the steric protection of the boron atoms by thebulky anthryl groups. The cyclodehydrogenation of 3 with anexcess of FeCl3 proceeded successfully to form 1a in 51%yield as a deep purple solid. As expected, the doubly B-dopednanographene 1a is stable enough to handle in air and wasisolated by column chromatography on silica gel without anyspecial precautions. Compound 1a is sufficiently soluble incommon organic solvents, such as chlorobenzene(4.8 mgmL!1) and ortho-dichlorobenzene (10.8 mg mL!1),thus demonstrating its processability in solution.
The structure of 1 was unambiguously characterized bymass spectrometry, NMR spectroscopy (Figure 1), and finallyX-ray crystallography (Figure 2). These analyses revealed 1ato be a single compound. High-resolution atmospheric
pressure chemical ionization time-of-flight (APCI-TOF) MSshowed a parent ion signal for 1a at m/z 1157.4942 (calcd forC84H63O4B2 [M+H]+, m/z 1157.4931; see the SupportingInformation). Two sets of coupled signals (Ha and Hb, andHc and Hd in Figure 1) were observed at 6.96 and 9.12 ppm,and 7.76 and 9.02 ppm, respectively in the 1H NMR spectrum
of 1a in [D2]tetrachloroethane at 353 K (Figure 1). Therelatively downfield chemical shifts of the Hb and Hd signalsare attributed to the deshielding effect by the ring current ofthe neighboring benzene rings in the cove region. The otherdeshielded singlet signal at 10.85 ppm corresponds to the He
atom, which reflects the close contact with the oxygen atoms(see the Supporting Information).[13] Variable-temperature1H NMR measurements from 193 to 353 K did not show anysignificant change. This temperature independency indicatesa large energy gap between the singlet closed-shell groundstate and a triplet excited state. The gap was calculatedtheoretically to be 34.9 kcalmol!1 for 1a at the B3LYP/6-31G* level, which is far larger than that of the parent undopednanographene 2 (1.5 kcalmol!1) with an open-shell groundstate (see the Supporting Information). The broad 11B NMRsignal of 1a at 58.0 ppm is typical of tricoordinated boroncompounds.
The single crystals were obtained by slow diffusion ofheptane into a solution of 1a in chlorobenzene. The X-raycrystallographic analysis revealed a contorted polycyclicskeleton of the B-doped nanographene 1a composed of 48sp2-hybridized C atoms and two tricoordinated B atoms(Figure 2).[15] Fifteen six-membered rings are fused to formthe nanographene sheet with four cove regions and two zigzagedges. As a consequence of steric overcrowding of the Hb andHd atoms in the cove regions, the p-conjugated core skeletonis distorted away from planarity. Although the distancebetween the most deviated C atoms and the C48B2 meanplane is 1.02 !, the dihedral angles between the mostcontorted benzene rings and the central C4B2 ring is 19.78.The doping positions of the two B atoms were determinedunambiguously, as the B!C bond lengths of 1.507(2), 1.531(2),and 1.535(2) ! are significantly longer than those of the otherC!C bonds (1.37–1.48 !). Notably, these B!C bonds aremuch shorter than those of the nonfused triphenylborane(1.57–1.59 !).[16] This structural characteristic has alreadybeen observed in the other planarized triarylboranes pre-
Scheme 1. Stepwise boron doping of an extended polyaromatic hydro-carbon.
Scheme 2. Synthesis of B-doped nanographene 1a. Reagents andconditions: a) 4, nBuLi, Et2O, from 0 8C to 25 8C, then 5, toluene, from0 8C to 25 8C; b) FeCl3, CH3NO2 and CH2Cl2.
Figure 1. 1H NMR spectrum of 1a in [D2]tetrachloroethane at 353 K.
AngewandteChemie
12207Angew. Chem. Int. Ed. 2012, 51, 12206 –12210 ! 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org
Yamaguchi, 2012
Fig. 9 Left: the structure of compound 17. Middle: A green OLED based on compound 15. Right: An orange OLED based on 17.
4. Devices incorporating 8 wt.% of this material doped intoCBP (4,4¢-bis(9-carbazolyl)biphenyl) showed remarkably highefficiency red phosphorescence, with maximum current, power andexternal quantum efficiencies (EQEs) of 10.31 cd A-1, 5.04 lm W-1
and 9.36%, respectively. It should be noted that, while much higherefficiencies have been achieved with the parent green phosphorIr(ppy)2(acac) using a similar device structure,8a,b the performanceof 4 is still very impressive as it is a red emitter and is expected tohave a much lower efficiency than the parent molecule accordingto the well-known energy gap law.
We have recently examined the impact of functionalization withtriarylboron on the performance of OLEDs containing platinumphosphors.11 Pt(II) complexes present a different challenge thancomplexes of Ir(III), as their square planar geometry increasesthe tendency of these materials for Pt–Pt stacking and exciplexemission. While in some situations this can be advantageous,especially in achieving white OLEDs,28 Pt(II) excimers generallyexhibit lower quantum efficiencies than the parent phosphors.Triarylborane-functionalized Pt(acac) compounds such as 14–16, however, have been found to be much less prone to excimerformation. In addition to 14–16, we have also examined a numberof other BMes2-functionalized NŸC-chelate Pt(acac) complexes.11
In all cases these boron-functionalized complexes were brightlyphosphorescent at room temperature in the solid state andsolution, due to a mixture of LC and MLCT phosphorescence.Furthermore, all of these complexes exhibited significantly higherUP than analogous complexes lacking a boron center.11 Consistentwith earlier studies, the presence of the triarylboron group greatlyincreased the intensity of the MLCT absorption band, and DFTcalculations indicate that the empty orbital on boron was a largecontributor to the LUMO in all cases. Indeed, complex 15 itselfwas found to exhibit an exceptionally high UP of 0.57 in the solidstate, and was evaluated as an emitter for OLEDs alongside theanalogous complex Pt(ppy)(acac), which lacked the boryl group.OLEDs using 15 as an emitter exhibited green emission (CIEcoordinates = 0.35, 0.61), with maximum efficiencies of 34.5 cdA-1, 29.8 lm W-1 and 8.9% EQE compared to 14.1 cd A-1, 11.7 lmW-1 and 6.9% EQE for those using Pt(ppy)(acac). Furthermore,the efficiency of devices containing 15 as the emitter were amongthe highest achieved using Pt(II) to date.29 The improved efficiencyof 15 OLEDs could be attributed to three factors: 1) higher internalquantum efficiency due to the improved UP of the borylatedphosphor itself, 2) reduced low-efficiency exciplex emission due tothe presence of the boryl group, and 3) improved electron injectionand mobility in the emissive layer.
Hole mobilities typically exceed electron mobilities in organicmaterials by 1–2 orders of magnitude,30 leading to charge im-balance in the device and reduced efficiency. For this reason,
improving electron mobility in the emissive layer is one strategythat can be used to achieve better carrier balance in OLEDs.31
To confirm that the BMes2 group indeed improves electrontransport in the device, we fabricated single-carrier devices32
capable of transporting electrons only from thin films of 15or Pt(ppy)(acac). Remarkably, the film of 15 was capable ofsupporting a current density 3–4 orders of magnitude higher thanthat of Pt(ppy)(acac), indicative of markedly improved electronmobility.11a Furthermore, this highlighted the bifunctional natureof the boron-functionalized materials, namely efficient electrontransport and phosphorescence.
Following the success of this system, we later extended thisconcept to a trifunctional material11b 17 (Fig. 9) designed asa phosphorescent successor to the highly fluorescent molecule7, which had been incorporated into efficient blue fluorescentOLEDs.7d Similar to 16 in structure, this material further containsthe N-phenyl-1-naphthyl group as a strong electron donor. Thismoiety, taken from the widely used hole transport materialNPB (N,N¢-di-[(1-naphthalenyl)-N,N¢-diphenyl]-(1,10-biphenyl)-4,4¢-diamine), should thus be able to efficiently support oxidationand hole-transport. Furthermore, incorporation of the NPBmoiety leads to bright ligand-centerd charge transfer phosphores-cence, facilitated by Pt(II). When used in a doped emissive layer inOLEDs, devices exhibiting bright orange electrophosphorescence(lEL = 581 nm, CIE = 0.52, 0.47) with efficiencies of 35.0 cd A-1,36.6 lm W-1 and 10.1% EQE have been achieved. This is quiteremarkable since the emission of 17 is much red-shifted, comparedto that of 15. Though to date only three reports on the subject havebeen published,11,16 these results give a promising outlook for theuse of triarylboron-containing metal complexes as phosphorescentmaterials in OLEDs.
5. Triarylboron-containing metal complexes as anion sensors
Many reports to date have focused on the use of triarylboroncompounds as chemical sensors for fluoride and cyanide, due to thehigh selectivity with which the BMes2 group binds these anions andthe unique colorimetric and luminescent color changes that resultfrom the anion binding event. Several excellent reviews have beenpublished recently on the use of triarylboranes as anion sensors,in which the reader will find further information on strategies foranion detection and improving the sensitivity of the chemosensorin organic and protic media.5b,c
Transition metal-containing triarylboranes offer the advantageof long lived phosphorescence that minimizes interference frombackground fluorescence or scattering in sensing applications. Inaddition, transition metal compounds offer new possibilities forredox-active sensors, in which the analyte binding event triggers
7810 | Dalton Trans., 2011, 40, 7805–7816 This journal is © The Royal Society of Chemistry 2011
Dow
nloa
ded
by H
okka
ido
Dai
gaku
on
19 N
ovem
ber 2
012
Publ
ishe
d on
20
May
201
1 on
http
://pu
bs.rs
c.or
g | d
oi:1
0.10
39/C
1DT1
0292
C
Fig. 9 Left: the structure of compound 17. Middle: A green OLED based on compound 15. Right: An orange OLED based on 17.
4. Devices incorporating 8 wt.% of this material doped intoCBP (4,4¢-bis(9-carbazolyl)biphenyl) showed remarkably highefficiency red phosphorescence, with maximum current, power andexternal quantum efficiencies (EQEs) of 10.31 cd A-1, 5.04 lm W-1
and 9.36%, respectively. It should be noted that, while much higherefficiencies have been achieved with the parent green phosphorIr(ppy)2(acac) using a similar device structure,8a,b the performanceof 4 is still very impressive as it is a red emitter and is expected tohave a much lower efficiency than the parent molecule accordingto the well-known energy gap law.
We have recently examined the impact of functionalization withtriarylboron on the performance of OLEDs containing platinumphosphors.11 Pt(II) complexes present a different challenge thancomplexes of Ir(III), as their square planar geometry increasesthe tendency of these materials for Pt–Pt stacking and exciplexemission. While in some situations this can be advantageous,especially in achieving white OLEDs,28 Pt(II) excimers generallyexhibit lower quantum efficiencies than the parent phosphors.Triarylborane-functionalized Pt(acac) compounds such as 14–16, however, have been found to be much less prone to excimerformation. In addition to 14–16, we have also examined a numberof other BMes2-functionalized NŸC-chelate Pt(acac) complexes.11
In all cases these boron-functionalized complexes were brightlyphosphorescent at room temperature in the solid state andsolution, due to a mixture of LC and MLCT phosphorescence.Furthermore, all of these complexes exhibited significantly higherUP than analogous complexes lacking a boron center.11 Consistentwith earlier studies, the presence of the triarylboron group greatlyincreased the intensity of the MLCT absorption band, and DFTcalculations indicate that the empty orbital on boron was a largecontributor to the LUMO in all cases. Indeed, complex 15 itselfwas found to exhibit an exceptionally high UP of 0.57 in the solidstate, and was evaluated as an emitter for OLEDs alongside theanalogous complex Pt(ppy)(acac), which lacked the boryl group.OLEDs using 15 as an emitter exhibited green emission (CIEcoordinates = 0.35, 0.61), with maximum efficiencies of 34.5 cdA-1, 29.8 lm W-1 and 8.9% EQE compared to 14.1 cd A-1, 11.7 lmW-1 and 6.9% EQE for those using Pt(ppy)(acac). Furthermore,the efficiency of devices containing 15 as the emitter were amongthe highest achieved using Pt(II) to date.29 The improved efficiencyof 15 OLEDs could be attributed to three factors: 1) higher internalquantum efficiency due to the improved UP of the borylatedphosphor itself, 2) reduced low-efficiency exciplex emission due tothe presence of the boryl group, and 3) improved electron injectionand mobility in the emissive layer.
Hole mobilities typically exceed electron mobilities in organicmaterials by 1–2 orders of magnitude,30 leading to charge im-balance in the device and reduced efficiency. For this reason,
improving electron mobility in the emissive layer is one strategythat can be used to achieve better carrier balance in OLEDs.31
To confirm that the BMes2 group indeed improves electrontransport in the device, we fabricated single-carrier devices32
capable of transporting electrons only from thin films of 15or Pt(ppy)(acac). Remarkably, the film of 15 was capable ofsupporting a current density 3–4 orders of magnitude higher thanthat of Pt(ppy)(acac), indicative of markedly improved electronmobility.11a Furthermore, this highlighted the bifunctional natureof the boron-functionalized materials, namely efficient electrontransport and phosphorescence.
Following the success of this system, we later extended thisconcept to a trifunctional material11b 17 (Fig. 9) designed asa phosphorescent successor to the highly fluorescent molecule7, which had been incorporated into efficient blue fluorescentOLEDs.7d Similar to 16 in structure, this material further containsthe N-phenyl-1-naphthyl group as a strong electron donor. Thismoiety, taken from the widely used hole transport materialNPB (N,N¢-di-[(1-naphthalenyl)-N,N¢-diphenyl]-(1,10-biphenyl)-4,4¢-diamine), should thus be able to efficiently support oxidationand hole-transport. Furthermore, incorporation of the NPBmoiety leads to bright ligand-centerd charge transfer phosphores-cence, facilitated by Pt(II). When used in a doped emissive layer inOLEDs, devices exhibiting bright orange electrophosphorescence(lEL = 581 nm, CIE = 0.52, 0.47) with efficiencies of 35.0 cd A-1,36.6 lm W-1 and 10.1% EQE have been achieved. This is quiteremarkable since the emission of 17 is much red-shifted, comparedto that of 15. Though to date only three reports on the subject havebeen published,11,16 these results give a promising outlook for theuse of triarylboron-containing metal complexes as phosphorescentmaterials in OLEDs.
5. Triarylboron-containing metal complexes as anion sensors
Many reports to date have focused on the use of triarylboroncompounds as chemical sensors for fluoride and cyanide, due to thehigh selectivity with which the BMes2 group binds these anions andthe unique colorimetric and luminescent color changes that resultfrom the anion binding event. Several excellent reviews have beenpublished recently on the use of triarylboranes as anion sensors,in which the reader will find further information on strategies foranion detection and improving the sensitivity of the chemosensorin organic and protic media.5b,c
Transition metal-containing triarylboranes offer the advantageof long lived phosphorescence that minimizes interference frombackground fluorescence or scattering in sensing applications. Inaddition, transition metal compounds offer new possibilities forredox-active sensors, in which the analyte binding event triggers
7810 | Dalton Trans., 2011, 40, 7805–7816 This journal is © The Royal Society of Chemistry 2011
Dow
nloa
ded
by H
okka
ido
Dai
gaku
on
19 N
ovem
ber 2
012
Publ
ishe
d on
20
May
201
1 on
http
://pu
bs.rs
c.or
g | d
oi:1
0.10
39/C
1DT1
0292
C Wang, 2010
■有機ホウ素化合物の高効率合成法の開発はますます重要になっている ●官能基許容性 ●光学活性隊の不斉合成 ●遷移金属フリー ●立体障害克服 ●低コスト
■ 有機ホウ素化合物そのものを医薬品としたもの
有機ホウ素化合物のニーズの高まり
NH
HN B
ON
N
Ph
OOH
OH
OB
F
OH
Bortezomib, 悪性リンパ腫治療薬 Tavaborole, 抗真菌剤
カルボン酸 の拮抗作用
糖鎖認識
■ 有機電子材料を志向した化合物
有機EL

