Coordination Chemistry Reviews 257 (2013) 42–63 Contents lists available at SciVerse ScienceDirect Coordination Chemistry Reviews journa l h o me page: www.elsevier.com/locate/ccr Review Mononuclear iron hydrogenase Subal Dey, Pradip K. Das, Abhishek Dey ∗ Department of Inorganic Chemistry, Indian Association for the Cultivation of Science, Kolkata 700032, India Contents 1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42 2. Isolation, characterisation of Hmd . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43 3. Crystallographic characterisation of the active site of Hmd. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45 4. Synthetic models of Hmd . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46 5. Electronic structure of Hmd and its ligand bound complexes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52 6. Catalytic activation of H 4 -MPT and catalytic activity of the active site: experimental and computational study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52 6.1. Electrophilic activation of the dihydrogen by H 4 MPT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53 6.2. Mononuclear non-heme iron mediated heterolysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56 6.2.1. Stereochemical requirement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56 6.2.2. Mechanism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57 7. Enzyme–substrate binary complex and insight into the catalytic inhibition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60 8. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62 References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62 a r t i c l e i n f o Article history: Received 15 February 2012 Received in revised form 6 April 2012 Accepted 14 April 2012 Available online 25 April 2012 Keywords: Hydrogenase Hmd a b s t r a c t The active site of the mononuclear Fe-hydrogenase (Hmd) is a unique non-heme Fe enzyme involved in the catalytic activation of molecular H 2 . Apart from the nitrile hydratases and the peptidyl deformylases it is the only non-heme enzyme that has a redox inactive Fe in its active site. Naturally it has caught the attention of biochemists, bio-physicists, synthetic inorganic chemists and computational chemists alike since its isolation in the early 1990s. Our renewed interest in alternative energy has fuelled research in understanding this simplest, in terms of active site organization, of the known hydrogenases over the last two decades. A significant amount of synthetic work has led to very successful small molecule structural mimics of the active site. In-depth computational studies have led to a few mechanistic proposals for the heterolytic H 2 cleavage reaction catalysed by this enzyme. There have been some recent developments in understanding its geometric and electronic structure and its contribution to its reactivity. This review provides and up-to-date overview of the research in this area. © 2012 Elsevier B.V. All rights reserved. 1. Introduction Abundant use of fossil fuel as the main source of energy for cen- turies has significantly depleted the natural oil reserves and has introduced significant amount of additional CO 2 into the atmo- sphere leading to significant changes in the environment. Hence, alternate fuels, like methane and hydrogen, are gaining importance due to economical and ecological reasons [1–6]. Methane is natu- rally produced by the methanogens which reduce CO 2 , which is their chief source of energy, to CH 4 [7]. While there are potentially large deposits of methane in the permafrost which envelops the ∗ Corresponding author. E-mail address: [email protected] (A. Dey). surface of the tundra and taiga regions, it can be also produced artificially via the chemical reduction of CO 2 by H 2 [8–10]. The use of molecular hydrogen either as a source of electrons neces- sary to reduce CO 2 to CH 4 [8,11,12] or directly in a O 2 /H 2 fuel cell has thus gained tremendous interest [13,6]. The use of molecular hydrogen as a source of reducing equivalent by anoxic organisms e.g. sulphate-reducing bacteria, methanogenic bacteria, acetogenic bacteria, nitrate-reducing bacteria, are well established. There are several microorganisms that can produce H 2 from H 2 O as well [14,15]. It has been estimated that these microorganisms produce or consume more than 200 million tons of H 2 per year [14]. More than 10 million tons of H 2 is also generated in the toxic habitats by aerobic and micro-aerophilic microorganisms as side product of N 2 fixation, (enzyme: nitrogenases) [16–18] or during formate dehy- drogenation (enzyme: formate dehydrogenases) [19,20]. Despite 0010-8545/$ – see front matter © 2012 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.ccr.2012.04.021

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

R
M
SD
C
a
ARRAA
KHH
1
tisadrtl
0h
Coordination Chemistry Reviews 257 (2013) 42– 63
Contents lists available at SciVerse ScienceDirect
Coordination Chemistry Reviews
journa l h o me page: www.elsev ier .com/ locate /ccr
eview
ononuclear iron hydrogenase
ubal Dey, Pradip K. Das, Abhishek Dey ∗
epartment of Inorganic Chemistry, Indian Association for the Cultivation of Science, Kolkata 700032, India
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 422. Isolation, characterisation of Hmd . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 433. Crystallographic characterisation of the active site of Hmd. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 454. Synthetic models of Hmd . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 465. Electronic structure of Hmd and its ligand bound complexes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 526. Catalytic activation of H4-MPT and catalytic activity of the active site: experimental and computational study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
6.1. Electrophilic activation of the dihydrogen by H4MPT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 536.2. Mononuclear non-heme iron mediated heterolysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
6.2.1. Stereochemical requirement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 566.2.2. Mechanism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
7. Enzyme–substrate binary complex and insight into the catalytic inhibition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 608. Conclusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
r t i c l e i n f o
rticle history:eceived 15 February 2012eceived in revised form 6 April 2012ccepted 14 April 2012vailable online 25 April 2012
a b s t r a c t
The active site of the mononuclear Fe-hydrogenase (Hmd) is a unique non-heme Fe enzyme involved inthe catalytic activation of molecular H2. Apart from the nitrile hydratases and the peptidyl deformylasesit is the only non-heme enzyme that has a redox inactive Fe in its active site. Naturally it has caught theattention of biochemists, bio-physicists, synthetic inorganic chemists and computational chemists alikesince its isolation in the early 1990s. Our renewed interest in alternative energy has fuelled research in
eywords:ydrogenasemd
understanding this simplest, in terms of active site organization, of the known hydrogenases over the lasttwo decades. A significant amount of synthetic work has led to very successful small molecule structuralmimics of the active site. In-depth computational studies have led to a few mechanistic proposals for theheterolytic H2 cleavage reaction catalysed by this enzyme. There have been some recent developmentsin understanding its geometric and electronic structure and its contribution to its reactivity. This reviewprovides and up-to-date overview of the research in this area.
. Introduction
Abundant use of fossil fuel as the main source of energy for cen-uries has significantly depleted the natural oil reserves and hasntroduced significant amount of additional CO2 into the atmo-phere leading to significant changes in the environment. Hence,lternate fuels, like methane and hydrogen, are gaining importanceue to economical and ecological reasons [1–6]. Methane is natu-
ally produced by the methanogens which reduce CO2, which isheir chief source of energy, to CH4 [7]. While there are potentiallyarge deposits of methane in the permafrost which envelops the∗ Corresponding author.E-mail address: [email protected] (A. Dey).
010-8545/$ – see front matter © 2012 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.ccr.2012.04.021
© 2012 Elsevier B.V. All rights reserved.
surface of the tundra and taiga regions, it can be also producedartificially via the chemical reduction of CO2 by H2 [8–10]. Theuse of molecular hydrogen either as a source of electrons neces-sary to reduce CO2 to CH4 [8,11,12] or directly in a O2/H2 fuel cellhas thus gained tremendous interest [13,6]. The use of molecularhydrogen as a source of reducing equivalent by anoxic organismse.g. sulphate-reducing bacteria, methanogenic bacteria, acetogenicbacteria, nitrate-reducing bacteria, are well established. There areseveral microorganisms that can produce H2 from H2O as well[14,15]. It has been estimated that these microorganisms produceor consume more than 200 million tons of H2 per year [14]. More
than 10 million tons of H2 is also generated in the toxic habitats byaerobic and micro-aerophilic microorganisms as side product of N2fixation, (enzyme: nitrogenases) [16–18] or during formate dehy-drogenation (enzyme: formate dehydrogenases) [19,20]. Despite
emistr
tmssm
cfHiheHlbi∼Pmaom
achnHiitccucon
tddhwoewnatTd
S. Dey et al. / Coordination Ch
hese large turnovers the steady state concentration of H2 in theseicro-organisms is very low (1–10 pa) [14]. This could be due to
low rates of H2 generation or poor H2 retention as it is sparinglyoluble in physiological pH’s (∼1 mM at 1 atm H2 pressure) andolecular H2, being a light gas, escapes easily into the space.A H2-based energy cycle using hydrogen as a clean, high effi-
iency and low cost energy vector is being sought after. Currentuel cells uses costly platinum or its alloys to electrolyse H2 or
2O reversibly [21]. As, hydrogen production from natural sources clearly not enough, alternative strategies e.g. – solar, nuclear,ydro, wind or wave energy-driven hydrogen production are beingxplored [22]. Such type of “hydrogen economy” requires efficient+ to H2 inter-conversion i.e. H+ + e− ↔ (1/2)H2. This requires cata-
ysts which enables this processes to proceed in the forward or theackward direction at diffusion controlled rates without demand-
ng a large driving force i.e. an over potential, which is greater than±150 mV from the thermodynamic equilibrium potential [23].latinum or its certain alloys do conform to these above require-ents but, the scarcity, cost and sustainability of platinum ups the
nte for a future hydrogen economy [21]. Thus the unavailabilityf practical catalysts for the H+ to H2 inter-conversion remains aajor problem [13,24–31].In 1931 Stephenson and Stickland showed that the facultative
naerobic colon bacterium Escherichia coli activate hydrogen as itan reduce methyl-viologen, thanks to the enzymes they termedydrogenases [32–34]. The possible structure of Fe–Fe hydroge-ase was reported by Warburg in 1946 [35]. He first observed that2 formation in growing culture of Clostridium butyricum, is inhib-
ted by carbon monoxide [36]. He also first identified the presence ofron, ligated to a sulphur ligand. These hydrogenases, enzymes con-aining redox active base metals like iron, nickel at their active sites,atalyses the H+ to H2 inter-conversion reversibly under ambientonditions without any over-potential in micro-organisms whichtilize the hydrogen as major energy source for various biochemi-al processes [2]. Thus, understanding and emulating the chemistryf the hydrogenases has become a priority to the scientific commu-ity.
Three types of hydrogenases have been identified in natureill now. Among them, Ni–Fe hydrogenase (containing nickel–ironinuclear centre) and Fe–Fe hydrogenase (containing iron–ironinuclear centre) are the most prevalent [3,30,37–45]. The Ni–Feydrogenase family includes the Se containing hydrogenases asell [10,30,39,42–44,46]. Much research effort has been focussed
n the theoretical and experimental modelling of these twonzymes in last few decades and these have been reviewed else-here [30,5,6,42]. There is a third type of hydrogenase which doesot contain any iron–sulphur cluster, discovered in 1990, known
s the iron–sulphur cluster free hydrogenase [47]. These con-ain a mono-nuclear non-heme iron catalytic centre [48,9,49–51].his enzyme is also known as H2-forming methylenetetrahy-romethanopterin (CH2 H4MPT) dehydrogenase (Hmd) [52,53].Scheme 1.1. Basic reactio
y Reviews 257 (2013) 42– 63 43
This third kind of hydrogenase enzyme is generally expressedunder Ni-limiting conditions and in the absence of light in themethanogens [54]. The three types of hydrogenases are phyloge-netically distinct. In contrast to the Fe–Fe hydrogenase or the Ni–Fehydrogenase, the catalytically active Fe centre in Fe-only hydro-genase is redox-inactive and catalyses heterolytic dissociation ofmolecular hydrogen only in the presence of the substrate, N5, N10-methenyltetrahydromethanopterin (CH H4MPT) 1 and reversiblytransfers the hydride in stereospecific manner to form N5, N10-methylenetetrahydromethanopterin (CH2 H4MPT) 2 and a proton(Scheme 1.1) [7,55,56]. The enzyme was initially believed to bemetal-free [8,8,9,57]. Shima et al. were the first to report the pres-ence of a redox-inactive iron centre in its catalytic active site [57]and subsequently the crystal structure was reported in 2008 [58]and revised in 2009 [59]. This enzyme is catalyses one of the severalsteps involved in the conversion of CO2 to CH4 in the methanogenicarchea.
The crystal structure of the photo-inactive cofactor, known asthe Fe-GP cofactor, is reported [60,61]. The apoenzyme (without theFe-GP cofactor and the substrate) and C176A mutated holoenzyme(with the Fe-GP cofactor but without the substrate) [58] struc-tures are also crystallographically characterised. The micro-biologyof these organisms and the biochemistry of these enzymes havebeen reviewed in details elsewhere [52,53]. This review is aimed atproviding a broad and up to date overview of the research on thisenzyme and its synthetic models. Along with the biochemistry ofthe enzyme, its structure and synthesis of related synthetic modelcomplexes, recent developments in understanding the geometricand electronic structure of the active site, its reactivity towardsdifferent ligands and details of the mechanistic investigations uti-lizing structural, spectroscopic, and computational techniques onthe enzyme and related synthetic model systems will be cata-logued.
2. Isolation, characterisation of Hmd
The difficulty behind the extraction of Hmd is the reactivityof this enzyme towards dioxygen and light. Hence, it is generallyexpressed under nickel-limiting conditions [57]. The availability ofhigh-purity enzyme material for detailed study using biophysicaltechniques [57–63] allowed its identification as a metaloenzyme,and subsequently our understanding of structure and functionhas dramatically expanded in the last two decades. In 1990Thauer et al. reported the presence of Hmd in Methanobacterimthermoautophicum, a thermophilic methanogen that reduces CO2to CH4 [47]. The hydrogenase activity was rapidly lost under
aerobic conditions or in the presence of dithiothreitol (DTT) ormercaptoethanol [64]. Hmd is a homodimer composed of only onetype of subunit that has an apparent molecular mass of 43 kDawith a specific activity about 1000 U mg−1. Determination of then catalysed by Hmd.