本日の講演内容
1. 銅触媒によるホウ素化反応:光学活性アルキルホウ素化合物の合成方法
3. メカノ応答性をもつ発光性金錯体
2. BBSホウ素化反応:遷移金属フリーでかつ官能基共存性のある芳香族ホウ素化合物の合成方法

B
H3C H
OH
H3C H
NR2
H3C H
R
H3C H
■光学活性アルキルホウ素化合物 は、各種光学活性化合物に 立体選択的に変換できるため 特に有用
光学活性アルキルホウ素化合物
触媒的不斉合成 官能基許容性
を満たすような 合成方法の確立 が課題

既存の合成方法では?
■ 触媒的不斉ホウ素化: 限定的(実は二三例しかない)
Ph
[Rh(cod)2]BF4 (1 mol %)(R)-BINAP (1 mol %)
–78°C, 6 h
OHB
O+ Ph
B(cat)
91%, 96.2 % ee
Hayashi, T.; Matsumoto, Y.; Ito, Y. J. Am. Chem. Soc. 1989, 111, 3426.!
■ 不斉ホウ素化合物を用いるヒドロホウ素化: 化学量論量の不斉源
BH2 +
H B(ipc)2 H OHoxidation
99% ee
OH+ 2
H B+H
B
■ ヒドロホウ素化:代表的な有機ホウ素化合物の合成方法の一つ

既存の合成方法では?■ 炭素求核剤+ホウ素求電子剤
R Li
R MgBr
+ R BR'2X BR'2
R R'
Li
✔ 官能許容性が大幅に低下 ✔ α-キラル金属→ラセミ化
N
Ph O N(i-Pr)2
O
(-)-sparteine
s-BuLi–78°C
Ph O N(i-Pr)2
OLi
H
N
Ph O N(i-Pr)2
OB
H
OO
Et
Et BO
O
MgBr2
PhBO
OEt
90%, 96% ee
R Li BX
■ 化学量論量の不斉源を利用 (Aggarwal, et al):頑張ればできる
R Li BX

ホウ素-銅触媒反応系の発見(2000年)
Segawa, Y.; Yamashita, M.; Nozaki, K. Science 2006, 314, 113.
NNBBr
iPr
iPr iPr
iPr
NNBLi
iPr
iPr iPr
iPrLi, naphthalene
THF
■ホウ素アニオンは合成が困難であった。
CuCl/KOAc: Takahashi, K.; Ishiyama, T.; Miyaura, N. Chem. Lett. 2000, 982. CuX/PR3: Ito, H.; Yamanaka, H.; Tateiwa, J.; Hosomi, A. Tetrahedron Lett. 2000, 41, 6821.
+
cat. CuX PR3
DMI, rt
H3O+
OO
BB B
O
OO
O
OO
87%
■エノンへのホウ素基の形式的求核付加
CuX
BB
Cu BL
L
ホウ素-銅触媒活性種•求核的な反応特性•選択性のコントロールが可能
銅触媒/ジボロン を用いた有機ホウ素化合物の合成法 を詳しく検討

・Xantphosがベストの配位子 ・高いγ-選択性 ・E体が生成
Ito, H.; Kawakami, C.; Sawamura, M. J. Am. Chem. Soc. 2005, 127, 16034.
Cu(O-t-Bu)/ ligand(5 mol %)
GC yield, %
dppf
100
3744
Xantphos
Ligand E : Z a
97 : 399 : 1
96 : 4> 99 : < 197 : 3 > 99 : < 1
11 62 : 38> 99 : < 1
γ : α
dppedppp
OPPh2Ph2P
+
2.0 equiv.
OB
O
OB
O
Xantphos:
Pd(dba)2 0 57 : 43
R
OCO2Me
R
B
R = (CH2)3Ph
O O
γ
THF, rt, 3 h
BCu
OR
L
B
R
OCO2Me
R
BO O
R
BOO+
57:43
Pd cat.
(pin)B–B(pin)
R
PdII
B(pin)
アリル位ホウ素置換反応の開発 (2005年~)

S R
2.4 equiv.
Bu
B(pin)MeO2CO Bu(pin)B B(pin)+
THF, 0 °C, 40 h
10 mol% Cu(O-t-Bu)ーXantphos
88%, 97% eeγ:α = >99:1, E:Z = >99:1
97% ee
Ito, H.; Kawakami, C.; Sawamura, M. J. Am. Chem. Soc. 2005, 127, 16034.
C CR2C
H R1R3
OR
HC C
B
R1R2
H
CR3H
C CR2C
H R1R3
OR
H
Cu BLL Cu B
■ SN2' 型反応により、さまざまなアリルホウ素化合物が合成可能
アリル位ホウ素置換反応の開発 (2005年~)
■ アリルホウ素化合物の触媒的不斉合成に初めて成功
Ito, H.; Ito, S.; Sasaki, Y.; Matsuura, K.; Sawamura, M. J. Am. Chem. Soc. 2007, 129, 14856.
ligand: QuinoxP* 今本恒雄先生(2005)THF, 0 ˚C R
B(pin)
(pin)B B(pin)+
R OCO2Me chiral ligand/ Cu(O-t-Bu)
5 mol%
20 h, 78%, 96% ee
N
N P
P
t-BuMe
t-Bu MeR = CH2CH2Ph

Ito, H.; Ito, S.; Sasaki, Y.; Matsuura, K.; Sawamura, M. J. Am. Chem. Soc. 2007, 129, 14856.
THF, 0 ˚C R
B(pin)
(pin)B B(pin)+
R OCO2Me Ligand/ Cu(O-t-Bu)5 mol%
■今本恒雄教授 千葉大学(2005)
R
RO
BCu
P
P
tBu
×触媒的不斉アリル位ホウ素置換反応
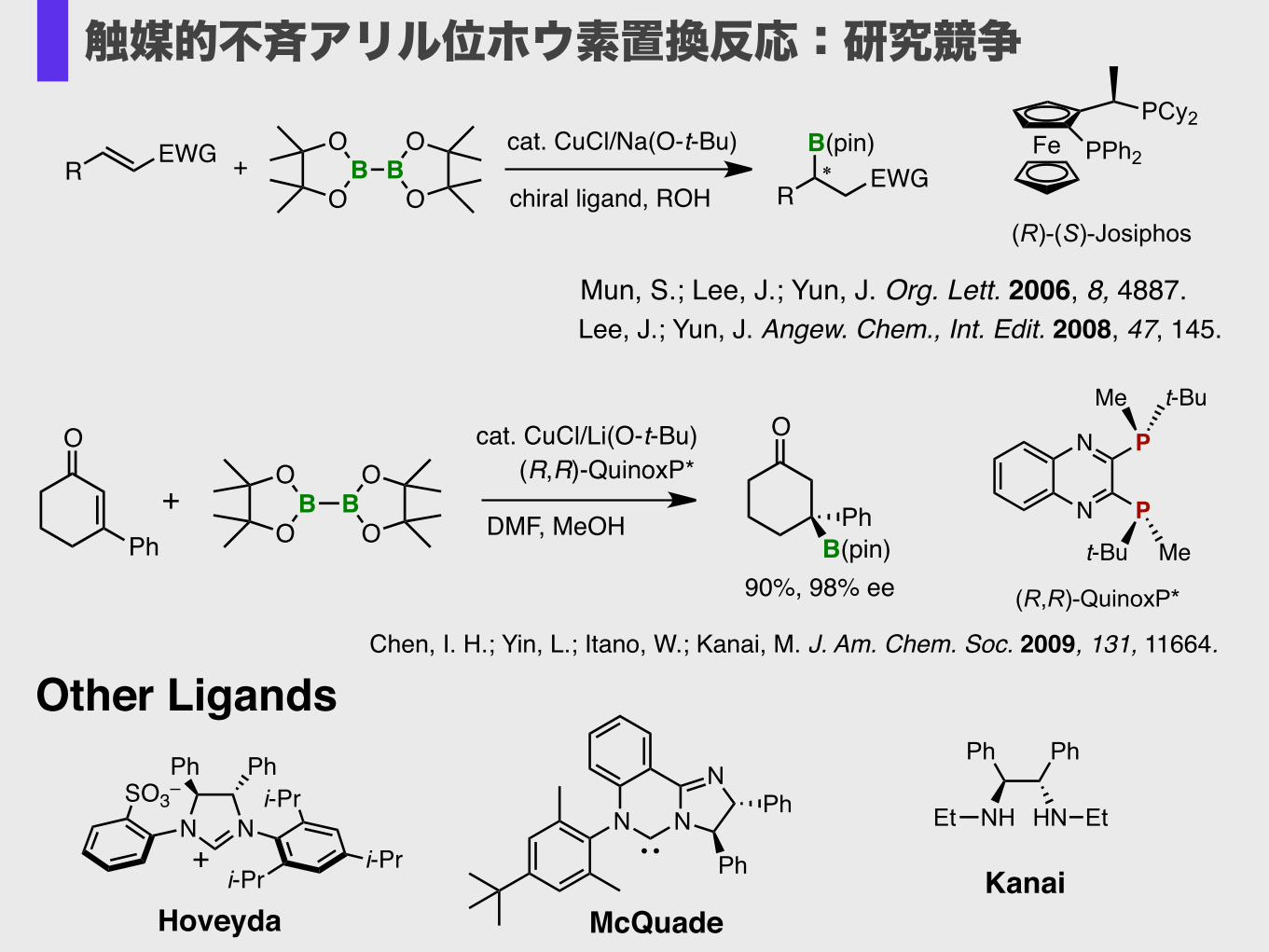
Mun, S.; Lee, J.; Yun, J. Org. Lett. 2006, 8, 4887.!Lee, J.; Yun, J. Angew. Chem., Int. Edit. 2008, 47, 145.�
R EWGOB
OB
O
O+
cat. CuCl/Na(O-t-Bu)
chiral ligand, ROH R∗∗ EWGB(pin) Fe PPh2
PCy2
(R)-(S)-Josiphos
O
Ph
cat. CuCl/Li(O-t-Bu)O
BO
OB
O+
(R,R)-QuinoxP*
DMF, MeOH
O
B(pin)Ph
90%, 98% ee
N
N P
P
t-BuMe
t-Bu Me
(R,R)-QuinoxP*
Chen, I. H.; Yin, L.; Itano, W.; Kanai, M. J. Am. Chem. Soc. 2009, 131, 11664.
N N
Ph Phi-Pr
i-Pri-Pr
SO3–
+
Other Ligands
Hoveyda
N N
N
Ph
Ph
McQuade
PhPh
NH HN EtEt
Kanai
触媒的不斉アリル位ホウ素置換反応:研究競争

Ito, H.; Okura, T.; Matsuura, K.; Sawamura, M. Angew. Chem., Int. Ed. 2010, 49, 560.
RO
(pin)Bdiboron
Cu(O-t-Bu)/(R,R)-QuinoxP* (5.0 mol %)
baseH2O
iPrOCO2H
Ph
OHPhCHO (1.0 eq.)
0 °C, 18 hrt, 2 hRO- = i-PrOCO2 87%, 97% ee
dr >99:1
ORRO
CCCBu
B(pin)
MeH
74%, 97% ee
10 mol %Cu(O-t-Bu)/Xantphos
THF, 50 °C, 5 h97% ee
Me C C C Bu
OCO2Me
H
(S)
(S)
2.0 equiv.
OB
O
OB
O+
■ 高い光学純度を持つアレニルホウ素化合物の合成に始めて成功
Ito, H.; Sasaki, Y.; Sawamura, M., J. Am. Chem. Soc. 2008, 130, 15774.
メソ化合物の非対称化•アレニルホウ素合成
OTIPSN
NN
N
NH2
Cl
抗ウィルス剤前駆体
H
OTBS
CO2Me
OH
H
97% ee, >98% ds複雑な構造の迅速不斉合成

Ito, H.; Okura, T.; Matsuura, K.; Sawamura, M. Angew. Chem., Int. Ed. 2010, 49, 560. Hot Paper
chiralCu cat.
ROH
OH
RElectrophile
(RCHO)
RO
(pin)B
diboron
RO
(pin)B
diboronCu(O-t-Bu)/(R,R)-QuinoxP* (5.0 mol %)
baseH2O
iPrOCO2H
Ph
OHPhCHO (1.0 eq.)
3a0 °C, 18 hrt, 2 h
OR = OCO2i-Pr
87%, 97% eedr >99:1
ORRO NuRONucleophile
meso-2-alkene--1,4-diol derivatives Pd-catalyzed AAA
RO
Pd(II)L*Pd(0)L* cat.
メソ化合物の非対称化

■アリルエステル
■エナンチオ選択的ホウ素化 &アルデヒド付加
■立体選択的ホウ素化 &アルデヒド付加
三段階で、二つの炭素炭素結合を形成しながら四つの不斉点を構築
OCO2i-Pri-PrOCO2
i-PrOCO2H
OR
CO2Me85%, 96% eeds >99:1
Cu(O-t-Bu)Xantphos(pin)B–B(pin)
H
OTBS
CO2Me
OH
H
78%, 97% ee, ds > 98%
1) 2) CHOPh
R = HR = OTBS
81%, ds > 95%
i-PrOCO2H
OHO
O
H
OTBSO
O
OH
H
Cu(O-t-Bu)Xantphos(pin)B–B(pin)
1) 2) PhCHO
R = HR = OTBS
85%,ds >95:5
メソ化合物の非対称化:迅速アセンブリ

Cu(O-t-Bu)/(S,S)-QuinoxP* (5 mol %)
OTIPSN
NN
N
NH231%(17% for total 3 steps)
HCHO aq. (5.0 equiv)Sc(OTf)3 (0.3 equiv)
Cl
(pin)B–B(pin) (1.5 equiv)THF, –20 °C, 16 h
–40°C, 96 h
NC PBu3 toluene, rt, 82 h
2-amino-6-chloropurine
MeOHrt, 39 h
OHOTIPS
OCO2i-Pr
B(pin)
K2CO3
85%
OCO2i-Pri-PrOCO2
OCO2iPrOTIPS
TIPSCl, imidazoleDMF, rt, 15 h
64%, 96% ee
OHN
NN
N
NH2(–)-Abacavir
HNOHNH
NN
N
NH2(–)-Carbovir
O
or2 steps
抗ウィルス剤前駆体の短ステップ合成

COOH NH
OO N
HNO O
HO OHFR-900848chrysantheric acid
■シクロプロパン骨格は多くの生理活性物質に含まれる
CB
R H
CR H
COH
R H
R''
R'
CCOOH
R H
光学活性シクロプロピルホウ素化合物:有用な合成ブロック
98% yielddr 80 : 20
H B
HOH
B
OH
OO
RR
Pd cat.
CH2N2
Pietruszka (1999)
シクロプロパン合成のための有機ホウ素化合物

Cu(I) cat.(pin)B–B(pin)
Cu BL
OR
RORE
RE
RORE
Cu BL
B(pin)
RERE = SiR3, Ar
CuB
RO
L
R
CuB L
R×■ σ(Cu–C)/σ*(C–O) 共役安定化
ORR
B(pin)Cu(I) cat.(pin)B–B(pin)
CuB
RO
L
RO
CuB L
R
R R
ORSi
Cu BL
■ σ(Cu–C)/σ*(Si–C) 共役安定化
より強力な配向基 により選択性制御
挿入の位置選択性の電子的置換基によるコントロール

Cu(I) cat.(pin)B–B(pin)
Cu BL
OR
RORE
RERO
RE
Cu BL B(pin)
RERE = SiR3, Arβ LCuOR+
(pin)B B(pin)
Cu(O-t-Bu) / ligand
R XTHF, 30 °C
B(pin)
RR = R3Si, Ar, HetAr
B(pin)
NBoc
B(pin)
S
B(pin)70%, 92% ee90%, 92% ee 70%, 92% ee
X = OCO2R, OPO(OR)2
B(pin)
Me3Si
94%, 94% ee
B(pin)
BnMe2Si
83%, 94% ee
Ito, H.; Kosaka, Y.; Nonoyama, K.; Sasaki, Y.; Sawamura, M. Angew. Chem., Int. Ed. 2008, 47, 7424.Zhong, C.; Kunii, S.; Kosaka, Y.; Sawamura, M.; Ito, H. J. Am. Chem. Soc. 2010, 132, 11440.
不斉環化ホウ素化反応の開発

Ito, H.; Toyoda, T.; Sawamura, M. J. Am. Chem. Soc., 2010, 132, 5990.
(pin)B–B(pin)
PhMe2SiOMs
cat. CuCl / dppp
K(O-t-Bu)/THF (1.0 equiv)(2.0 equiv)
rt, THF, 20 h
93%trans/cis >99:1E/Z 1:>99
PhMe2SiOMs PhMe2Si B(pin)
76%trans/cis 5:95E/Z 95:5
PhMe2Si B(pin)
B(pin) B(pin)PhMe2Si B(pin)PhMe2Si B(pin)
89% 63% 68% 78%
■ 四員環•五員環化合物も立体選択的に合成可能
さまざまな選択的環化ホウ素化への展開
O
NSO O
Ph
NN
Kubota, K.; Yamamoto, E.; Ito, H. J. Am. Chem. Soc. 2013, 135, 2635. Histamine H3 Receptor Ligand
■ アルケニルハライドの環化B(pin)
(pin)B
Br
Br
CuCl (5 mol%)Xantphos (5 mol%)K(O-t-Bu) (1.2 equiv)
diboron (1.2 equiv)THF, 30 °C 88% 医薬品候補化合物の迅速合成

vBrown, H. C.; Jadhav, P. K.; Bhat, K. S. J. Am. Chem. Soc. 1985, 107, 2564.
(ipc)2B 94% eeH(ipc)2BH
【背景】1,3-ジエンでは、1,4-ヒドロホウ素化のみ報告例あり
不斉ヒドロ(プロト)ホウ素化反応の開発
Ph (pin)B B(pin)
Cu(O-t-Bu)/Xantphos (5 mol %)
THF, rt, 26 h+ Ph
B(pin)B(pin)
73%
Ph (pin)B B(pin)
Cu(O-t-Bu)/Xantphos (5 mol %)
THF, rt, 1 h+ Ph
DB(pin)
73%+ D2O
Ph
CuB(pin)
日本化学会春季年会 2006, unpublished result.
N N
Ph Phi-Pr
i-Pri-Pr
SO3–
Ph (pin)B B(pin)+cat.CuCl/K(O-t-Bu)
THF, –50°C, 48 hMeOH, 2.0 equiv
+
Ph B(pin)
80%, 98% eeLee, Y.; Hoveyda, A. J. Am. Chem. Soc. 2009, 131, 3160.