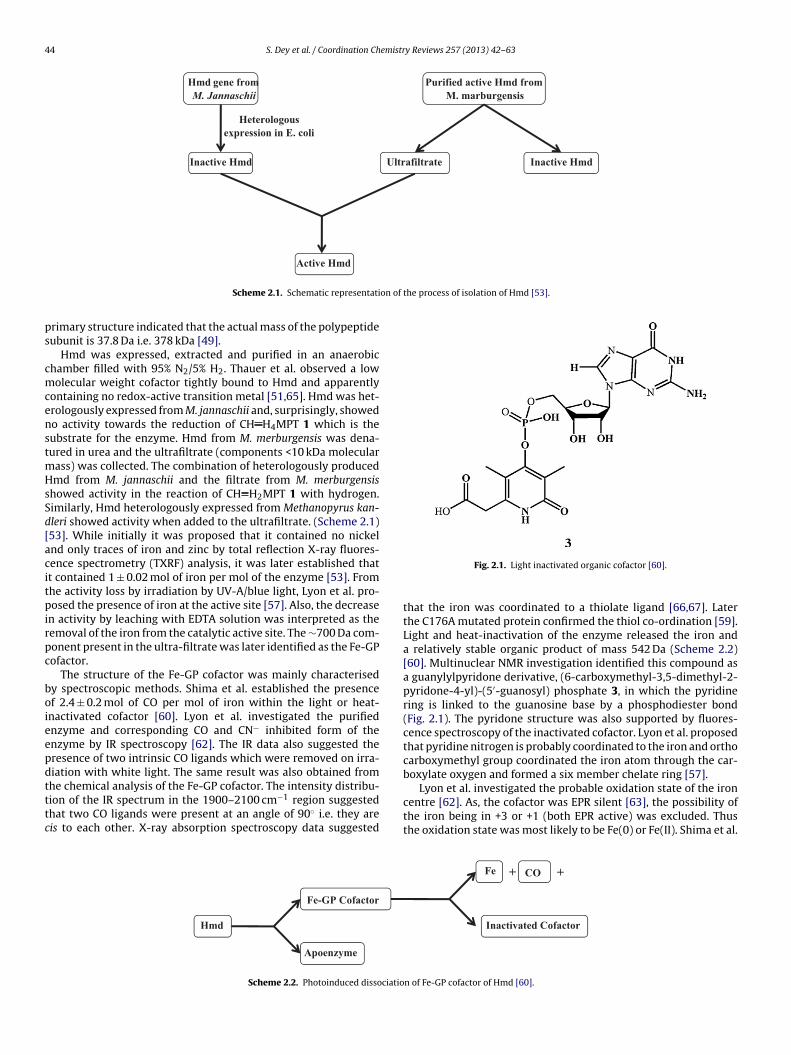
44 S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63
gene from Hmd
M. Jannaschii
Inactive Hmd
Heterologous
expression in E. coli
Purified active Hmd from
M. marburgensis
Ultrafiltrate Inactive Hmd
Active Hmd
on of the process of isolation of Hmd [53].
ps
cmcenstmHsSd[acitpirpc
boieepdtttc
Scheme 2.1. Schematic representati
rimary structure indicated that the actual mass of the polypeptideubunit is 37.8 Da i.e. 378 kDa [49].
Hmd was expressed, extracted and purified in an anaerobichamber filled with 95% N2/5% H2. Thauer et al. observed a lowolecular weight cofactor tightly bound to Hmd and apparently
ontaining no redox-active transition metal [51,65]. Hmd was het-rologously expressed from M. jannaschii and, surprisingly, showedo activity towards the reduction of CH H4MPT 1 which is theubstrate for the enzyme. Hmd from M. merburgensis was dena-ured in urea and the ultrafiltrate (components <10 kDa molecular
ass) was collected. The combination of heterologously producedmd from M. jannaschii and the filtrate from M. merburgensis
howed activity in the reaction of CH H2MPT 1 with hydrogen.imilarly, Hmd heterologously expressed from Methanopyrus kan-leri showed activity when added to the ultrafiltrate. (Scheme 2.1)53]. While initially it was proposed that it contained no nickelnd only traces of iron and zinc by total reflection X-ray fluores-ence spectrometry (TXRF) analysis, it was later established thatt contained 1 ± 0.02 mol of iron per mol of the enzyme [53]. Fromhe activity loss by irradiation by UV-A/blue light, Lyon et al. pro-osed the presence of iron at the active site [57]. Also, the decrease
n activity by leaching with EDTA solution was interpreted as theemoval of the iron from the catalytic active site. The ∼700 Da com-onent present in the ultra-filtrate was later identified as the Fe-GPofactor.
The structure of the Fe-GP cofactor was mainly characterisedy spectroscopic methods. Shima et al. established the presencef 2.4 ± 0.2 mol of CO per mol of iron within the light or heat-nactivated cofactor [60]. Lyon et al. investigated the purifiednzyme and corresponding CO and CN− inhibited form of thenzyme by IR spectroscopy [62]. The IR data also suggested theresence of two intrinsic CO ligands which were removed on irra-iation with white light. The same result was also obtained from
he chemical analysis of the Fe-GP cofactor. The intensity distribu-ion of the IR spectrum in the 1900–2100 cm−1 region suggestedhat two CO ligands were present at an angle of 90◦ i.e. they areis to each other. X-ray absorption spectroscopy data suggestedHmd
Apoenzyme
Fe-GP Cofactor
Scheme 2.2. Photoinduced dissociatio
Fig. 2.1. Light inactivated organic cofactor [60].
that the iron was coordinated to a thiolate ligand [66,67]. Laterthe C176A mutated protein confirmed the thiol co-ordination [59].Light and heat-inactivation of the enzyme released the iron anda relatively stable organic product of mass 542 Da (Scheme 2.2)[60]. Multinuclear NMR investigation identified this compound asa guanylylpyridone derivative, (6-carboxymethyl-3,5-dimethyl-2-pyridone-4-yl)-(5′-guanosyl) phosphate 3, in which the pyridinering is linked to the guanosine base by a phosphodiester bond(Fig. 2.1). The pyridone structure was also supported by fluores-cence spectroscopy of the inactivated cofactor. Lyon et al. proposedthat pyridine nitrogen is probably coordinated to the iron and orthocarboxymethyl group coordinated the iron atom through the car-boxylate oxygen and formed a six member chelate ring [57].
Lyon et al. investigated the probable oxidation state of the iron
centre [62]. As, the cofactor was EPR silent [63], the possibility ofthe iron being in +3 or +1 (both EPR active) was excluded. Thusthe oxidation state was most likely to be Fe(0) or Fe(II). Shima et al.Fe + CO +
Inactivated Cofactor
n of Fe-GP cofactor of Hmd [60].

emistry Reviews 257 (2013) 42– 63 45
rttCsattbwoifMwtotLaoam
3H
MimgtsCcmes
ofTmsw
S. Dey et al. / Coordination Ch
eported the Mössbauer data of the 57Fe labelled Hmd and proposedhe presence of a low spin Fe(II). Further the Mössbauer data ofhe CO, CN− inhibited enzyme indicated that exogenous CO andN− ligands could bind the iron at an exchangeable sixth ligandite [63]. However, addition of H2 or CH H2MPT 1 did not lead tony significant changes in the Mössbauer spectrum. This revealedhat H2 or CH H2MPT 1 were possibly not interacting directly withhe iron centre. This is surprising as even if CH H4MPT 1 does notind to the iron, it may be expected that hydrogen, its substrate,ould need to bind to or interact with the iron centre directly in
rder to be activated for a heterolytic cleavage. Dey addressed thisssue using DFT calculations and provided a plausible explanationor this unusual observation (vida infra) [68]. Quantification of the
össbauer signal intensities showed that 1.14 ± 0.25 mol of ironas present per mole of enzyme. This was in good agreement with
he value obtained from TXRF experiments. Thus cumulative databtained from IR, Mössbauer and magnetic measurement indicatedhe iron in the active site was most likely a low-spin Fe(II) [69].ater, synthetic modelling of the cofactor, their spectral featuresnd parallel DFT calculations unanimously supported the presencef a low-spin Fe(II) at the active site. This was confirmed by X-raybsorption near-edge spectroscopy of the enzyme and syntheticodel complexes [70].
. Crystallographic characterisation of the active site ofmd
Pilak et al. reported the crystal structure of the apoenzyme from. jannaschii and M. kandleri in 2006 [61]. The homodimeric protein
s subdivided into three domains – two terminal peripheral unitsade up of N-terminal domains of each monomer and a central
lobular domain composed of intertwined C-terminal domains ofhe two monomers. Interestingly, in the modelled Hmd-cofactortructure, the pyridine moiety of the cofactor is located near theys176 residue, which indicated that only Cys176 is essential foratalysis (Fig. 3.1). The authors modelled the CH2 H4MPT enzy-atic prosthetic group in the structure and this revealed that the
lectrophilic C14a centre is 6 A away from the iron centre, providingufficient space to bind a molecular hydrogen.
Detailed crystallographic characterisation of the active site wasbtained by reconstituting the Fe-GP cofactor into the apoenzymerom M. jannaschii, heterologously expressed in E. coli (Fig. 3.2) [58].
he Fe-GP cofactor was anchored to the enzyme by the guanosylonophosphate moiety. Unexpectedly, while the holoenzymetructure was obtained in an open conformation, the apoenzymeas crystallized in a closed conformation. The Fe-GP cofactor
Fig. 3.1. Initial proposal for the active site structure [58].
Fig. 3.2. X-ray crystallographically characterised DTT-bound active site structure[59].
binding led to small conformational changes at the N-terminaldomain in addition to the overall closed to open conformationalchange.
From the apoenzyme structure it was observed that a mononu-cleotide binding site was present at the solvent exposed cleft. Thecrystal structure obtained by reconstituting the Fe-GP cofactor withthe apoenzyme revealed that this cleft indeed accommodated theorganic nucleotidic part of the Fe-GP cofactor. The structure ofthe GMP cofactor was already determined from NMR analysis. Thehighest electron density was assigned iron and the pyridinol nitro-gen was proposed the one of the ligand to the iron centre. Pyridinolnitrogen was thought to bind in sp2 hybridised form rather thansp3 hybridised state in pyridone tautomeric form as the �-backaccepting ability is higher in the pyridine form. Electron densityalso indicated that Cys176 and an intrinsic CO ligand are bound tothe Fe centre trans to each other.
In the first crystallographic report Shima et al. thought initiallythat the carboxymethyl group was present at a cis orientation rel-ative to the pyridinol nitrogen and trans relative to an unknownligand 4 [58]. The electron density between iron and pyridinol ringdid not fit to that of the carboxymethyl group and could be reason-ably interpreted as the second intrinsic CO ligand. So, they revisedthe geometry and modelled the pyridinol ring at the opposite sideof the iron centre (Fig. 3.1), later. The lack of electron density of thegroup carboxylate was explained by the flexibility of the carboxy-late group.
Assuming the iron centre of the active site to be distorted octa-hedral, the ligand trans to the pyridinol nitrogen was interpreted asan weakly bound solvent molecule as the electron density is 2.7 Aaway from the iron centre. Also, the electron density trans to thesecond CO ligand could not be interpreted, and this ligand thereforeremained unknown in this report [58]. Overall a square-pyramidalor distorted octahedral iron complex with two intrinsic CO ligands,the Cys176-thiol/thiolate, a sp2-hybridised pyridinol nitrogen andan unknown ligand was predicted [70]. This structure did not pro-vide enough information regarding the protonation state of the2-hydroxyl group of the pyridinol ligand and the thiol ligand as
well as the orientation of the carboxymethyl group. An increasein the electron density at the site occupied by the unknown lig-and, by soaking with 3 mM KCN, suggested that it is probably anexchangeable ligand binding site.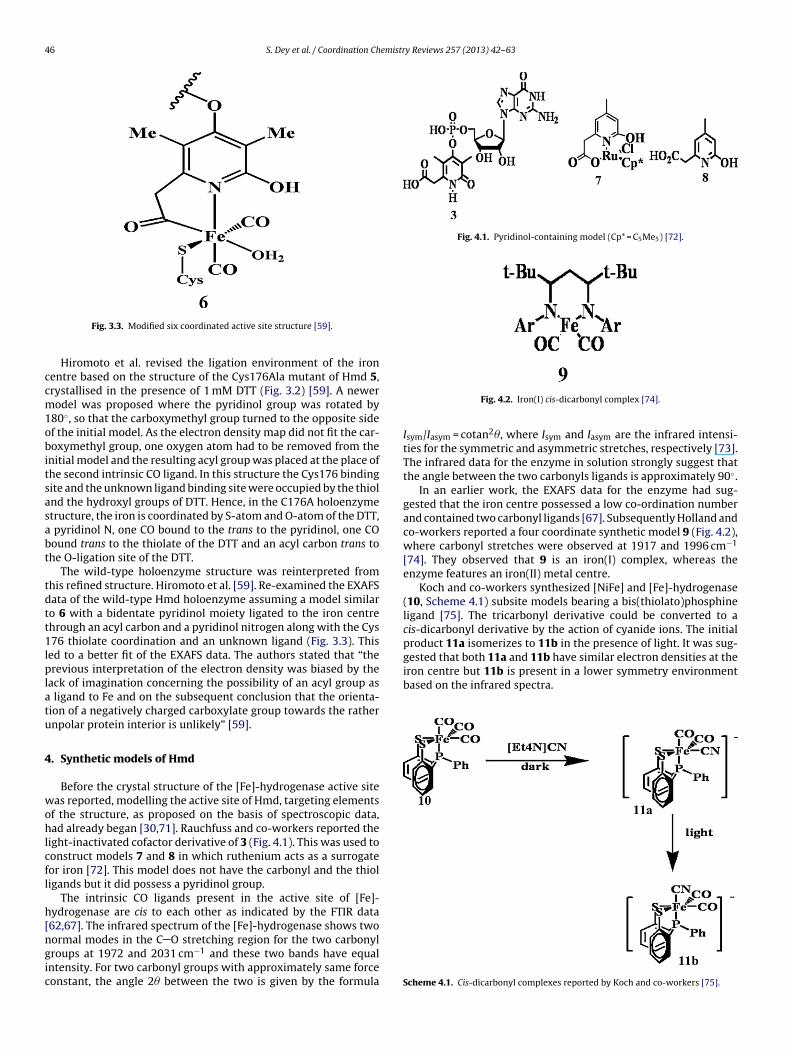
46 S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63
ccm1obitsasabt
tdtt1lplatu
4
wohlcfl
h[ngic
Fig. 4.1. Pyridinol-containing model (Cp* = C5Me5) [72].
gested that both 11a and 11b have similar electron densities at theiron centre but 11b is present in a lower symmetry environmentbased on the infrared spectra.