v
■ 触媒的1,2-不斉ヒドロホウ素化に初めて成功
Sasaki, Y.; Zhong, C.; Sawamura, M.; Ito, H. J. Am. Chem. Soc. 2010, 132, 1226.
(pin)B (pin)B+
THF, MeOH (2.0 equiv)–40°C, 24.5h
Cu(O-t-Bu)–(R,R)-Me-DuPhos(5.0 mol %)(pin)B B(pin) (1.5 equiv)
96%, 96% ee, dr >99:1H
v MeMe
BuB(pin) B(pin)
BuMe
Bucat. Cu(OtBu)/diphosphine
(pin)B B(pin)THF, MeOH
cat. Cu(OtBu)/PPh3
(pin)B B(pin)THF, MeOHup to 84% ee
■ 1,3-enyne の選択的ヒドロホウ素化を実現した
Sasaki, Y.; Horita, Y.; Zhong, C.; Sawamura, M.; Ito, H. Angew. Chem., Int. Edit. 2011, 50, 2778.
不斉ヒドロ(プロト)ホウ素化反応の開発
(pin)B (pin)B 96 % eechiral Cu catalystB2(pin)2(1.5 equiv)
THF, t-BuOH (5.0 equiv)room temp.77% (dr 92:8)
chiral Cu catalystB2(pin)2(1.5 equiv)
THF, MeOH (5.0 equiv)–40°C 87% (dr 7:93)

Ito, H; Kunii, S; Sawamura, M. Nature Chemistry, 2010, 2, 972.
Cu(O-t-Bu)(R,R)-QuinoxP*(5.0 mol %)
(pin)B B(pin)(1.5 equiv)Et2O, 24 h
OCH3Ph
racemic 98% yield97% ee
Ph
BO
O
PhCHO
Ph
B
CO
HPh Ph
HO
Ph
85% yield, 97% ee
直接エナンチオ収束反応:人工触媒で初の発見
OCH3Ph
CH3OPh
OCH3Ph
CH3O Phracemic
CuB
L*
CuB
L*
Ph
BO
O
anti-SN2'
syn-SN2'
■ 一つの不斉触媒が二つのエナンチオ選択的反応を進行させる

Racemic
Kinetic Resolution conv.< 50%
Optically Active Products
Chiral Catalyst
Dynamic Kinetic Resolution
SA
SB
PA
PA
I
Dynamic Kinetic Transformation
Direct Enantio-‐‑‒Convergent Reaction
Ito, H; Kunii, S; Sawamura, M. Nature Chemistry, 2010, 2, 972.
ラセミ体の原料から光学活性化合物を得る方法
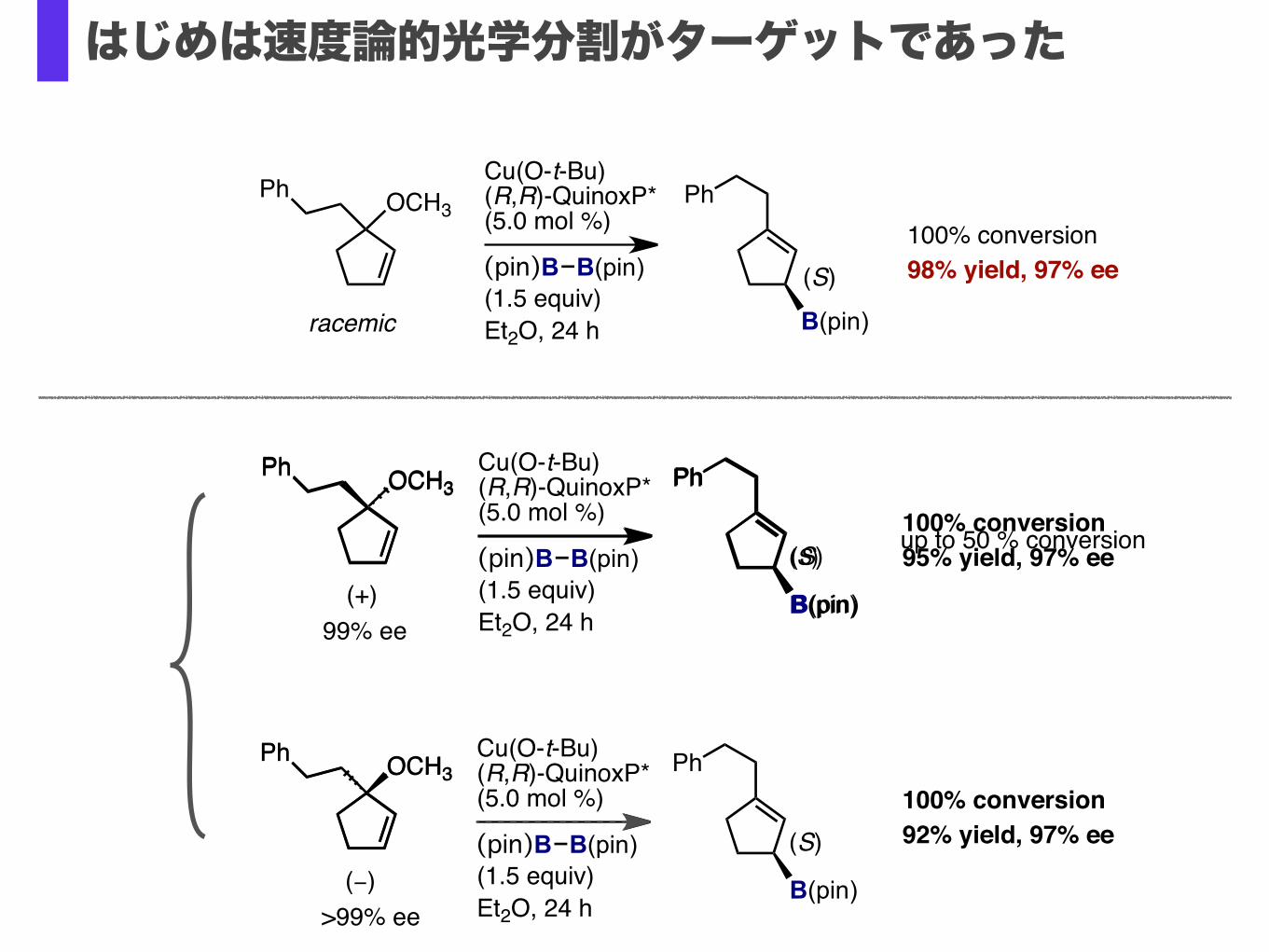
Cu(O-t-Bu)(R,R)-QuinoxP*(5.0 mol %)
(pin)B B(pin)(1.5 equiv)Et2O, 24 h
OCH3Ph
racemic
100% conversion98% yield, 97% ee
Ph
B(pin)
(S)
(+)
OCH3Ph Cu(O-t-Bu)
(R,R)-QuinoxP*(5.0 mol %)
(pin)B B(pin)(1.5 equiv)Et2O, 24 h
Ph
B(pin)
(S)100% conversion95% yield, 97% ee
99% ee
(−)
OCH3Ph Cu(O-t-Bu)
(R,R)-QuinoxP*(5.0 mol %)
(pin)B B(pin)(1.5 equiv)Et2O, 24 h
Ph
B(pin)
(S)100% conversion92% yield, 97% ee
>99% ee
OCH3Ph Ph
B(pin)
(S) up to 50 % conversion
OCH3Ph
はじめは速度論的光学分割がターゲットであった

Cu(O-t-Bu)(R,R)-QuinoxP*(5.0 mol %)
(1.5 equiv)
OCH2Ph
B(pin)
(S)-1, 93% ee (S)-2, 88%, 99% ee
(pin)B B(pin)
B(pin)
(S)-2, 46%, 99% ee
OCH2Ph
(S)-1, 89% ee(0.6 equiv)(pin)B B(pin)
(0.6 equiv) B(pin)
(S)-2, 43%, 86% ee
OCH2Ph
(R)-1, 98% ee
(pin)B B(pin)
Cu(O-t-Bu)(R,R)-QuinoxP*(5.0 mol %)
(1.5 equiv)
OCH2Ph
B(pin)(R)-1, 93% ee (S)-2, 91%, 88% ee
(pin)B B(pin)Racemization was not found.
基質のラセミ化が進行しているかどうか?

Nu
–Pd0L*
+Pd0L*
X
XPdII
X L* Nu
racemic
OTroc
OTroc
OTroc
OTroc
OTroc
OTroc
OTroc
XLPd
Pd2(dba)3Trost ligand
H
NO2PhO2S
OH
OTroc
OTrocOTrocracemic
NO2PhO2S
OH
OHOH
(−)-cyclophelitiolIntermediate withsymmetric structure
OCH3Ph
Ph
M
中間体の対称化がおこっているか?

CH
B(pin)(L*)Cu(pin)B
anti-SN2'
OCH2Ph
D D
CPhCH2O
(L*)Cu
(pin)B
syn-SN2'D
H
B(pin)
D
OCH2Ph
(S)-1d
D
B(pin)
Dcat. Cu(I) /(R,R)-QuinoxP*
diboron
94%, 99% ee
OCH2Ph
(R)-1d
D
B(pin)
Dcat. Cu(I) /(R,R)-QuinoxP*
diboron
92%, 88% ee
D化した基質を用いた実験:二つの反応経路

CH OCH2Ph
(xantphos)Cu
(pin)B
anti-attack
Cu(xantphos)
B(pin)
syn-attack
> 99 %ee
OPh2P PPh2
Xantphos:
syn-SN2'
B(pin)
B(pin)
anti-SN2'58.5 : 41.5
アキラルな触媒では?

CPhCH2O H
CH OCH2Ph
(L*)Cu
(pin)B
anti-attack
B(pin)
anti-SN2'
syn-SN2'
B(pin)
enantioselectivity>>stereoselectivity
Cu(L*)
B(pin)
anti-attack
stereoselectivity
(L*)Cu
(pin)B
syn-attack
enantioselectivity
N
N P
P
t-BuMe
t-Bu Me
エナンチオ面選択が立体選択を上回るのが7

TSA !+16.4 kcal/mol�
(a)�
TSB !+18.9 kcal/mol�
(b)�
(c)�
TSC!+15.9 kcal/mol�
TSD!+17.3 kcal/mol�
(d)�
Cu PP
BO O(S)-1
OCH3HIII
III IV
Cu PP
BO O(R)-1
III
III IV
H OCH3
Cu PP
BO O(R)-1
H3CO HIII
III IV
Cu PP
BO O
HCH3O
(S)-1
III
III IV
CH
B(pin)
anti-SN2'OCH2Ph
(L*)Cu
(pin)B
anti-attack
Cu(L*)
B(pin)
syn-attack
Transition State Structures DFT (M052X/6-31G)
N
N P
P
t-BuMe
t-Bu Me
QuinoxP*

TSA !+16.4 kcal/mol�
(a)�
TSB !+18.9 kcal/mol�
(b)�
(c)�
TSC!+15.9 kcal/mol�
TSD!+17.3 kcal/mol�
(d)�
Cu PP
BO O(S)-1
OCH3HIII
III IV
Cu PP
BO O(R)-1
III
III IV
H OCH3
Cu PP
BO O(R)-1
H3CO HIII
III IV
Cu PP
BO O
HCH3O
(S)-1
III
III IV
CPhCH2O
Cu(L*)
B(pin)
syn-SN2'H
B(pin)
anti-attack
(L*)Cu
(pin)B
syn-attack
Transition State Structures

Ito, H; Kunii, S; Sawamura, M. Nature Chemistry, 2010, 2, 972.
不斉第四級炭素を含む化合物の構築

K. Kubota
C(sp3)ーX への求核的ホウ素置換反応
Br +
CuCl / Xantphos (3 mol %)K(O-t-Bu) (1.0 equiv)
THF, rtB(pin)Alkyl AlkylB B
O
OO
O
1.2 equiv
B(pin)B(pin)
4 h, 85% 5 h, 91%
B(pin)
5 h, 90%
B(pin)
44 h, 0%
B(pin)
48 h, 17%
B(pin)
5 h, 51%
B(pin)
B(pin)
24 h, 62%a
B(pin)B(pin)
30 h, 68%a
aReaction was carried out at 40°C with 15 mol % of catalyst, 2.2 equiv of B2pin2 and 2.0 equiv of base.
B(pin)
5 h, 84%
B(pin)
4 h, 92%
Alkyl X Alkyl BCu cat.
B BO
OO
O+
X = Cl, Br, I O
O
base
Alkyl MgX or LiXB(pin)
CuCl / Xantphos: Ito, H.; Kubota, K. Org. Lett. 2012, 14, 890. CuI / PPh3: Yang, C.-T.; Steel, P. G.; Marder, T. B.; Liu, L. et al. Angew. Chem., Int. Ed. 2012, 51, 528.
Ni catalyst: Dudnik, A. S.; Fu, G. C. J. Am. Chem. Soc. 2012, 134, 10693.Pd catalyst: Joshi-Pangu, A.; Ma, X.; Diane, M.; Iqbal, S.; Kribs, R. J.; Huang, R.; Wang, C.-Y.; Biscoe, M. R. J. Org. Chem. 2012, 77, 6629.Pd, Ni catalyst: Yi, J.; Liu, J. H.; Liang, J.; Dai, J. J.; Yang, C.-T.; Fu, Y.; Liu, L.
Adv. Synth. Catal. 2012, 354, 1685.Fe catalyst: Atack, T. C.; Lecker, R. M.; Cook, S. P. J. Am. Chem. Soc. 2014, ASAP.Zn catalyst: Bose, S. K.; Fucke, K.; Liu, L.; Steel, P. G.; Marder, T. B. Angew. Chem., Int. Edit. 2014, 53, 1799.
競争が激しい研究対象

O
O
B(pin)
t-Bu O
O
B(pin)3
TIPSO B(pin)3
MsO B(pin)3
OB(pin)
5 h, 86% 6 h, 51%
24 h, 80%a 5 h, 80% 24 h, 82%a
B(pin)
5 h, 96%
官能基許容性と立体選択性
OMgBr
OMgBr
+
Cl Bror
CuCl / Xantphos (10 mol%)K(O-t-Bu) (2.0 equiv)
(pin)B-B(pin), THF, rt B(pin) X = Cl, 31 h, 85%, d.r. >99:1Br, 30 h, 81%, d.r. >99:1
Epimerization
PhBr
>99% ee
PhB(pin)
93%, 0% ee
RacemizationCuCl / Xantphos (5 mol%)K(O-t-Bu) (1.2 equiv)
(pin)B-B(pin), THF, rt, 24 h

CuCl / Xantphos: Ito, H.; Kubota, K. Org. Lett. 2012, 14, 890.
Br+
CuCl / Xantphos (3 mol %)K(O-t-Bu)(1.0 equiv)
THF, rt, 5 h
B(pin)B B
O
OO
O
1.2 equiv 91%
CuI / PPh3: Yang, C.-T.; Steel, P. G.; Marder, T. B.; Liu, L. et al. Angew. Chem., Int. Ed. 2012, 51, 528.
CuI (10 mol %)PPh3 (13 mol %)B B
O
OO
O+
Br
Li(O-t-Bu) (2.0 equiv)DMF, 37°C, 24 h
B(pin)
79% yield
·CuI/PPh3 Catalyst System·Highy Catalyst Loding
1.5 equiv
性能比較
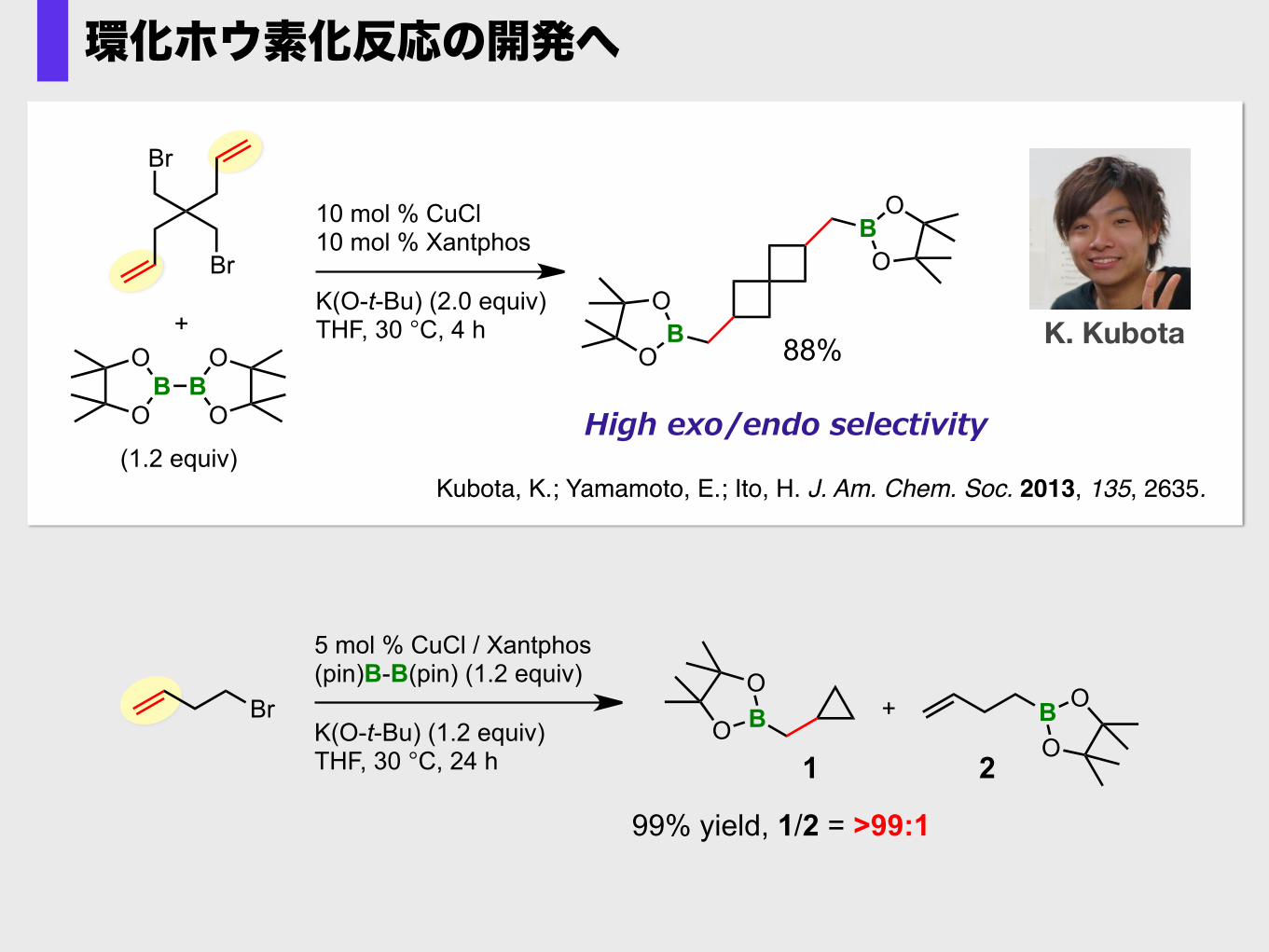
K. Kubota
Kubota, K.; Yamamoto, E.; Ito, H. J. Am. Chem. Soc. 2013, 135, 2635.
B
B 88%
O
O
O
O
OB
OB
O
O
(1.2 equiv)
Br
Br
+
10 mol % CuCl10 mol % Xantphos
K(O-t-Bu) (2.0 equiv)THF, 30 °C, 4 h
High exo/endo selectivity
環化ホウ素化反応の開発へ
5 mol % CuCl / Xantphos(pin)B-B(pin) (1.2 equiv)
K(O-t-Bu) (1.2 equiv)THF, 30 °C, 24 h
BO
O+Br B
O
O
1 2
99% yield, 1/2 = >99:1

B
Cu
Br
OPh2P PPh2
5 mol % CuCl / Xantphos(pin)B-B(pin) (1.2 equiv)
K(O-t-Bu) (1.2 equiv)THF, 30 °C, 24 h
BO
O+Br B
O
O
1 2
99% yield, 1/2 = >99:1
3 mol % CuCl / Xantphos(pin)B-B(pin) (1.2 equiv)
K(O-t-Bu) (1.0 equiv)THF, rt, 4 h
Br BO
O
85% yield
アルケンが存在すると環化が優先する
Kubota, K.; Yamamoto, E.; Ito, H. J. Am. Chem. Soc. 2013, 135, 2635.

Br n
CuB
Lt-BuO I–K+
LCu(O-t-Bu)
B CuL
Br
B
LCuBr
+
n
substitution insertion
+ K(O-t-Bu)– KBr
nL = Xantphos+ K(O-t-Bu)
oxidativeaddition
n
CuB
Lt-BuO III
reductiveelimination
nB
CuCl
+ K(O-t-Bu) – KCl
B B+BOR–– KBr
想定される反応機構
Kubota, K.; Yamamoto, E.; Ito, H. J. Am. Chem. Soc. 2013, 135, 2635.

a The endo-cyclization product was detected (7%).
(pin)B
4 h, 86%
(pin)B
MeMe
4 h, 83%
(pin)B
Me
Me
4 h, 90%
(pin)B
4 h, 95%d.r. = 1.4:1
(pin)B
4 h, 84%
(pin)B
6 h, 87%d.r. = 1.1:1
Si(pin)BMe
Me
4 h, 74%a
5 mol % CuCl5 mol % Xantphos(pin)B-B(pin) (1.2 equiv)
K(O-t-Bu) (1.2 equiv)THF, 30 °C
nCC
Cu
(pin)B – CuBrC n
n = 1−3 n = 1−3CC
BrBr C
(pin)B
L
b The six-membered ring product was detected. (4%)
complex mixtureb
Br
反応の適用範囲