Fig. 3.3. Modified six coordinated active site structure [59].
Hiromoto et al. revised the ligation environment of the ironentre based on the structure of the Cys176Ala mutant of Hmd 5,rystallised in the presence of 1 mM DTT (Fig. 3.2) [59]. A newerodel was proposed where the pyridinol group was rotated by
80◦, so that the carboxymethyl group turned to the opposite sidef the initial model. As the electron density map did not fit the car-oxymethyl group, one oxygen atom had to be removed from the
nitial model and the resulting acyl group was placed at the place ofhe second intrinsic CO ligand. In this structure the Cys176 bindingite and the unknown ligand binding site were occupied by the thiolnd the hydroxyl groups of DTT. Hence, in the C176A holoenzymetructure, the iron is coordinated by S-atom and O-atom of the DTT,
pyridinol N, one CO bound to the trans to the pyridinol, one COound trans to the thiolate of the DTT and an acyl carbon trans tohe O-ligation site of the DTT.
The wild-type holoenzyme structure was reinterpreted fromhis refined structure. Hiromoto et al. [59]. Re-examined the EXAFSata of the wild-type Hmd holoenzyme assuming a model similaro 6 with a bidentate pyridinol moiety ligated to the iron centrehrough an acyl carbon and a pyridinol nitrogen along with the Cys76 thiolate coordination and an unknown ligand (Fig. 3.3). This
ed to a better fit of the EXAFS data. The authors stated that “therevious interpretation of the electron density was biased by the
ack of imagination concerning the possibility of an acyl group as ligand to Fe and on the subsequent conclusion that the orienta-ion of a negatively charged carboxylate group towards the rathernpolar protein interior is unlikely” [59].
. Synthetic models of Hmd
Before the crystal structure of the [Fe]-hydrogenase active siteas reported, modelling the active site of Hmd, targeting elements
f the structure, as proposed on the basis of spectroscopic data,ad already began [30,71]. Rauchfuss and co-workers reported the
ight-inactivated cofactor derivative of 3 (Fig. 4.1). This was used toonstruct models 7 and 8 in which ruthenium acts as a surrogateor iron [72]. This model does not have the carbonyl and the thioligands but it did possess a pyridinol group.
The intrinsic CO ligands present in the active site of [Fe]-ydrogenase are cis to each other as indicated by the FTIR data62,67]. The infrared spectrum of the [Fe]-hydrogenase shows two
ormal modes in the C O stretching region for the two carbonylroups at 1972 and 2031 cm−1 and these two bands have equalntensity. For two carbonyl groups with approximately same forceonstant, the angle 2� between the two is given by the formulaFig. 4.2. Iron(I) cis-dicarbonyl complex [74].
Isym/Iasym = cotan2�, where Isym and Iasym are the infrared intensi-ties for the symmetric and asymmetric stretches, respectively [73].The infrared data for the enzyme in solution strongly suggest thatthe angle between the two carbonyls ligands is approximately 90◦.
In an earlier work, the EXAFS data for the enzyme had sug-gested that the iron centre possessed a low co-ordination numberand contained two carbonyl ligands [67]. Subsequently Holland andco-workers reported a four coordinate synthetic model 9 (Fig. 4.2),where carbonyl stretches were observed at 1917 and 1996 cm−1
[74]. They observed that 9 is an iron(I) complex, whereas theenzyme features an iron(II) metal centre.
Koch and co-workers synthesized [NiFe] and [Fe]-hydrogenase(10, Scheme 4.1) subsite models bearing a bis(thiolato)phosphineligand [75]. The tricarbonyl derivative could be converted to acis-dicarbonyl derivative by the action of cyanide ions. The initialproduct 11a isomerizes to 11b in the presence of light. It was sug-
Scheme 4.1. Cis-dicarbonyl complexes reported by Koch and co-workers [75].
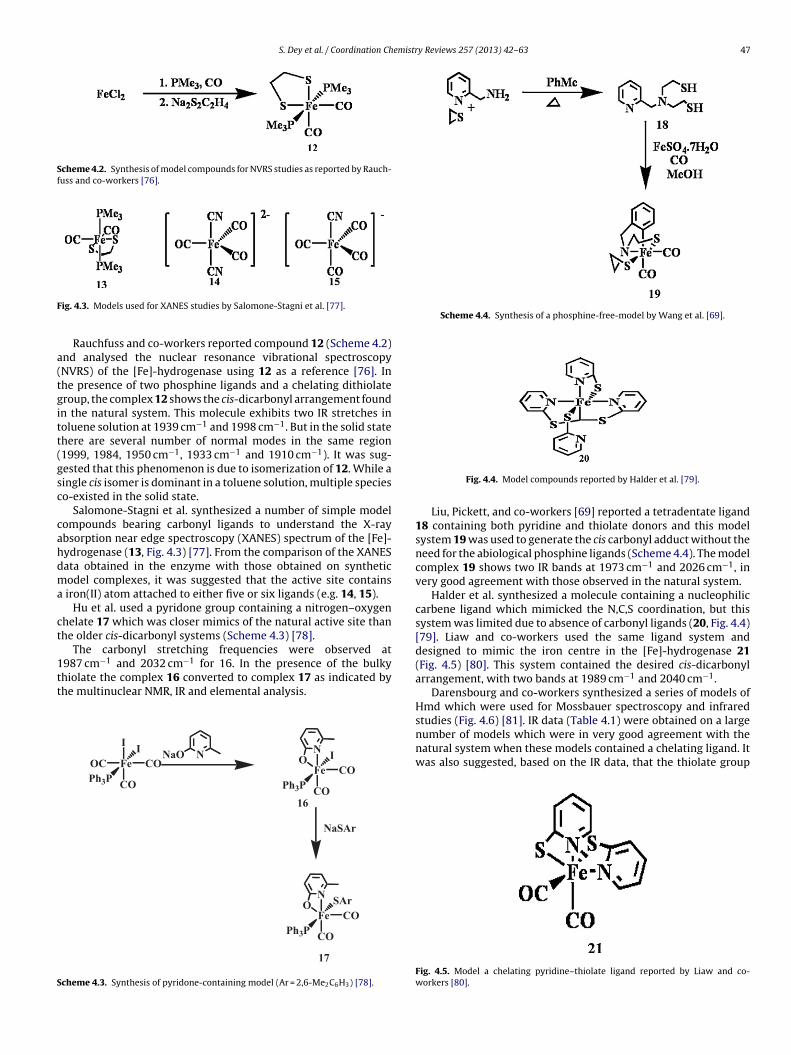
S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63 47
Scheme 4.2. Synthesis of model compounds for NVRS studies as reported by Rauch-fuss and co-workers [76].
F
a(tgitt(gsc
cahdma
ct
1tt
S
Scheme 4.4. Synthesis of a phosphine-free-model by Wang et al. [69].
ig. 4.3. Models used for XANES studies by Salomone-Stagni et al. [77].Rauchfuss and co-workers reported compound 12 (Scheme 4.2)nd analysed the nuclear resonance vibrational spectroscopyNVRS) of the [Fe]-hydrogenase using 12 as a reference [76]. Inhe presence of two phosphine ligands and a chelating dithiolateroup, the complex 12 shows the cis-dicarbonyl arrangement foundn the natural system. This molecule exhibits two IR stretches inoluene solution at 1939 cm−1 and 1998 cm−1. But in the solid statehere are several number of normal modes in the same region1999, 1984, 1950 cm−1, 1933 cm−1 and 1910 cm−1). It was sug-ested that this phenomenon is due to isomerization of 12. While aingle cis isomer is dominant in a toluene solution, multiple specieso-existed in the solid state.
Salomone-Stagni et al. synthesized a number of simple modelompounds bearing carbonyl ligands to understand the X-raybsorption near edge spectroscopy (XANES) spectrum of the [Fe]-ydrogenase (13, Fig. 4.3) [77]. From the comparison of the XANESata obtained in the enzyme with those obtained on syntheticodel complexes, it was suggested that the active site contains
iron(II) atom attached to either five or six ligands (e.g. 14, 15).Hu et al. used a pyridone group containing a nitrogen–oxygen
helate 17 which was closer mimics of the natural active site thanhe older cis-dicarbonyl systems (Scheme 4.3) [78].
The carbonyl stretching frequencies were observed at−1 −1
987 cm and 2032 cm for 16. In the presence of the bulkyhiolate the complex 16 converted to complex 17 as indicated byhe multinuclear NMR, IR and elemental analysis.
Fe
I
COOC
CO
I
Ph3P
NNaO
Fe
N
CO
CO
I
Ph3P
O
NaSAr
Fe
N
CO
CO
SAr
Ph3P
O
16
17
cheme 4.3. Synthesis of pyridone-containing model (Ar = 2,6-Me2C6H3) [78].
Fig. 4.4. Model compounds reported by Halder et al. [79].
Liu, Pickett, and co-workers [69] reported a tetradentate ligand18 containing both pyridine and thiolate donors and this modelsystem 19 was used to generate the cis carbonyl adduct without theneed for the abiological phosphine ligands (Scheme 4.4). The modelcomplex 19 shows two IR bands at 1973 cm−1 and 2026 cm−1, invery good agreement with those observed in the natural system.
Halder et al. synthesized a molecule containing a nucleophiliccarbene ligand which mimicked the N,C,S coordination, but thissystem was limited due to absence of carbonyl ligands (20, Fig. 4.4)[79]. Liaw and co-workers used the same ligand system anddesigned to mimic the iron centre in the [Fe]-hydrogenase 21(Fig. 4.5) [80]. This system contained the desired cis-dicarbonylarrangement, with two bands at 1989 cm−1 and 2040 cm−1.
Darensbourg and co-workers synthesized a series of models ofHmd which were used for Mossbauer spectroscopy and infraredstudies (Fig. 4.6) [81]. IR data (Table 4.1) were obtained on a large
number of models which were in very good agreement with thenatural system when these models contained a chelating ligand. Itwas also suggested, based on the IR data, that the thiolate groupFig. 4.5. Model a chelating pyridine–thiolate ligand reported by Liaw and co-workers [80].

48 S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63
zed b
pMs
iadtic
TE
Fig. 4.6. Model compounds synthesi
lays a key role in tuning the electronic structures of these models.össbauer data of complex 26 matches the most with the active
ite.Both Liaw and Darensbourg extended the concept of a chelat-
ng nitrogen–sulphur ligand. Liaw and co-workers synthesizedn aniline-derived thiol (complex 30) in place of the pyridine-erived ligand in 21 (Fig. 4.7) [82,83]. The carbonyl stretches for
he anionic complex (at 1929 cm−1 and 1992 cm−1) are unsurpris-ngly shifted from those of the [Fe]-subsite and the neutral modelomplexes. The CN− bound stretching frequency for the compoundable 4.1xperimental IR stretching frequencies of the model complexes [81,83].
Model � (CO) cm−1
1 22a 2018, 2040, 20872 22b 2023, 2034, 20823 23 2019, 2034, 20824 24 2003, 20445 25 2002, 20436 26 2041, 2050, 20957 27 2022, 2047, 20938 28a 2017, 2031, 20819 28b 2032, 2045, 2093
10 28c 2053, 210411 29a 1971, 201912 29b 1983, 203113 31a 1927, 198514 31b 1956, 201215 31c 1942, 200216 Hmd 1944, 201117 9 1917, 199618 34 1954, 2013, 209419 41 1958, 202620 42a 1962, 202921 42b 1961, 202622 47 2032, 198723 48 2039, 198824 49 2035, 200125 50 2026, 197426 51 2021, 196727 52 2025, 198528 53 2021, 197329 54 2028, 198630 55 2018, 196831 56 2031, 198041 57 1998, 193242 59 1950 cm−1, 2013 cm−1
y Darensbourg and co-workers [81].