HOMO level of I with Xantphos (−4.49 eV) was consider-
ably higher than those of the PPh3 (−5.20 eV) and NHC
(−4.71 eV) complexes, indicating that the Xantphos com-
plex had a stronger back donation ability to alkenes,
which is considered to be important for the addition of
borylcopper(I) to alkenes.12
To understand the ligand ef-
fect, distortion/interaction analysis was also performed.21
When the structures of the borylcopper(I) complexes (I)
with PPh3 and NHC were distorted to the structure in the
transition states, the additional free energies were needed
by 16.2 and 18.6 kcal/mol, respectively (Supporting Infor-
mation). Contrary, the Xantphos complex only required
11.7 kcal/mol for the conformation change from I to TS,
indicating the pre-activation nature of the Xantphos
complex (I) in the addition to alkenes.
Table 4. DFT Calculations (B3PW91/cc-pVDZ) of Al-kene Addition Step in Copper(I)-Catalyzed Boryla-tion
C CH
HH
H
CuLB
+
I
O
O
II
C CH
HHH
CuB L
III
O
O CuB
L
C CH
HHH
O
O
TS P
C C
H
B CuL
HH
H
OO
∆G (298 K, 1.0 atm, gas-phase)a / kcal mol
-1
L I+II III TS P
Xantphos 0 7.1 (–6.5) 17.6 (2.1) –11.4 (–24.9)
PPh3 0 3.5 (–10.4) 19.0 (3.6) –16.2 (–30.5)
IMes 0 7.3 (–8.1) 18.9 (3.0) –14.2 (–30.1)
aElectronic energies are shown in parentheses.
DFT calculations revealed that the activation barrier
difference is a key factor for this regioselectivity (Scheme
8). In the proposed alkylcopper intermediate, the less
bulky Cu(xantphos) moiety is placed at the sterically con-
gested internal carbon. Based on the structure of the ad-
dition product, this seems to be unfavorable. DFT calcula-
tions with propene substrate for the two diastereomeric
pathways were conducted. Path A can afford the major
product for the addition of borylcopper(I), whereas path
B corresponds to the formation of the minor product. The
activation free energy for path A was lower than that of
path B by 1.94 kcal/mol. Contrary, π-complex IIIP and the
alkylcopper product PP were more stabilized in path B
than in path A. In the transition state, the C1 carbon,
which will bind to boron atom in the product, formed a
transient five-coordinated geometry with highly congest-
ed environment. The substituent on the C1 atom thus
causes destabilization of the transition state. This can
explain the transition state in path A has the lower barrier
as compared to those in path B.
Scheme 8. DFT calculations (B3PW91/cc-pVDZ) for Two Diastereomeric Pathways.a
C CH
HH3CH
CuB L
IIIPB +8.9 (–5.4)
O
O CuB
L
C CH
H3CHH
O
O
TSPB +24.0 (+7.0) PPB –7.9 (–22.9)
C C
H
B CuL
HH3C
H
OO
L = XantphosC C
H
H
H
CH3+I
IIP
C CH
CH3HH
CuB L
IIIPA +10.7 (–3.7)
O
O CuB
L
C CH
HHCH3
O
O
TSPA +22.0 (+5.8) PPA –6.5 (–21.5)
C C
H
B CuL
CH3
HH
OO
majorproduct
minorproduct
path A
path B
1 2
21
(eg)BCuL
0 (0)
a
Relative G value (kcal mol-1) at 298 K, 1.0 atm, gas-phase.
Electronic energies are shown in parentheses.
We have proposesd a mechanism for the process, as
shown in Scheme 9. The copper(I) alkoxide (A) formed
via the reaction of the CuCl, ligand, and K(O-t-Bu) mix-
ture initially reacts with diboron to form the
borylcopper(I) intermediate (B). When Xantphos was
used as the ligand, the borylcopper(I) intermediate pos-
sessed the ability to add to the C−C double bond of the
substrate 4 (path a) to form the alkylcopper(I) species (C)
with concomitant formation of an ate complex (D) by
coordination of the alkoxide. Subsequent sequential oxi-
dative addition and elimination of bromide with inversion
of the stereochemistry gives the cyclic copper(III) inter-
mediate (E), in a manner similar to that of the SN2 reac-
tion postulated for the alkyl substitution of alkyl halides
with cuprates.22
Subsequent reductive elimination of the
copper moiety from the E produces the cyclization prod-
uct 5, as well as reproducing A. The cyclization of six
membered rings would not proceed according to this
mechanism because the seven membered ring intermedi-
ate (E, n = 4) appeared to be unstable (Table 3, entry 14).
When a monophosphine were used as the ligand, the re-
activity of the borylcopper(I) towards alkene addition
would be less favourable (path b), with boryl substitution
(n = 1,2) or radical cyclization proceeding (n = 3) instead.
Scheme 9. Proposed Mechanism for the Copper(I)-Catalyzed Borylative Cyclization
CuL
+ K(O-t-Bu)
B
n
X
a b
XB
CuL
B
Cu
Lt -BuOIII
LCu(O-t-Bu)
Bn
n
Bnn
LCuXB B
− KX
−+oxidativeaddition
reductiveelimination
L = Xantphos L = PPh3
− KX
A
B
46, n = 1, 2C
E
5
B OR
B
5, n = 3or
n = 1–3
nX
B
Cut-BuO L
K+I
−
D
+ K(O-t -Bu)
CONCLUSION
In summary, we have identified an unprecedented
reactivity of borylcopper(I) toward unactivated terminal
Page 5 of 7
ACS Paragon Plus Environment
Journal of the American Chemical Society
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

(pin)B
4 h, 92%
(pin)B
4 h, 90%
N O
O
Br
Br
+
10 mol % CuCl10 mol % Xantphos(pin)B-B(pin) (2.2 equiv)
K(O-t-Bu) (2.0 equiv)THF, 30 °C, 4 h
B
B 88%
O
O
O
O
(pin)B
4 h, 80%
NS
O O
Me
Br 5 mol % CuCl / Xantphos(pin)B-B(pin) (1.2 equiv)
K(O-t-Bu) (1.2 equiv)THF, 30 °C, 4 h
B
90%
O
O
Kubota, K.; Yamamoto, E.; Ito, H. J. Am. Chem. Soc. 2013, 135, 2635.
スピロ環化合物の合成

1. NaBO3/4H2O THF/H2O, rt, 1 h
2. Jones Reagent acetone, 0 °C, 1 h 64% (2 steps)
NHN
HBTU, iPrNEtDMF, rt, 2 h, 91%
C-‐‑‒O Bond Formation
Condensation
Histamine H3 Receptor Ligand
O
NSO O
HO
O
NSO O
NN
5 mol % CuCl / Xantphos(pin)B-B(pin) (1.2 equiv)
t-BuOK (1.2 equiv)THF, 30 °C, 4 h, 82%
B(pin)
NS
O O
NS
O O
Br
Astrazeneca, US 2010/0130477, May 27, 2010.
医薬品候補化合物の合成

過去に合成できなかった有機ホウ素化合物の新合成方法
early study:Tetrahedron Lett. 2000
BOO
J. Am. Chem. Soc. 2005J. Am. Chem. Soc. 2007
B
OR
OO
Angew. Chem., Int. Ed. 2010
BR OO
J. Am. Chem. Soc. 2010
(rac)-
R
BO
O
Angew. Chem., Int. Ed. 2008J. Am. Chem. Soc. 2010
C C CB
BuMe
H
OO
J. Am. Chem. Soc. 2008
BO
O
J. Am. Chem. Soc. 2010
BO
O
or
B
B
O
O
O
OJ. Am. Chem. Soc. 2013
BO
O
Org. Lett. 2012
RB
O
O
Nature Chem. 2010
Bu
BO
O
Angew. Chem., Int. Ed. 2011
B BO
OO
O
LCu X+ LCu B
O
OX B(pin)–
� ����������������������� ����

Allyboronate Ester from Various Substrates
ORR'
OHOR R'CHO 1
234
ORR'
OH
R'CHO OR
anti-1,2-diolsyn-1,2-diol
BB
**
B
OR2
B
TMSOMe
Soderquist, J. A.
B OTBSO
OR
R
MiyauraBrown, H. C.
R = CO2i-Pr
Lee, J. C. H.; Hall, D. G. J. Am. Chem. Soc. 2010, 132, 5544.Lessard, S.; Peng, F.; Hall, D. J. Am. Chem. Soc. 2009, 131, 9612.
R'
OHR'CHO
syn-1,2-diolderivatives
O
B(pin)
O
Hall's Methods
O(pin)B
OTfO
OEt
R
Cr catalyst
Pd catalyst
H–B(pin)R
(Z)-γ-alkoxyallyl- boronates

Enantioselecitve Borylation of Alkenylacetals
Y. Takenouchicatalyst (5 mol %)
OMe
B(pin)
PhK(O-t-Bu) (1.0 equiv)THF, 0°C(Z)-1a (S,E)-2a
Ph
P
P
Me t-Bu
t-Bu Me
(R,R)-BenzP*
N
N P
P
Me t-Bu
t-Bu Me
(R,R)-QuinoxP*
P
P
Me
MeMe
Me(R,R)-Me-Duphos
OMeMeO(pin)B–B(pin) (1.5 equiv)
O
O
O
O
PPh2PPh2
(R)-Segphos95%, 97% ee 63%, 93% ee 14%, 73% ee 38%, 21% ee
Yamamoto, E.; Takenouchi, Y.; Ozaki, T.; Miya, T.; Ito, H. J. Am. Chem. Soc. 2014, 136, 47, 16515

Enantioselecitve Borylation of Alkenylacetals
Takenouchi, Yamamoto, Ito, et al, submittedISHC 2014, Poster(Tues) 0272
(5 mol %)
R OR
B(pin)
K(O-t-Bu) (1.0 equiv), THF, 0°C(Z)-1 (S,E)-2
R
ORRO
CuCl/(R,R)-BenzP*(pin)B–B(pin) (1.5 equiv)
OMe
B(pin)TBSO
(R,E)-2j, 4 h86%, 97% eeb
OBn
B(pin)
(S,E)-2b, 3 h94%, 98% ee
OBn
B(pin)
(S,E)-2c, 4 h88%, 96% ee
OBn
B(pin)
(S,E)-2d, 4 h85%, 97% ee
5
OBn
B(pin)
(S,E)-2e, 4 h79%, 96% ee
CyOBn
B(pin)
(S,E)-2f, 8 h91%, 95% ee
OBn
B(pin)
(S,E)-2h, 8 h83%, 97% ee
OAc
OBn
B(pin)
(S,E)-2g, 8 h81%, 97% ee
MOMO
Ph
OBn
B(pin)
(S,E)-2k, 11 h, 62c(97)d%, 98% eec
OBn
B(pin)
(S,E)-2l, 11 h, 88e(98)d%, 96% eee(S,E)-2i, 4 h93%, 97% ee
OBn
B(pin)
TBSO
O
O
NMe
O
O
NC

Sequential Bolylation/Aldehyde Addition from Ketal
1) TBSCl, imidazole DMF, rt, 72%2) CuCl/Xantphos (5 mol %) B2(pin)2 (1.5 equiv) K(O-t-Bu) (1.0 equiv) THF, rt, 87%
TBSO Ph
B(pin)
OTBSOH
Ph
(1S,2R,3R)-6o72%, 96% ee, single isomer
(1S,3'R)-5o97% ee, 90:10 dr
4-BrC6H4CHO(2.0 equiv)
PhCHO (5.0 equiv)
60 oC, 4 d
60% (2 steps), 97% eesingle isomer
OHO Ph
OOO OB(pin)
B(pin)1o
OHCuCl/(R,R)-BenzP*(5 mol %) (pin)B–B(pin)(1.5 equiv)
Br
toluene 30 oC, 48 h
K(O-t-Bu)(1.0 equiv)THF, 0 oC, 11 h
(1R,1'S)-4o
Yamamoto, E.; Takenouchi, Y.; Ozaki, T.; Miya, T.; Ito, H. J. Am. Chem. Soc. 2014, 136, 47, 16515

Borylation of Aldehydes
R H
O
R = Alkyl, Ary
cat. Cu(I) / ICy
benzene, rtB B
O
O O
O+
R B(pin)
OB(pin)
J. P. Sadighi (2006)
Racemic
isolated in glove box
cat. Cu(I) / ICy
alcoxide base, 2MeOH, toluene
R B(pin)
OH
G. A. Molander (2012)
KHF2
R BF3K
OH
RacemicR H
OB B
O
O O
O+
Laitar, D. S.; Tsui, E. Y.; Sadighi, J. P. J. Am. Chem. Soc. 2006, 128, 11036.
Molander, G. A.; Wisniewski, S. R. J. Am. Chem. Soc. 2012, 134, 16856.
Ph BO
OCy
Cy
OBn
Ph BF3K+
OBn
Ph Ar
OBn
> 99% ee > 99–97% ee
Pd cat.
Ar-Cl

Product Protection for Isolation and HPLC Analysis
R H
O
R = Alkyl, Ary
B BO
O O
O+
cat. Cu(I) / L*
R H
B(pin)HO
Can be asymmetric catalysis?
O
H
1.5 mol % ICyCuClB2pin2 (1.0 equiv)
3 mol % Na(O-t-Bu)MeOH (2.0 equiv)toluene, rt, 1 h1a
protection
H
B(pin)PO
H
B(pin)BnO
BnBr, NaH, THF
H
B(pin)PhOCO
PhCOOH, EDC, DMAP
H
B(pin)BnMe2SiO
BnMe2SiCl, imidazole
isolated yield (%)rac-3
conditions: conditions:33% 8% 72%
conditions:
K. Kubota
Kubota, K.; Yamamoto, E.; Ito, H. J. Am. Chem. Soc. 2015 in press.