30 is 2101 cm−1 whereas the C N stretch is observed at 2091 cm−1
for the CN− bound Hmd.Darensbourg and co-workers reported less strained complexes
of the same aniline ligand (Scheme 4.5 and Table 4.1) [83]. Thesefive-coordinate complexes have a vacant site which is the “solvent”binding site in the active site of [Fe]-hydrogenase. Out of these only31a showed a tendency to bind carbon monoxide under ambientconditions. By the action of a strong acid like HBF4·Et2O, 31a wasprotonated under a CO atmosphere and formed a new compoundwith IR stretches at 2046 cm−1 and 2102 cm−1 which was assignedas the meridional tricarbonyl. Treatment of this species with baseregenerated 31a.
Rauchfuss and co-workers used a thioester containing a phos-phine ligand for chelation (Scheme 4.6) [84]. The oxidative additionyields the desired acyl ligation with a bound thiolate in an elegantsingle step. The tricarbonyl 32 lose CO reversibly to form com-pound 33 which is exists as two isomers (axial–equatorial andequatorial–axial). Compound 32 showed the carbonyl stretches at1981 cm−1, 2020 cm−1, and 2075 cm−1 very similar to the bandsobserved for the CO inhibited form of the enzyme (2001 cm−1,2025 cm−1, and 2074 cm−1). Compound 32 reacted with CN− toform 34, which exhibits FTIR stretches very similar to thoseobtained for the CN− inhibited form of the enzyme (1954 cm−1,2013 cm−1, and 2094 cm−1 for 34 vs. 1956 cm−1, 2020 cm−1, and2090 cm−1 for the CN− inhibited enzyme).
Hu and co-workers have utilized this acyl-containing precursor35 and reacted it with the sodium salt of 2-mercapto-6-methylpyridine to obtain the dimeric product 36 (Scheme 4.7) [85].
In the presence of CN− and CO, 36 yields compounds 37 (whereL = CN−, CO). It is noted that these complexes contain relatively tightnitrogen–sulphur chelates, which is absent in the natural system.Fig. 4.7. Model of aniline–thiolate ligand reported by Liaw and co-workers [82].
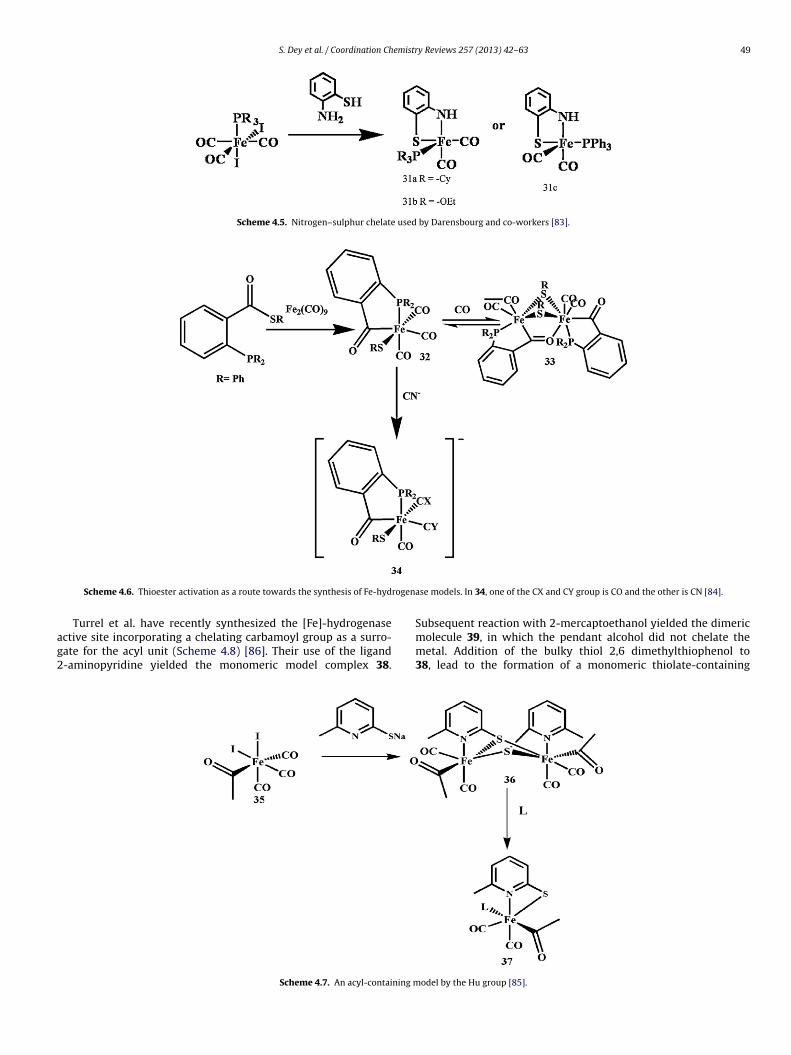
S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63 49
Scheme 4.5. Nitrogen–sulphur chelate used by Darensbourg and co-workers [83].
rogen
ag2
Scheme 4.6. Thioester activation as a route towards the synthesis of Fe-hyd
Turrel et al. have recently synthesized the [Fe]-hydrogenasective site incorporating a chelating carbamoyl group as a surro-ate for the acyl unit (Scheme 4.8) [86]. Their use of the ligand-aminopyridine yielded the monomeric model complex 38.
Scheme 4.7. An acyl-containing m
ase models. In 34, one of the CX and CY group is CO and the other is CN [84].
Subsequent reaction with 2-mercaptoethanol yielded the dimericmolecule 39, in which the pendant alcohol did not chelate themetal. Addition of the bulky thiol 2,6 dimethylthiophenol to38, lead to the formation of a monomeric thiolate-containing
odel by the Hu group [85].
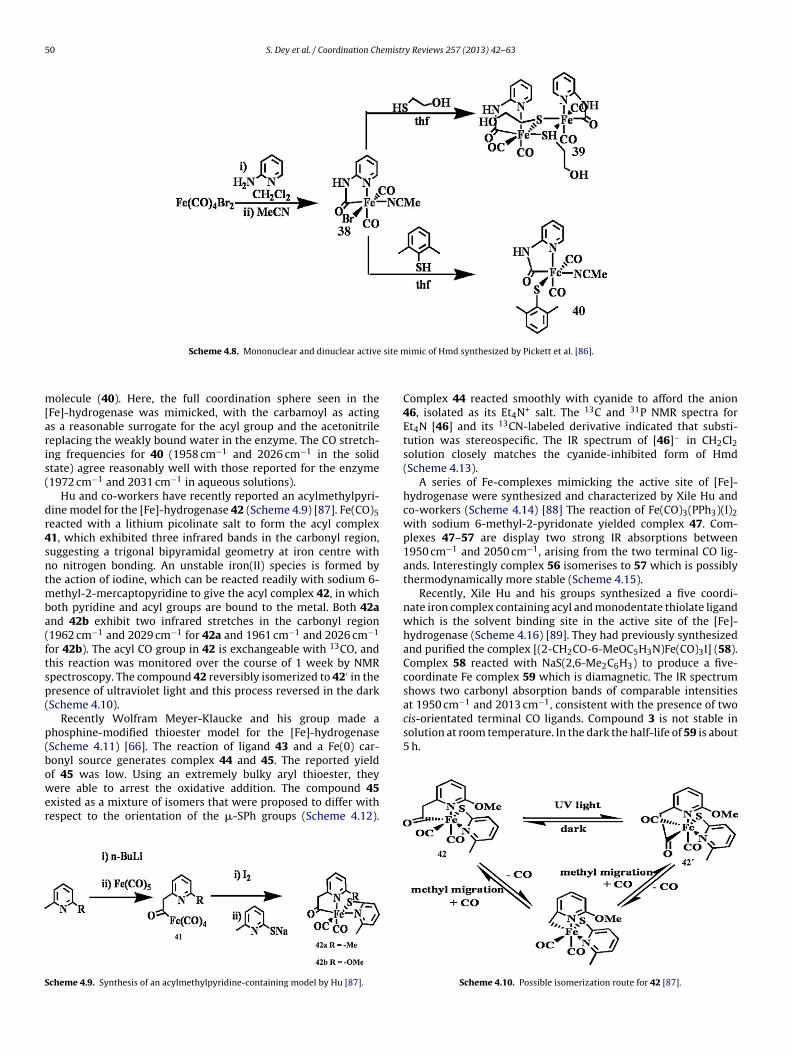
50 S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63
site m
m[aris(
dr4sntmba(ftsp(
p(bower
S
at 1950 cm and 2013 cm , consistent with the presence of twocis-orientated terminal CO ligands. Compound 3 is not stable insolution at room temperature. In the dark the half-life of 59 is about5 h.
Scheme 4.8. Mononuclear and dinuclear active
olecule (40). Here, the full coordination sphere seen in theFe]-hydrogenase was mimicked, with the carbamoyl as actings a reasonable surrogate for the acyl group and the acetonitrileeplacing the weakly bound water in the enzyme. The CO stretch-ng frequencies for 40 (1958 cm−1 and 2026 cm−1 in the solidtate) agree reasonably well with those reported for the enzyme1972 cm−1 and 2031 cm−1 in aqueous solutions).
Hu and co-workers have recently reported an acylmethylpyri-ine model for the [Fe]-hydrogenase 42 (Scheme 4.9) [87]. Fe(CO)5eacted with a lithium picolinate salt to form the acyl complex1, which exhibited three infrared bands in the carbonyl region,uggesting a trigonal bipyramidal geometry at iron centre witho nitrogen bonding. An unstable iron(II) species is formed byhe action of iodine, which can be reacted readily with sodium 6-
ethyl-2-mercaptopyridine to give the acyl complex 42, in whichoth pyridine and acyl groups are bound to the metal. Both 42and 42b exhibit two infrared stretches in the carbonyl region1962 cm−1 and 2029 cm−1 for 42a and 1961 cm−1 and 2026 cm−1
or 42b). The acyl CO group in 42 is exchangeable with 13CO, andhis reaction was monitored over the course of 1 week by NMRpectroscopy. The compound 42 reversibly isomerized to 42′ in theresence of ultraviolet light and this process reversed in the darkScheme 4.10).
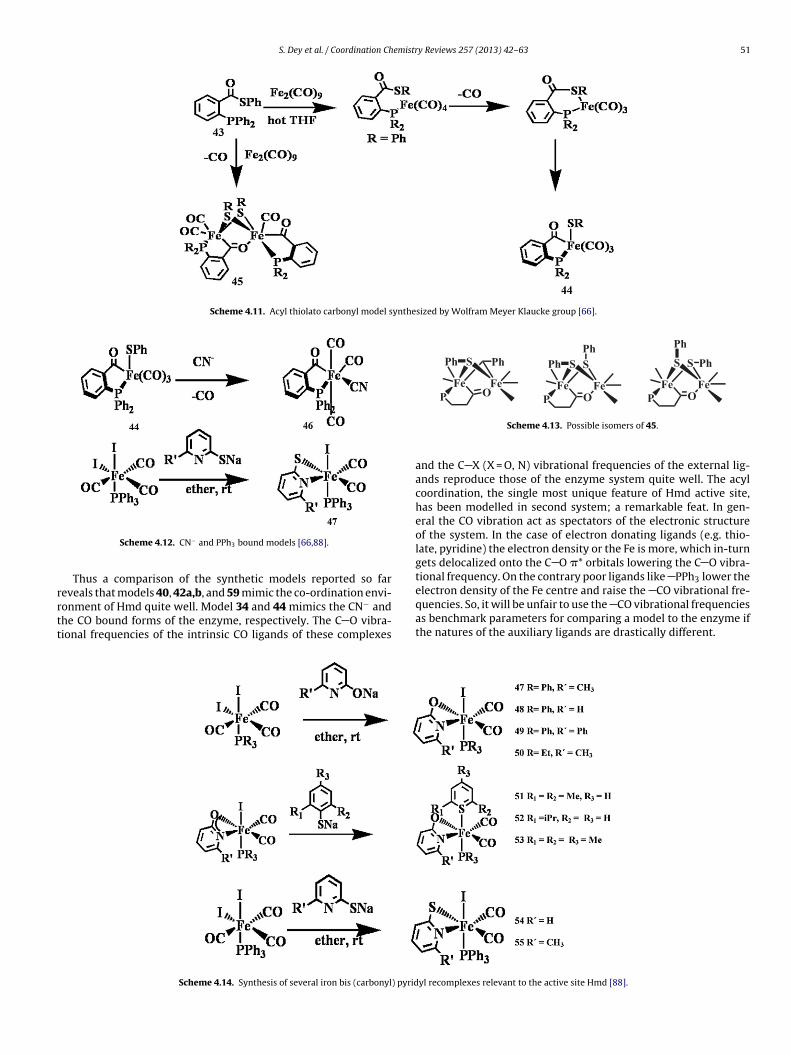
Recently Wolfram Meyer-Klaucke and his group made ahosphine-modified thioester model for the [Fe]-hydrogenaseScheme 4.11) [66]. The reaction of ligand 43 and a Fe(0) car-onyl source generates complex 44 and 45. The reported yieldf 45 was low. Using an extremely bulky aryl thioester, they
ere able to arrest the oxidative addition. The compound 45xisted as a mixture of isomers that were proposed to differ withespect to the orientation of the �-SPh groups (Scheme 4.12).
cheme 4.9. Synthesis of an acylmethylpyridine-containing model by Hu [87].
imic of Hmd synthesized by Pickett et al. [86].
Complex 44 reacted smoothly with cyanide to afford the anion46, isolated as its Et4N+ salt. The 13C and 31P NMR spectra forEt4N [46] and its 13CN-labeled derivative indicated that substi-tution was stereospecific. The IR spectrum of [46]− in CH2Cl2solution closely matches the cyanide-inhibited form of Hmd(Scheme 4.13).
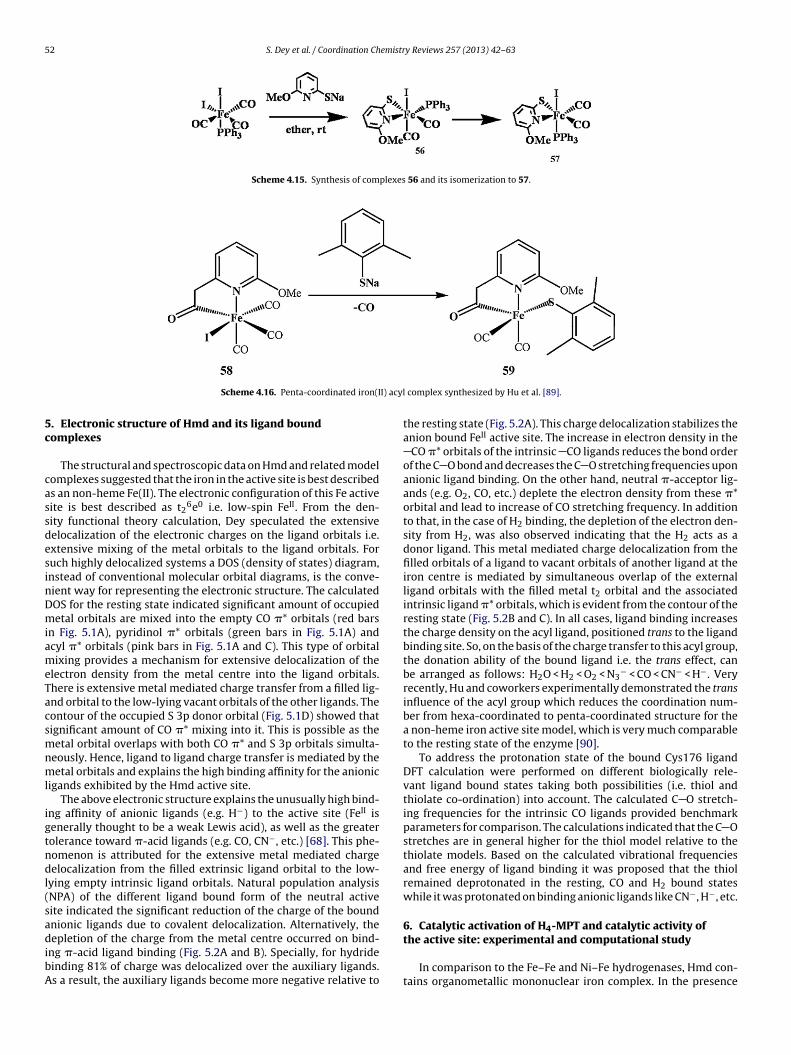
A series of Fe-complexes mimicking the active site of [Fe]-hydrogenase were synthesized and characterized by Xile Hu andco-workers (Scheme 4.14) [88] The reaction of Fe(CO)3(PPh3)(I)2with sodium 6-methyl-2-pyridonate yielded complex 47. Com-plexes 47–57 are display two strong IR absorptions between1950 cm−1 and 2050 cm−1, arising from the two terminal CO lig-ands. Interestingly complex 56 isomerises to 57 which is possiblythermodynamically more stable (Scheme 4.15).
Recently, Xile Hu and his groups synthesized a five coordi-nate iron complex containing acyl and monodentate thiolate ligandwhich is the solvent binding site in the active site of the [Fe]-hydrogenase (Scheme 4.16) [89]. They had previously synthesizedand purified the complex [(2-CH2CO-6-MeOC5H3N)Fe(CO)3I] (58).Complex 58 reacted with NaS(2,6-Me2C6H3) to produce a five-coordinate Fe complex 59 which is diamagnetic. The IR spectrumshows two carbonyl absorption bands of comparable intensities
−1 −1
Scheme 4.10. Possible isomerization route for 42 [87].
S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63 51
Scheme 4.11. Acyl thiolato carbonyl model synthesized by Wolfram Meyer Klaucke group [66].
rrtt
FeO
Fe
P
PhSPh
FeO
Fe
S
P
SPh
Ph
FeO
Fe
S
P
PhS
Ph
Scheme 4.12. CN− and PPh3 bound models [66,88].
Thus a comparison of the synthetic models reported so far
eveals that models 40, 42a,b, and 59 mimic the co-ordination envi-onment of Hmd quite well. Model 34 and 44 mimics the CN− andhe CO bound forms of the enzyme, respectively. The C O vibra-ional frequencies of the intrinsic CO ligands of these complexesScheme 4.14. Synthesis of several iron bis (carbonyl) pyrid
Scheme 4.13. Possible isomers of 45.
and the C X (X = O, N) vibrational frequencies of the external lig-ands reproduce those of the enzyme system quite well. The acylcoordination, the single most unique feature of Hmd active site,has been modelled in second system; a remarkable feat. In gen-eral the CO vibration act as spectators of the electronic structureof the system. In the case of electron donating ligands (e.g. thio-late, pyridine) the electron density or the Fe is more, which in-turngets delocalized onto the C O �* orbitals lowering the C O vibra-tional frequency. On the contrary poor ligands like PPh3 lower theelectron density of the Fe centre and raise the CO vibrational fre-
quencies. So, it will be unfair to use the CO vibrational frequenciesas benchmark parameters for comparing a model to the enzyme ifthe natures of the auxiliary ligands are drastically different.yl recomplexes relevant to the active site Hmd [88].
52 S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63
Scheme 4.15. Synthesis of complexes 56 and its isomerization to 57.
I) acyl
5c
cassdesinDmiameTacsmnml
igtndl(sadibA
Scheme 4.16. Penta-coordinated iron(I
. Electronic structure of Hmd and its ligand boundomplexes
The structural and spectroscopic data on Hmd and related modelomplexes suggested that the iron in the active site is best describeds an non-heme Fe(II). The electronic configuration of this Fe activeite is best described as t2
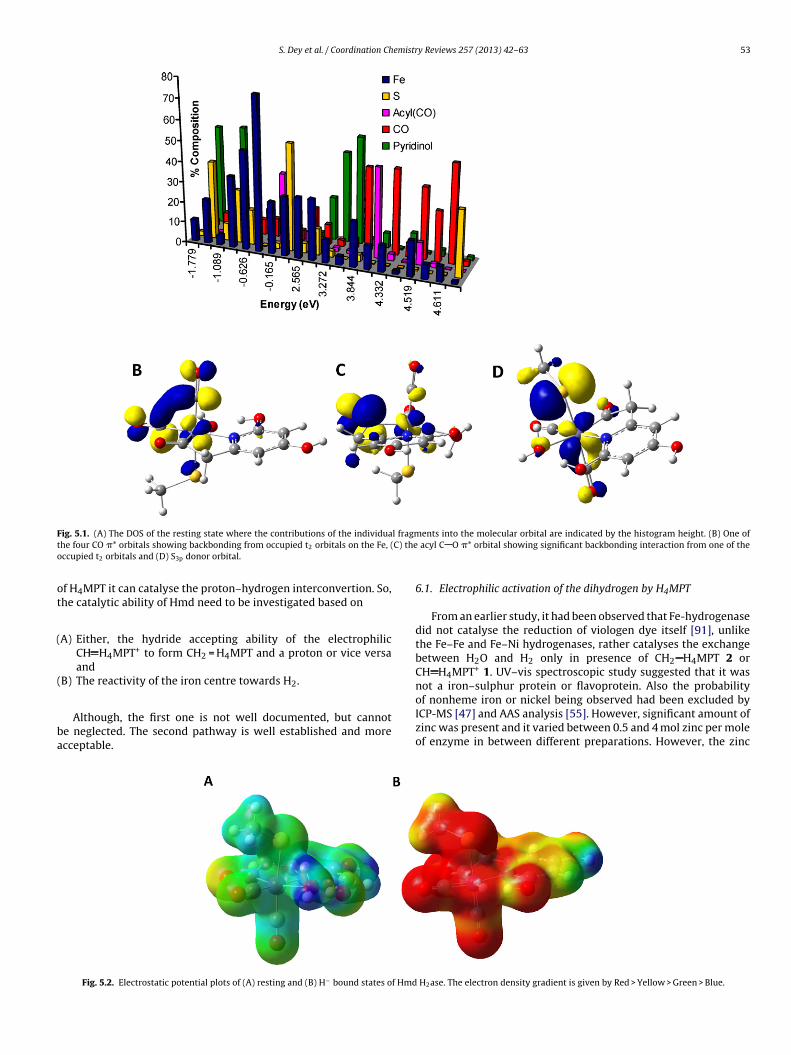
6e0 i.e. low-spin FeII. From the den-ity functional theory calculation, Dey speculated the extensiveelocalization of the electronic charges on the ligand orbitals i.e.xtensive mixing of the metal orbitals to the ligand orbitals. Foruch highly delocalized systems a DOS (density of states) diagram,nstead of conventional molecular orbital diagrams, is the conve-ient way for representing the electronic structure. The calculatedOS for the resting state indicated significant amount of occupiedetal orbitals are mixed into the empty CO �* orbitals (red bars
n Fig. 5.1A), pyridinol �* orbitals (green bars in Fig. 5.1A) andcyl �* orbitals (pink bars in Fig. 5.1A and C). This type of orbitalixing provides a mechanism for extensive delocalization of the
lectron density from the metal centre into the ligand orbitals.here is extensive metal mediated charge transfer from a filled lig-nd orbital to the low-lying vacant orbitals of the other ligands. Theontour of the occupied S 3p donor orbital (Fig. 5.1D) showed thatignificant amount of CO �* mixing into it. This is possible as theetal orbital overlaps with both CO �* and S 3p orbitals simulta-
eously. Hence, ligand to ligand charge transfer is mediated by theetal orbitals and explains the high binding affinity for the anionic
igands exhibited by the Hmd active site.The above electronic structure explains the unusually high bind-
ng affinity of anionic ligands (e.g. H−) to the active site (FeII isenerally thought to be a weak Lewis acid), as well as the greaterolerance toward �-acid ligands (e.g. CO, CN−, etc.) [68]. This phe-omenon is attributed for the extensive metal mediated chargeelocalization from the filled extrinsic ligand orbital to the low-
ying empty intrinsic ligand orbitals. Natural population analysisNPA) of the different ligand bound form of the neutral activeite indicated the significant reduction of the charge of the boundnionic ligands due to covalent delocalization. Alternatively, the
epletion of the charge from the metal centre occurred on bind-ng �-acid ligand binding (Fig. 5.2A and B). Specially, for hydrideinding 81% of charge was delocalized over the auxiliary ligands.s a result, the auxiliary ligands become more negative relative to
complex synthesized by Hu et al. [89].
the resting state (Fig. 5.2A). This charge delocalization stabilizes theanion bound FeII active site. The increase in electron density in the
CO �* orbitals of the intrinsic CO ligands reduces the bond orderof the C O bond and decreases the C O stretching frequencies uponanionic ligand binding. On the other hand, neutral �-acceptor lig-ands (e.g. O2, CO, etc.) deplete the electron density from these �*orbital and lead to increase of CO stretching frequency. In additionto that, in the case of H2 binding, the depletion of the electron den-sity from H2, was also observed indicating that the H2 acts as adonor ligand. This metal mediated charge delocalization from thefilled orbitals of a ligand to vacant orbitals of another ligand at theiron centre is mediated by simultaneous overlap of the externalligand orbitals with the filled metal t2 orbital and the associatedintrinsic ligand �* orbitals, which is evident from the contour of theresting state (Fig. 5.2B and C). In all cases, ligand binding increasesthe charge density on the acyl ligand, positioned trans to the ligandbinding site. So, on the basis of the charge transfer to this acyl group,the donation ability of the bound ligand i.e. the trans effect, canbe arranged as follows: H2O < H2 < O2 < N3
− < CO < CN− < H−. Veryrecently, Hu and coworkers experimentally demonstrated the transinfluence of the acyl group which reduces the coordination num-ber from hexa-coordinated to penta-coordinated structure for thea non-heme iron active site model, which is very much comparableto the resting state of the enzyme [90].
To address the protonation state of the bound Cys176 ligandDFT calculation were performed on different biologically rele-vant ligand bound states taking both possibilities (i.e. thiol andthiolate co-ordination) into account. The calculated C O stretch-ing frequencies for the intrinsic CO ligands provided benchmarkparameters for comparison. The calculations indicated that the C Ostretches are in general higher for the thiol model relative to thethiolate models. Based on the calculated vibrational frequenciesand free energy of ligand binding it was proposed that the thiolremained deprotonated in the resting, CO and H2 bound stateswhile it was protonated on binding anionic ligands like CN−, H−, etc.
6. Catalytic activation of H4-MPT and catalytic activity of
the active site: experimental and computational studyIn comparison to the Fe–Fe and Ni–Fe hydrogenases, Hmd con-tains organometallic mononuclear iron complex. In the presence
S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63 53
F l fragmt C) theo
ot
(
(
ba
ig. 5.1. (A) The DOS of the resting state where the contributions of the individuahe four CO �* orbitals showing backbonding from occupied t2 orbitals on the Fe, (ccupied t2 orbitals and (D) S3p donor orbital.
f H4MPT it can catalyse the proton–hydrogen interconvertion. So,he catalytic ability of Hmd need to be investigated based on
A) Either, the hydride accepting ability of the electrophilicCH H4MPT+ to form CH2 = H4MPT and a proton or vice versaand
B) The reactivity of the iron centre towards H2.