Searching Optimal Ligand
O1. CuCl / L* (5 mol %), K(O-t-Bu) (10 mol %) (pin)B-B(pin) 2 (1.0 equiv) MeOH (2.0 equiv, THF, 30 °C, 6 h
2. BnMe2SiCl, imidazole CH2Cl2, 3 h1a
H
(S)-3a
H
BnMe2SiO B(pin)
P
P
Me t-Bu
t-Bu Me
(R,R)-BenzP*68%, 60% ee
N
N P
P
Me t-Bu
t-Bu Me
(R,R)-QuinoxP*61%, 69% ee
O
O
O
O
PPh2PPh2
PPh2PPh2
(R)-SEGPHOS74%, 24% ee
(R)-BINAP66%, 69% ee
O
O
O
O
PP
Me
MeMe
Me
2
2(R)-DM-SEGPHOS, 71%, 32% ee
O
O
O
O
PP
tBuOMe
tButBu
OMetBu
2
2
(R)-DTBM-SEGPHOS, 72%, 96% ee

Substrate Scope
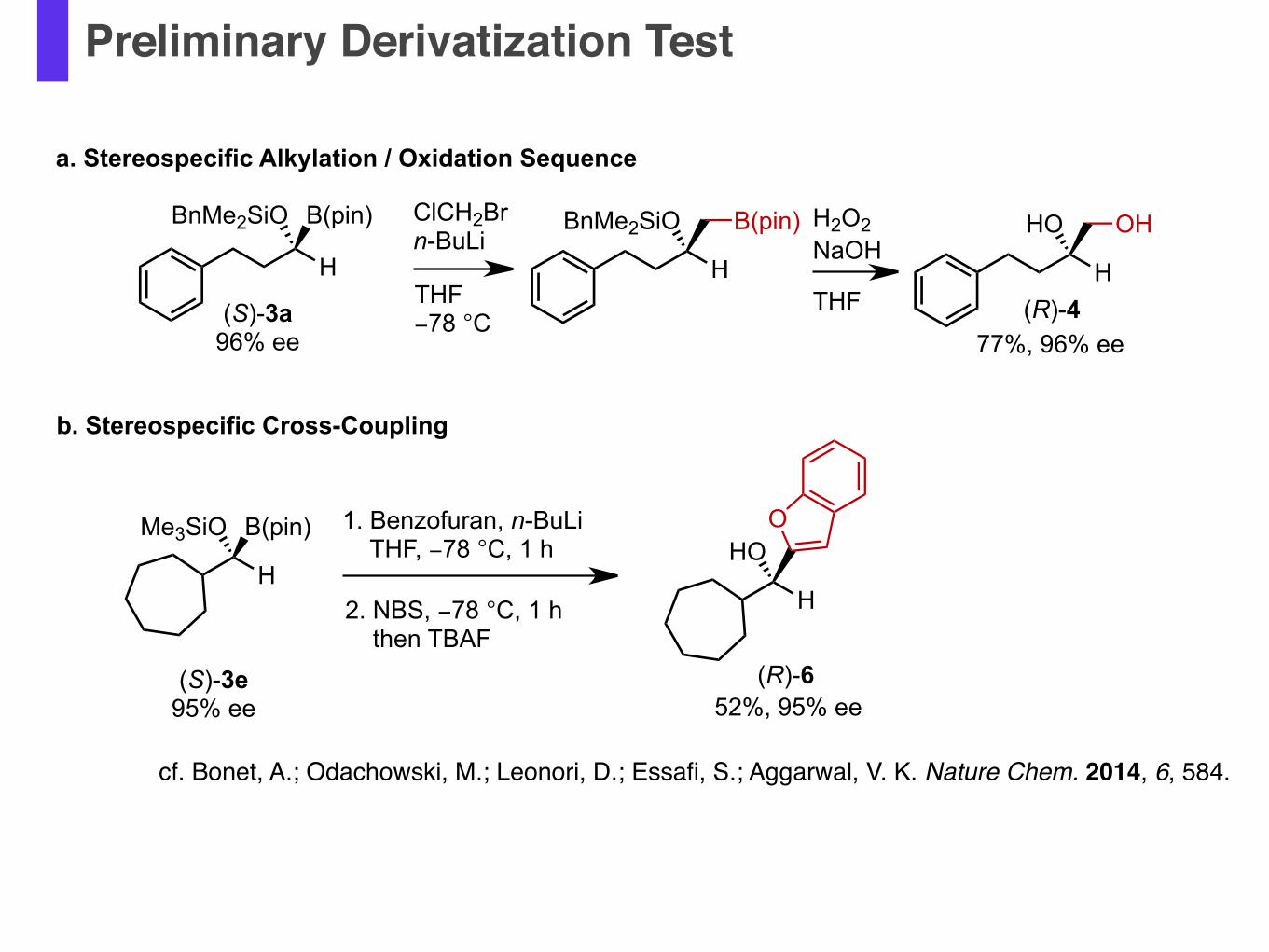
Preliminary Derivatization Test
cf. Bonet, A.; Odachowski, M.; Leonori, D.; Essafi, S.; Aggarwal, V. K. Nature Chem. 2014, 6, 584.
(S)-3a96% ee
ClCH2Brn-BuLi
THF−78 °C
H
BnMe2SiO
(R)-4
B(pin)
H
HO OH
77%, 96% ee
H2O2NaOH
THF
H
Me3SiO B(pin) 1. Benzofuran, n-BuLi THF, −78 °C, 1 h
H
HOO
2. NBS, −78 °C, 1 h then TBAF
(S)-3e95% ee 52%, 95% ee
(R)-6
b. Stereospecific Cross-Coupling
a. Stereospecific Alkylation / Oxidation Sequence
H
BnMe2SiO B(pin)

本日の講演内容
1. 銅触媒によるホウ素化反応:光学活性アルキルホウ素化合物の合成方法
3. メカノ応答性をもつ発光性金錯体
2. BBSホウ素化反応:遷移金属フリーでかつ官能基共存性のある芳香族ホウ素化合物の合成方法

■シリルボラン/塩基法 遷移金属フリーのホウ素化
92% yield
(1.5 equiv)KOMe (1.2 equiv)
DME, 30 °C, 1 h
Si BO
O
Br BO
O
Ph
Yamamoto, E.; Izumi, K.; Horita, Y.; Ito, H. J. Am. Chem. Soc. 2012, 134, 19997.
山本英治博士
堀田優子 泉 清孝
■ 有機リチウムやGrignard試薬利用
B
X LiBuLi
BX
官能基許容性に問題あり
■ 宮浦パラジウム触媒法
XPd cat.
B B
B
パラジウム触媒の残存 立体障害に弱い、反応が遅い
C(sp2)ーX へのホウ素置換反応

シリルボランの性質シリルボランは塩基による活性化でケイ素求核剤となる
OB
OSiMes
MesOMe Si
Mes
MesOMeLi
BuLi (2 equiv)toluene–78 °C, 1 h
Me3SiCl (2.45 equiv)
SiMes
MesOMeMe3Si
88% yield
Kawachi, A.; Minamimoto, T.; Tamao, K. Chem. Lett. 2001, 1216.
Chiral NHC (10 mol%)
OB
OSi Ph R1
O
R2
1
R1
O
R2
Si
up to 98% yieldup to 98:2 er
R1 = alkyl, aryl, OMe, H
DBU (15 mol%)
Ph
N N
Ph Ph
Ph Ph
Me Me
Chiral NHCO'Brien, J. M.; Hoveyda, A. H. J. Am. Chem. Soc. 2011, 133, 7712.
Si BO
OPh + RO–M+
Si BO
OPhOR
Si BO
OPhOR

Brbase (1.2 equiv)
solvent, 30 oC , 1 h
B(pin)
MeO MeO
SiMe2Ph1
MeO
OB
OSi Ph
(1.5 equiv)
なぜか芳香族ハロゲン化物のホウ素化が進行!
Yamamoto, E.; Izumi, K.; Horita, Y.; Ito, H. J. Am. Chem. Soc. 2012, 134, 19997.
baseLiOMeNaOMeKOMeKOtBuK2CO3
KFDBU
B/Si
80:2095:573:27
total yield (%)0819266000
bSolvent: DME
“tBuO”では選択性・収率低下
ナトリウムでは選択性低下
アルコキシド塩基: カリウムメトキシドがベスト
BBS法と名付けた base promoted borylation with silylborane

63Yamamoto, E.; Izumi, K.; Horita, Y.; Ito, H. J. Am. Chem. Soc. 2012, 134, 19997–20000.
良好な収率で反応が進行する。早い(一時間以内完結)

Yamamoto, E.; Izumi, K.; Horita, Y.; Ito, H. J. Am. Chem. Soc. 2012, 134, 19997–20000.
官能基や立体障害があってもスムーズに反応が進行する

manuscript under preparation
BBS法はヘテロ環化合物についても適用可能
3k, 52% 3l, (87)%
B(pin)
S
3h, (77)%
3i, 74(85)%
OB(pin)
NEt
B(pin)
3j, 61(75)%S
B(pin)
3p, 58(63)%
NNBn
B(pin)
ON
B(pin)
S B(pin)
NN
B(pin)N
O
B(pin)
Ph Ph
N
SEt B(pin)
N
N
B(pin)N
N
B(pin)
N
N
B(pin)
MeO
N
3m, 59(68)%
3n, 67%c 3o, 51d(74)% 3q, 46(68)%
3r, not detected 3s, (38)%
3t, 73%
B(pin)
3c, R' = H, 64(85)%
B(pin)
3a, 50(84)%OS
B(pin)
3b, 63(78)%R'
3d, R' = Ph, 62(87)%
3u, (66)%
N
B(pin)
3v, 58%
N
B(pin)

manuscript under preparation
アルケニルハライドは立体特異的に変換される。
B(pin)Cy
B(pin)
B(pin)Ph
B(pin)
OMOM
B(pin)
B(pin)
B(pin)
68(86)%c 72% 64%
(60)%d
B(pin)O 5
B(pin)O
B(pin)
Ph
Ph
64% 74%
43%
O
OBu
58% 53%
51(70)%d,e
O
B(pin)
Cy
68%e
(E)-6a, X = I (Z)-6b, X = I (Z)-6c, X = I
(Z)-6g, X = I (Z)-6h, X = I
6d, X = Br
(E)-6e, X = I 6f, X = Br
6j, X = Br6i, X = Br 6k, X = Br
OPh
O
5

BBS法の反応機構は?
なぜ求核的な芳香族置換反応が進行するのか?
なぜケイ素でなくホウ素へ置換されるのか?
官能基許容性はなぜ生じるのか?
なぜ立体障害に強いのか?