Although, the first one is not well documented, but cannote neglected. The second pathway is well established and morecceptable.
Fig. 5.2. Electrostatic potential plots of (A) resting and (B) H− bound states of Hmd
ents into the molecular orbital are indicated by the histogram height. (B) One of acyl C O �* orbital showing significant backbonding interaction from one of the
6.1. Electrophilic activation of the dihydrogen by H4MPT
From an earlier study, it had been observed that Fe-hydrogenasedid not catalyse the reduction of viologen dye itself [91], unlikethe Fe–Fe and Fe–Ni hydrogenases, rather catalyses the exchangebetween H2O and H2 only in presence of CH2 H4MPT 2 orCH H4MPT+ 1. UV–vis spectroscopic study suggested that it wasnot a iron–sulphur protein or flavoprotein. Also the probability
of nonheme iron or nickel being observed had been excluded byICP-MS [47] and AAS analysis [55]. However, significant amount ofzinc was present and it varied between 0.5 and 4 mol zinc per moleof enzyme in between different preparations. However, the zincH2ase. The electron density gradient is given by Red > Yellow > Green > Blue.

54 S. Dey et al. / Coordination Chemistr
S
c[suaciiottowsCfwwT
action of the molecular hydrogen with the model imidazolidine
cheme 6.1. Isotope exchange experiments with CH2 H4MPT and solvent [53,93].
ontent did not correlate with the specific activity of the enzyme55]. On the other hand, activity towards different ligands had beentudied. It was observed that presence of CO (5–50%) and CN− (5%)p to a certain amount did not cause activity loss. Also, nitrite, azidend acetylene were not inhibitors under the conditions of the assayondition. Isotope exchange studies with solvents are summarisedn Scheme 6.1 [53]. Titrium exchange or deuterium exchange studyn the presence and absence of CH H4MPT+ 1 showed that 0.5 molf 3H/D was incorporated into the CH2 H4MPT 2, which is relevanto 62 (Scheme 6.2), or double displacement to form 60 and onhe reverse reaction i.e. the release of the corresponding 0.5 molf 3H/D into the aqueous medium from the C3H2/D2 H4MPT 60,as also observed [56,65,92,8]. Other isotope exchange results
howed that CD2 H4MPT 60 in H2O produces HD and H2 whereasH2 H4MPT 2 in D2O produces HD and D2. Production of H2
rom CD H MPT 2 in H O (D from CH H MPT in D O) [93]
2 4 2 2 2 4 2as explained by the exchange between HD and the solvent,hich was caused by CH H4MPT+-assisted Hmd catalysis [94].his indicated that only one hydrogen from CH2 H4MPT 1 was
Scheme 6.2. NMR-labelling studies showing
y Reviews 257 (2013) 42– 63
included in the dihydrogen (Scheme 6.2) formed. Similar resultswere also obtained from 2D NMR analysis of the solution of Hmdand CH H4MPT+ 1 in D2O where the signal of the methenylhydrogen gradually disappeared suggesting a H/D exchange [56].The formation of CD2 H4MPT 60 in the reaction of CH2 H4MPT 2with D2 in D2O could also be explained by H/D exchange. Although,no discussion of its mechanism have appeared in the literature, theenhanced rate of proton exchange between 1 and 2 likely involvesthe deprotonation of 1 leading to the formation of N-heterocycliccarbene 66 or zwitterionic form 65, relevant to the biologicalthiamine salts [95]. Determination of the stereochemistry showedthat hydride was transferred from hydrogen to take up the pro-Rposition of C14a in CH2 H4MPT 2 (Scheme 6.3).
Berkessel and Thauer invoked the super-electrophilic activa-tion of dihydrogen by the CH H4MPT+ 1 [65,96,97]. Bindingof CH H4MPT+ 1 to the enzyme initiates the heterolytic cleav-age of dihydrogen. Based on the work by Olah et al. [98,99]on the activation of the alkanes for hydride exchange in thepresence of superacids, they described the phenomenon as theformation of the dicationic species on protonation at the nitro-gen atoms, 70. This idea was further supported by the enhancedrate increase of the H/D exchange on increasing the pH of themedium. This dicationic species may increase the electrophilicnature of the C14a of CH H4MPT+ 1, allowing it to abstract ahydride from molecular hydrogen to form CH2 H4MPT+ (73 or74). In contrast, ab initio calculation showed that weak inter-
[65,100]. A van der Waals ternary complex of the H2 with imida-zolidine 71, similar to the linear transition state in Scheme 4.6, canform.
stereochemistry of the reaction [56].
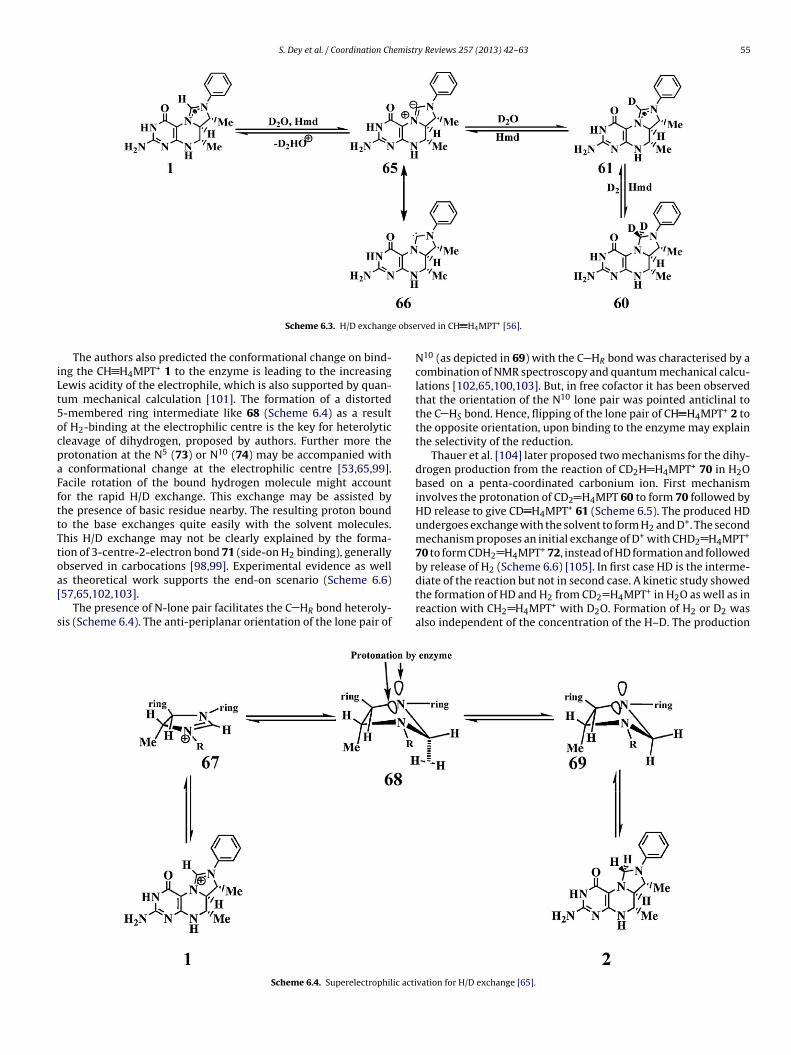
S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63 55
e obse
iLt5ocpaFfttTtoa[
s
Scheme 6.3. H/D exchang
The authors also predicted the conformational change on bind-ng the CH H4MPT+ 1 to the enzyme is leading to the increasingewis acidity of the electrophile, which is also supported by quan-um mechanical calculation [101]. The formation of a distorted-membered ring intermediate like 68 (Scheme 6.4) as a resultf H2-binding at the electrophilic centre is the key for heterolyticleavage of dihydrogen, proposed by authors. Further more therotonation at the N5 (73) or N10 (74) may be accompanied with
conformational change at the electrophilic centre [53,65,99].acile rotation of the bound hydrogen molecule might accountor the rapid H/D exchange. This exchange may be assisted byhe presence of basic residue nearby. The resulting proton boundo the base exchanges quite easily with the solvent molecules.his H/D exchange may not be clearly explained by the forma-ion of 3-centre-2-electron bond 71 (side-on H2 binding), generallybserved in carbocations [98,99]. Experimental evidence as well
s theoretical work supports the end-on scenario (Scheme 6.6)57,65,102,103].The presence of N-lone pair facilitates the C HR bond heteroly-is (Scheme 6.4). The anti-periplanar orientation of the lone pair of
Scheme 6.4. Superelectrophilic acti
rved in CH H4MPT+ [56].
N10 (as depicted in 69) with the C HR bond was characterised by acombination of NMR spectroscopy and quantum mechanical calcu-lations [102,65,100,103]. But, in free cofactor it has been observedthat the orientation of the N10 lone pair was pointed anticlinal tothe C HS bond. Hence, flipping of the lone pair of CH H4MPT+ 2 tothe opposite orientation, upon binding to the enzyme may explainthe selectivity of the reduction.
Thauer et al. [104] later proposed two mechanisms for the dihy-drogen production from the reaction of CD2H H4MPT+ 70 in H2Obased on a penta-coordinated carbonium ion. First mechanisminvolves the protonation of CD2 H4MPT 60 to form 70 followed byHD release to give CD H4MPT+ 61 (Scheme 6.5). The produced HDundergoes exchange with the solvent to form H2 and D+. The secondmechanism proposes an initial exchange of D+ with CHD2 H4MPT+
70 to form CDH2 H4MPT+ 72, instead of HD formation and followedby release of H2 (Scheme 6.6) [105]. In first case HD is the interme-
diate of the reaction but not in second case. A kinetic study showedthe formation of HD and H2 from CD2 H4MPT+ in H2O as well as inreaction with CH2 H4MPT+ with D2O. Formation of H2 or D2 wasalso independent of the concentration of the H–D. The productionvation for H/D exchange [65].
56 S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63
Scheme 6.5. Proposed pathway 1 for the formation of H2 by CH2 H4MPT+ [53,65,99].
optc
Scheme 6.6. Proposed pathway 2 for H2 heterolysis [98,99].
f H–D was assumed to be unlikely. Hence the second pathway is
referred and the only transition state 71 able to exchange pro-on stereospecifically. This mechanism precipitated a number ofomputational studies that involves the amidine ions [102,57,103].Scheme 6.7. Protonation coupled dual pathways for su
Scheme 6.8. Stereochemical views of the iron-hydrogenase (left) and Fe–Fe hydro-genase (right).
6.2. Mononuclear non-heme iron mediated heterolysis
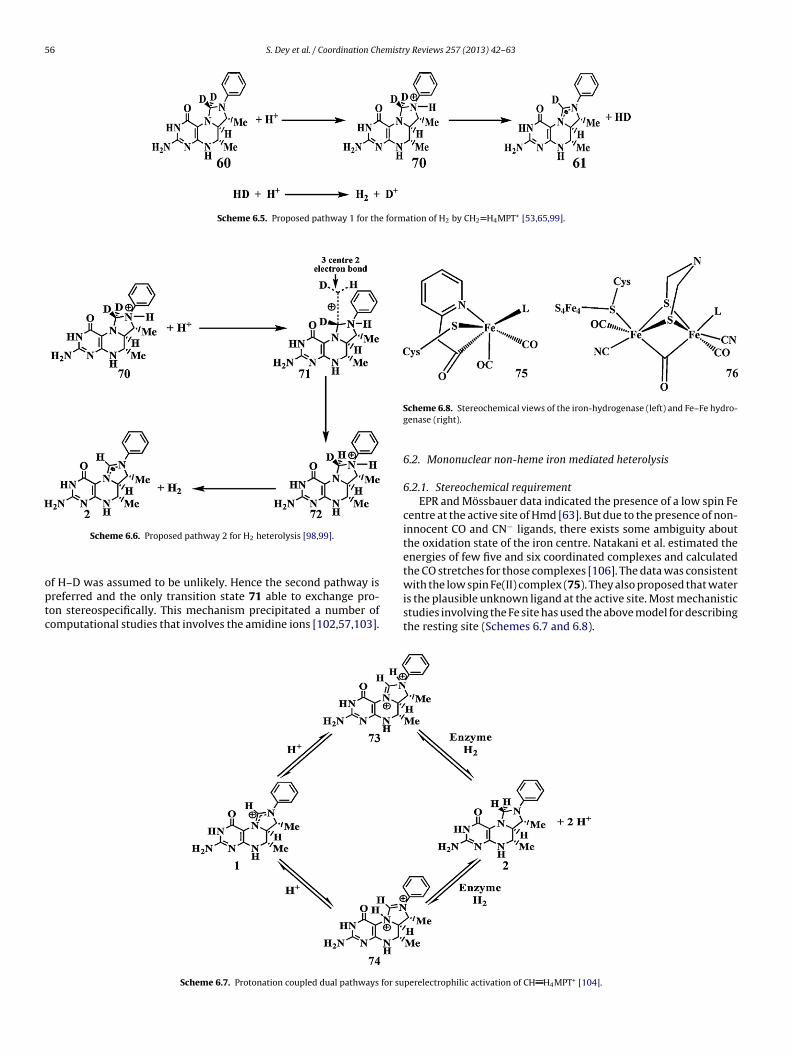
6.2.1. Stereochemical requirementEPR and Mössbauer data indicated the presence of a low spin Fe
centre at the active site of Hmd [63]. But due to the presence of non-innocent CO and CN− ligands, there exists some ambiguity aboutthe oxidation state of the iron centre. Natakani et al. estimated theenergies of few five and six coordinated complexes and calculatedthe CO stretches for those complexes [106]. The data was consistentwith the low spin Fe(II) complex (75). They also proposed that water
is the plausible unknown ligand at the active site. Most mechanisticstudies involving the Fe site has used the above model for describingthe resting site (Schemes 6.7 and 6.8).perelectrophilic activation of CH H4MPT+ [104].