遷移金属の触媒作用ではない
68
Pd, Cu, Fe, Ni, Rh, Ag, Co, Pt, Ru, Ir の添加による加速効果は見られず
KOMe 試薬のICP発光分析遷移金属は 検出限界以下
Yamamoto, E.; Izumi, K.; Horita, Y.; Ito, H. J. Am. Chem. Soc. 2012, 134, 19997–20000.
反応機構に関する実験:遷移金属コンタミネーション?
そもそもこのような反応は、どのような遷移金属の触媒反応においても、知られていない

69Yamamoto, E.; Izumi, K.; Horita, Y.; Ito, H. J. Am. Chem. Soc. 2012, 134, 19997–20000.
ラジカル反応の可能性は?:関与の可能性は低い

70
AFIR自動反応経路探索: 北海道大学 前田・武次先生
Ref: 前田理、畑中美穂、植松遼平、武次徹也、諸熊奎治人工力誘起反応法による化学反応経路の自動探索:有機合成化学への応用と展望有機合成化学協会誌, 2014, 5, 567.
S. Maeda T. Taketsugu
RO– M+
Si BO
OPh X

J. Am. Chem. Soc. 2015, revised.
ハロゲンへのアタックとアリールアニオン生成が鍵
RO– M+
Si BO
OPh
OBO
OR
SiPh
M
BrAr
ArBr
Si BrPh
OBO
OR
Ar
M
BO
OAr
OBO
OR
SiPh
M
–MBrB O
O
O
R
M
Ar
Br
SiMe
MePh
–ROSiMe2Ph
A
B C
R = Me, EtM = K, Na
なぜケイ素でなくホウ素へ置換されるのか? 官能基許容性はなぜ生じるのか? なぜ立体障害に強いのか?
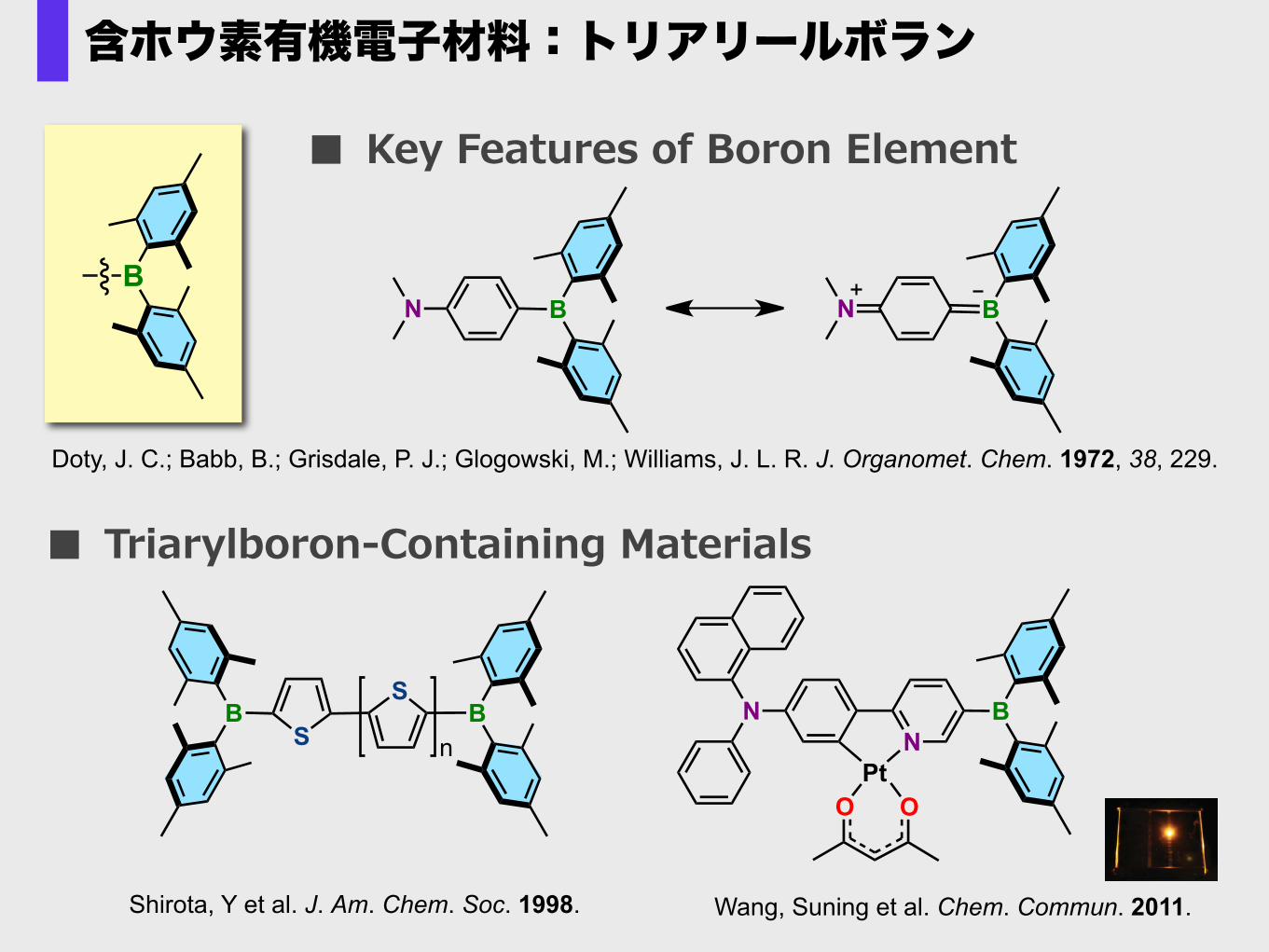
■ Key Features of Boron Element
NN
PtOO
B
Wang, Suning et al. Chem. Commun. 2011.
■ Triarylboron-‐‑‒Containing Materials
N B N BB
BS
SB
n
Shirota, Y et al. J. Am. Chem. Soc. 1998.
Doty, J. C.; Babb, B.; Grisdale, P. J.; Glogowski, M.; Williams, J. L. R. J. Organomet. Chem. 1972, 38, 229.
含ホウ素有機電子材料:トリアリールボラン

K. IzumiE. Yamamoto
Me SiPh
PhMeBrMe SiMe
Ph
Ph
1.5 equiv
B Me B
101% yield, B/Si ratio = 92:8
NaOtBu(1.2 equiv)
1,4-dioxane/hexane(1/3)50 °C, 24 h
BBS法をトリアリールボランに適用
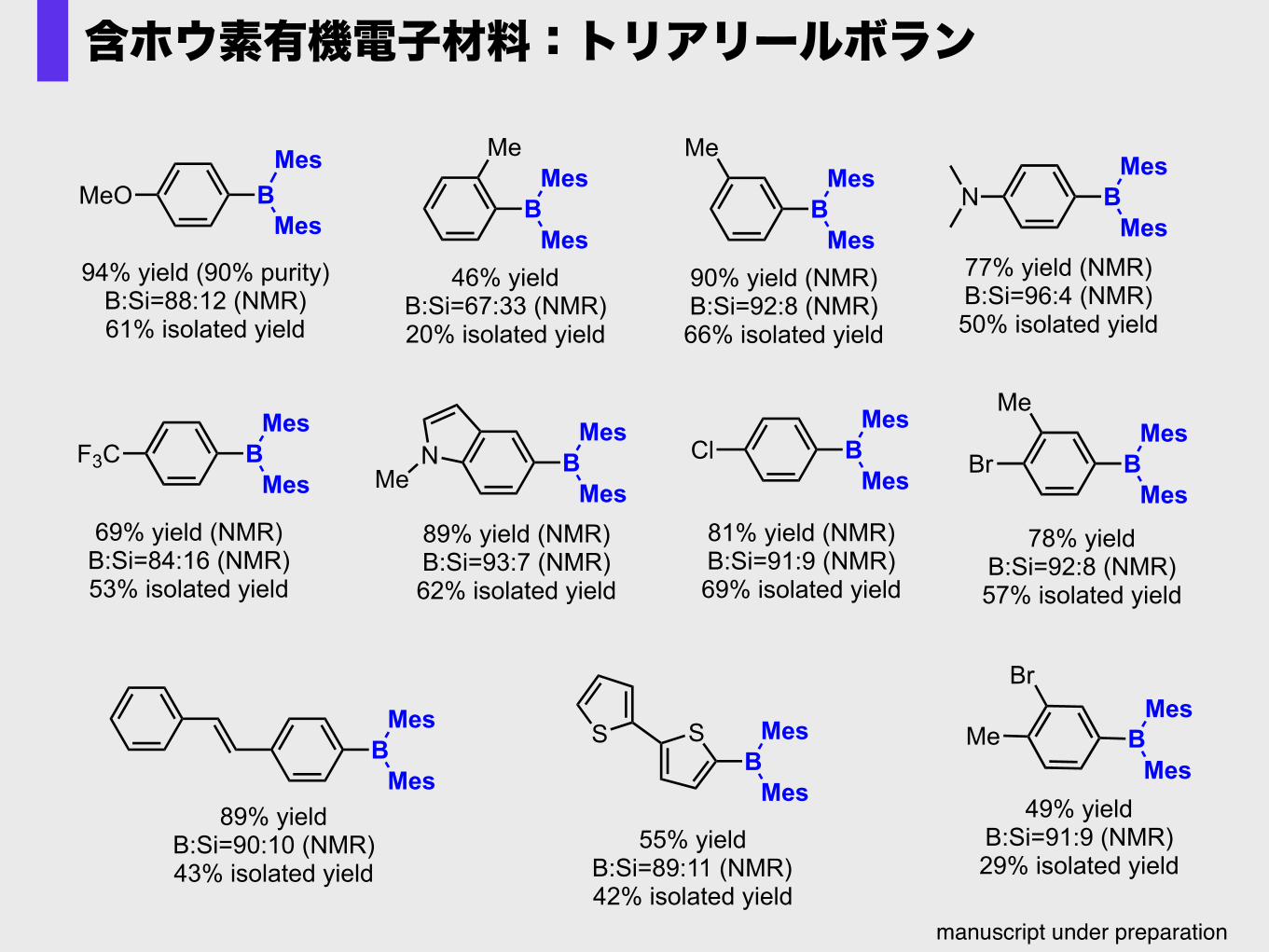
含ホウ素有機電子材料:トリアリールボラン
MeO BMes
Mes
94% yield (90% purity)B:Si=88:12 (NMR)61% isolated yield
B
Me
Mes
Mes
90% yield (NMR)B:Si=92:8 (NMR)66% isolated yield
F3C BMes
Mes
69% yield (NMR)B:Si=84:16 (NMR)53% isolated yield
B
Me
Mes
Mes
46% yieldB:Si=67:33 (NMR)20% isolated yield
N BMes
Mes
77% yield (NMR)B:Si=96:4 (NMR)50% isolated yield
NMe B
Mes
Mes89% yield (NMR)B:Si=93:7 (NMR)62% isolated yield
Cl BMes
Mes
81% yield (NMR)B:Si=91:9 (NMR)69% isolated yield
BMes
Mes
89% yieldB:Si=90:10 (NMR)43% isolated yield
BSS Mes
Mes
55% yieldB:Si=89:11 (NMR)42% isolated yield
Me
Br BMes
Mes
78% yieldB:Si=92:8 (NMR)57% isolated yield
Me
Br
BMes
Mes
49% yieldB:Si=91:9 (NMR)29% isolated yield
manuscript under preparation

本日の講演内容
1. 銅触媒によるホウ素化反応:光学活性アルキルホウ素化合物の合成方法
2. BBSホウ素化反応:遷移金属フリーでかつ官能基共存性のある芳香族ホウ素化合物の合成方法

Related Documents