S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63 57
states
hwn�lbhti
6
gcaTiT2tolfatgHtep
Scheme 6.9. Proposed resting
Stiebritz and Reiher reported that for both Fe-Hmd and Fe–Feydrogenase the ligands trans to the H2 binding site in active formas a CO ligand i.e. acyl in Hmd and bridged CO in Fe–Fe hydroge-ase. Other four ligands are arranged in a plane such that the two-donor ligands were at one side and the two strong �-acceptor
igands were on the other side [107,108]. They suggested this toe the necessary geometric structure for both Fe-only and Fe–Feydrogenase where in the latter case only one Fe contributes tohe reactivity while the other contributes to the structure predom-nantly.
.2.2. MechanismBased on the first proposed crystal structure of the Hmd hydro-
enase by Shima et al. [58], Yang and Hall predicted, using DFTalculation, a trigger mechanism that invokes a heterolytic cleav-ge of molecular hydrogen at the iron centre assisted by MPT+[109].hey proposed two possible resting states 81 and 82 which arenter-convertable by protonation or deprotonation (Scheme 6.9).hese structures are quite different from the actual structure. The-hydroxyl group was shifted to the 3-position of the pyridinol ringo avoid strong interaction with the CO ligand or the sulphur atomf the cysteine ligand and the O-atom of the carboxylate ligand wasigated to the iron atom. The calculated CO stretching frequenciesor the deprotonated state were closer to the experimental valuesnd thus this was considered as the active site model. The H2 activa-ion without the assistance of MPT+, which requires the carboxylateroup assistance, was energetically unfavourable (Scheme 6.10).
ence, a MPT+ assisted catalysis (Scheme 6.11) was proposed. Ini-iation of the cycle involves a weak interaction of MPT+ (via itslectrophilic C atom) with the Fe centre which partially or tem-orarily reduces the pyridinol–Fe interaction and the rotation of the
Scheme 6.10. Enthalpies for direct H2 sp
for trigger mechanism [109].
carboxylate group (83). This reduces the H2O/H2 ligand exchangebarrier and forms 84 (Scheme 6.11). H2 splitting was took place eas-ily when MPT+ was close to Fe and resulting in 85 (Scheme 6.11).One hydride is adopted by MPT+ to form HMPT which then disso-ciates from the iron centre leaving the H+ ion bound to the iron.Simultaneously, H2O occupied the position trans to pyridinol N-atom and shifts the H+ to occupy the position vacated by HMPT, 86.The resting state was achieved by proton transfer steps followedby carboxylate ligation, similar to the 87. The other possibility forregenerating the resting state is proton release before H2O coordi-nation and carboxylate binding. Here, change in donor capabilityand of the pyridine ring as well as MPT+ binding triggers the het-erolytic cleavage of H2. The strong donor ability of the pyridine ringprevents the direct cleavage of the hydrogen which is only possi-ble in the presence of MPT+. One unique feature of this proposedmechanism is direct protonation of an Fe(II) centre (86) which isunprecedented in previous literature.
Yang and Hall modified the proposed catalytic mechanism afterthe report of the revised crystal structure of the enzyme where the
COOH coordination in (81) was replaced by an acyl coordination in88 [110]. The structural parameters and the CO vibrational frequen-cies of the H2O bound active site were similar to the experimentalvalues. The H2O/H2 ligand exchange reaction was energeticallyneutral and presumably rapid, 89. This ligand exchange was lattercalculated by Dey to be an uphill process. After H2O/H2 exchange,Cys176 assisted splitting of H2 takes place. The transition state(TS90) for this step is 6.6 kcal/mol higher in energy than the H2
bound complex 89 and the final complex 91 is 3.4 cal/mol morestable than 88 and exhibits a strong Fe Hı−· · ·Hı+ O dihydrogenbond (Scheme 6.12). The calculated vibrational frequencies for thecis-CO ligands in 91 are 2007 and 1949 cm−1 which are similar tolitting by 82 without MPT+ [109].
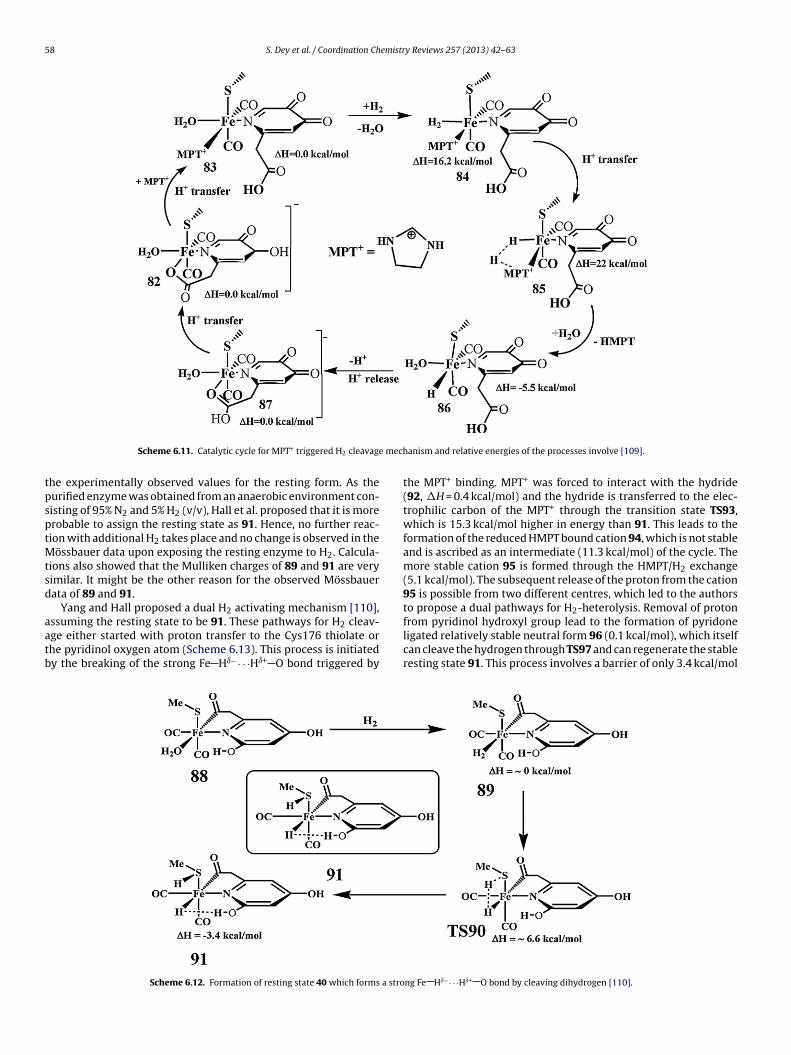
58 S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63
mech
tpsptMtsd
aatb
Scheme 6.11. Catalytic cycle for MPT+ triggered H2 cleavage
he experimentally observed values for the resting form. As theurified enzyme was obtained from an anaerobic environment con-isting of 95% N2 and 5% H2 (v/v), Hall et al. proposed that it is morerobable to assign the resting state as 91. Hence, no further reac-ion with additional H2 takes place and no change is observed in the
össbauer data upon exposing the resting enzyme to H2. Calcula-ions also showed that the Mulliken charges of 89 and 91 are veryimilar. It might be the other reason for the observed Mössbauerata of 89 and 91.
Yang and Hall proposed a dual H2 activating mechanism [110],
ssuming the resting state to be 91. These pathways for H2 cleav-ge either started with proton transfer to the Cys176 thiolate orhe pyridinol oxygen atom (Scheme 6.13). This process is initiatedy the breaking of the strong Fe Hı−· · ·Hı+ O bond triggered byScheme 6.12. Formation of resting state 40 which forms a stro
anism and relative energies of the processes involve [109].
the MPT+ binding. MPT+ was forced to interact with the hydride(92, �H = 0.4 kcal/mol) and the hydride is transferred to the elec-trophilic carbon of the MPT+ through the transition state TS93,which is 15.3 kcal/mol higher in energy than 91. This leads to theformation of the reduced HMPT bound cation 94, which is not stableand is ascribed as an intermediate (11.3 kcal/mol) of the cycle. Themore stable cation 95 is formed through the HMPT/H2 exchange(5.1 kcal/mol). The subsequent release of the proton from the cation95 is possible from two different centres, which led to the authorsto propose a dual pathways for H2-heterolysis. Removal of proton
from pyridinol hydroxyl group lead to the formation of pyridoneligated relatively stable neutral form 96 (0.1 kcal/mol), which itselfcan cleave the hydrogen through TS97 and can regenerate the stableresting state 91. This process involves a barrier of only 3.4 kcal/molng Fe Hı−· · ·Hı+ O bond by cleaving dihydrogen [110].
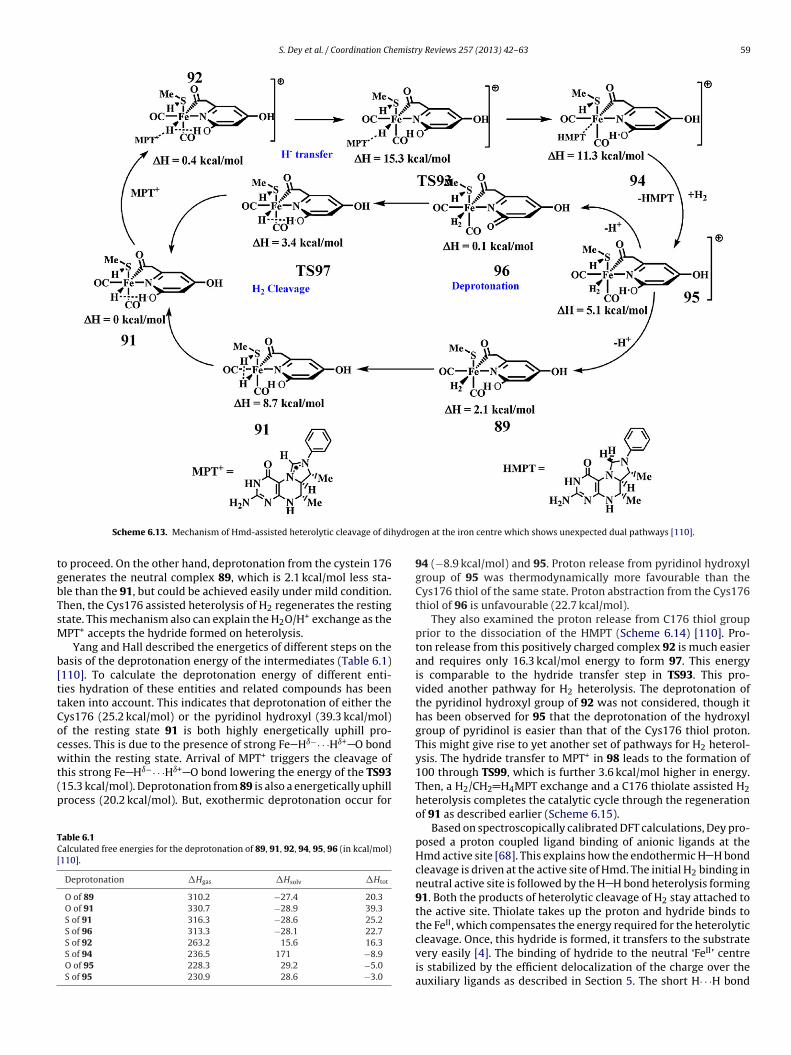
S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63 59
ydrog
tgbTsM
b[ttCocwt(p
TC[
Scheme 6.13. Mechanism of Hmd-assisted heterolytic cleavage of dih
o proceed. On the other hand, deprotonation from the cystein 176enerates the neutral complex 89, which is 2.1 kcal/mol less sta-le than the 91, but could be achieved easily under mild condition.hen, the Cys176 assisted heterolysis of H2 regenerates the restingtate. This mechanism also can explain the H2O/H+ exchange as thePT+ accepts the hydride formed on heterolysis.Yang and Hall described the energetics of different steps on the
asis of the deprotonation energy of the intermediates (Table 6.1)110]. To calculate the deprotonation energy of different enti-ies hydration of these entities and related compounds has beenaken into account. This indicates that deprotonation of either theys176 (25.2 kcal/mol) or the pyridinol hydroxyl (39.3 kcal/mol)f the resting state 91 is both highly energetically uphill pro-esses. This is due to the presence of strong Fe Hı−· · ·Hı+ O bond
+
ithin the resting state. Arrival of MPT triggers the cleavage ofhis strong Fe Hı−· · ·Hı+ O bond lowering the energy of the TS9315.3 kcal/mol). Deprotonation from 89 is also a energetically uphillrocess (20.2 kcal/mol). But, exothermic deprotonation occur forable 6.1alculated free energies for the deprotonation of 89, 91, 92, 94, 95, 96 (in kcal/mol)110].
Deprotonation �Hgas �Hsolv �Htot
O of 89 310.2 −27.4 20.3O of 91 330.7 −28.9 39.3S of 91 316.3 −28.6 25.2S of 96 313.3 −28.1 22.7S of 92 263.2 15.6 16.3S of 94 236.5 171 −8.9O of 95 228.3 29.2 −5.0S of 95 230.9 28.6 −3.0
en at the iron centre which shows unexpected dual pathways [110].
94 (−8.9 kcal/mol) and 95. Proton release from pyridinol hydroxylgroup of 95 was thermodynamically more favourable than theCys176 thiol of the same state. Proton abstraction from the Cys176thiol of 96 is unfavourable (22.7 kcal/mol).
They also examined the proton release from C176 thiol groupprior to the dissociation of the HMPT (Scheme 6.14) [110]. Pro-ton release from this positively charged complex 92 is much easierand requires only 16.3 kcal/mol energy to form 97. This energyis comparable to the hydride transfer step in TS93. This pro-vided another pathway for H2 heterolysis. The deprotonation ofthe pyridinol hydroxyl group of 92 was not considered, though ithas been observed for 95 that the deprotonation of the hydroxylgroup of pyridinol is easier than that of the Cys176 thiol proton.This might give rise to yet another set of pathways for H2 heterol-ysis. The hydride transfer to MPT+ in 98 leads to the formation of100 through TS99, which is further 3.6 kcal/mol higher in energy.Then, a H2/CH2 H4MPT exchange and a C176 thiolate assisted H2heterolysis completes the catalytic cycle through the regenerationof 91 as described earlier (Scheme 6.15).
Based on spectroscopically calibrated DFT calculations, Dey pro-posed a proton coupled ligand binding of anionic ligands at theHmd active site [68]. This explains how the endothermic H H bondcleavage is driven at the active site of Hmd. The initial H2 binding inneutral active site is followed by the H H bond heterolysis forming91. Both the products of heterolytic cleavage of H2 stay attached tothe active site. Thiolate takes up the proton and hydride binds tothe FeII, which compensates the energy required for the heterolytic
cleavage. Once, this hydride is formed, it transfers to the substratevery easily [4]. The binding of hydride to the neutral ‘FeII’ centreis stabilized by the efficient delocalization of the charge over theauxiliary ligands as described in Section 5. The short H· · ·H bond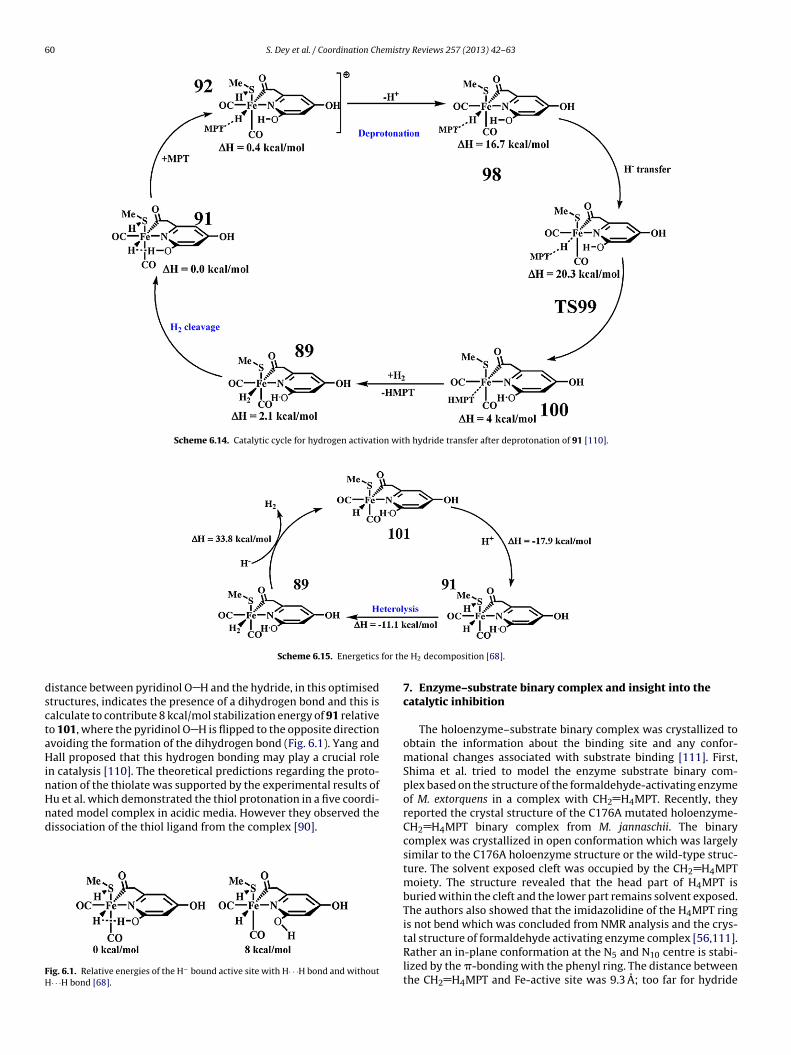
60 S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63
Scheme 6.14. Catalytic cycle for hydrogen activation with hydride transfer after deprotonation of 91 [110].
for th
dsctaHinHnd
FH
Scheme 6.15. Energetics
istance between pyridinol O H and the hydride, in this optimisedtructures, indicates the presence of a dihydrogen bond and this isalculate to contribute 8 kcal/mol stabilization energy of 91 relativeo 101, where the pyridinol O H is flipped to the opposite directionvoiding the formation of the dihydrogen bond (Fig. 6.1). Yang andall proposed that this hydrogen bonding may play a crucial role
n catalysis [110]. The theoretical predictions regarding the proto-ation of the thiolate was supported by the experimental results of
u et al. which demonstrated the thiol protonation in a five coordi-ated model complex in acidic media. However they observed theissociation of the thiol ligand from the complex [90].ig. 6.1. Relative energies of the H− bound active site with H· · ·H bond and without· · ·H bond [68].
e H2 decomposition [68].
7. Enzyme–substrate binary complex and insight into thecatalytic inhibition
The holoenzyme–substrate binary complex was crystallized toobtain the information about the binding site and any confor-mational changes associated with substrate binding [111]. First,Shima et al. tried to model the enzyme substrate binary com-plex based on the structure of the formaldehyde-activating enzymeof M. extorquens in a complex with CH2 H4MPT. Recently, theyreported the crystal structure of the C176A mutated holoenzyme-CH2 H4MPT binary complex from M. jannaschii. The binarycomplex was crystallized in open conformation which was largelysimilar to the C176A holoenzyme structure or the wild-type struc-ture. The solvent exposed cleft was occupied by the CH2 H4MPTmoiety. The structure revealed that the head part of H4MPT isburied within the cleft and the lower part remains solvent exposed.The authors also showed that the imidazolidine of the H4MPT ringis not bend which was concluded from NMR analysis and the crys-
tal structure of formaldehyde activating enzyme complex [56,111].Rather an in-plane conformation at the N5 and N10 centre is stabi-lized by the �-bonding with the phenyl ring. The distance betweenthe CH2 H4MPT and Fe-active site was 9.3 A; too far for hydride
S. Dey et al. / Coordination Chemistry Reviews 257 (2013) 42– 63 61
cleava
tfdtb
mawsttuatsppsoagpc
oToci
Scheme 7.1. Proposed binary complex assisted H2
ransfer. It’s seemed likely that to catalyse the reaction, the activeorm of CH2 H4MPT i.e. nonplanar conformation of the imidazoli-ine ring, could be achieved through the interaction of the Re face ofhe imidazolidine ring and the iron centre which could be achievedy rotating the phenyl ring.
To model the closed form i.e. the catalytically active confor-ation of the enzyme with CH2 H4MPT bound, Shima et al. took
dvantage of crystallizing the apoenzyme structure of M. jannaschii,hich was crystallised in the closed form before. The apoenzyme
tructure and the open C176A-holoenzyme–substrate complex ofhese two systems are almost identical. Open to closed conforma-ional change is accompanied by the movement of the peripheralnit upon substrate binding. The distance between CH2 H4MPTnd Fe-GP cofactor was decreased to 3 A in the closed form, enougho allow molecular hydrogen binding and H− transfer. Also, thistructure showed that the C14a methylene-H4MPT occupied theosition trans to the acyl binding site of the metal centre and theroposed site for H2 binding and cleavage. The substrate binding isupposedly stabilized by the interaction between pyridinol groupf the Fe-GP and phenyl ring of the H4MPT, the hydroxyl groupnd the N10 atom and also that between the CO ligand and C4a oxoroup. The structure in the closed conformation also indicated theresence of a narrow hydrophobic channel which could be the gashannel directing H2 towards the active site.
Based on the open/closed conformational transition, Shima et al.utlined a hypothetical catalytic mechanism (Scheme 7.1) [111].
he catalysis was initiated upon binding of the CH H4MPT to thepen conformation of the enzyme that induces the closure of theleft to yield the closed conformation, 103. This closure may alsonduce a conformational change in CH H4MPT that enhances itsge pathway as described by Thauer et al. [53,111].
electrophilic character at C14a. H2 is then introduced to the activesite through the hydrophobic gas channel and bind the iron centreat the open binding site. Then, the polarised hydrogen is cleaved bythe adjacent carbocation, 104 (C14a of CH H4MPT). The hydride isthen transferred to the CH H4MPT and the resulting H+ is adoptedby a base which may be the Cys176 residue or the pyridinol oxygen,105. This ensures the formation of CH2 H4MPT and H+. No role ofthe metal was invoked here.
At high concentration, CO and CN− are known inhibitors of Hmd[57]. This was also indicated from the calculated free energy of bind-ing of these ligands. Interestingly for O2, a known inhibitor for otherhydrogenases, the calculated binding energy is unfavourable. Thiswas due to the presence of the non-innocent CO and acyl ligandswhich depleted the Fe(II) centre of its electron density making ita good Lewis acid. This increased its binding affinity for electrondonating anionic ligands but weakened its affinity for other elec-tron withdrawing ligands like O2. It was suggested that all theseligands (including O2) have better binding affinity to the FeII cen-tre than the substrate H2. The conformational change from open toclosed form (vide supra) reduces the distance of CH H4MPT fromthe iron centre from 9.3 A to 3 A. The space-filling models for thedifferent ligand-bound state suggested that in the closed confor-mation the narrow vacant space between the iron centre and theCH H4MPT is not sufficient for accommodating a H2O, CO or CN−
species for steric reasons. But, a small H2 molecule can be accom-modated within the space easily. In the absence of CH H4MPT
the H2 binding to the Fe(II) is not favourable energetically [110]because this involves displacement of a bound H2O. Dey proposedthat H2O may, in fact, act as inhibitor of the Hmd. The bind-ing of CH H4MPT probably dissociates the H2O molecule (due to
6 emistr
sr
8
shsudtspipngtae
R
2 S. Dey et al. / Coordination Ch
teric congestion) and allows the binding of H2, thus triggering theeaction.
. Conclusion
The iron site of Hmd is a new domain of the non-heme Fe land-cape. Significant amount of biochemical and spectroscopic dataave been accumulated on this unique non-heme Fe enzyme. Theynthesis and DFT calculations, reproducing these data, have beensed to analyse the electronic structure of the resting site and itsifferent inhibited forms. Assimilation of these experimental andheoretical data has lead to a deeper understanding of this activeite and, naturally, a few plausible reaction mechanisms have beenroposed for Hmd. However, a lot remains to be explored; key
ntermediates involved in the enzymatic cycle and H+/D+ exchangerocesses needs to be trapped and identified. These intermediateseed to be modelled in small molecules to obtain insight into theeometric and electronic structure contribution towards the reac-ivity of this hydrogenase. These efforts may or may not result in
cheap catalyst to generate H2 from H2O, but they will definitelynhance our understanding of the H+ → H2 process.
eferences
[1] C. Winter, Int. J. Hydrogen Energy 30 (2005) 1371.[2] R. Cammack, in: R. Cammack, M. Frey, R. Robson (Eds.), Hydrogen as a Fuel:
Learning from Nature, Taylor & Francis, London, UK, 2001, pp. 159–180.[3] F.A. Armstrong, J.C. Fontecilla-Camps, Science 321 (2008) 498.[4] G.J. Kubas, Chem. Rev. 107 (2007) 4152.[5] P.E.M. Siegbahn, J.W. Tye, M.B. Hall, Chem. Rev. 107 (2007) 4414.[6] K.A. Vincent, A. Parkin, F.A. Armstrong, Chem. Rev. 107 (2007) 4366.[7] R.K. Thauer, Microbiology 144 (1998) 2377.[8] R.K. Thauer, A.R. Klein, G.C. Hartmann, Chem. Rev. 96 (1996) 3031.[9] S. Shima, R.K. Thauer, Chem. Rec. 7 (2007) 37.
[10] R.K. Thauer, A.-K. Kaster, M. Goenrich, M. Schick, T. Hiromoto, S. Shima, Annu.Rev. Biochem. 79 (2010) 507.
[11] L. Zhang, T. Happe, A. Melis, Planta 214 (2002) 552.[12] R.K. Thauer, A.-K. Kaster, H. Seedorf, W. Buckel, R. Hedderich, Nat. Rev. Micro-
biol. 6 (2008) 579.[13] A. Le Goff, V. Artero, B. Jousselme, P.D. Tran, N. Guillet, R. Métayé, A. Fihri, S.
Palacin, M. Fontecave, Science 326 (2009) 1384.[14] A.J. Richardson, C.S. Stewart, in: J.P. Elaich, M. Bruschi, J.L. Garcia (Eds.),
Microbiology and Biochemistry of Strict Anaerobes Involved in InterspeciesHydrogen Transfer, Plenum Press, New York, 1990, pp. 463–466.
[15] R. Conrad, Microbiol. Rev. 60 (1996) 609.[16] P.C. Dos Santos, D.R. Dean, Y. Hu, M.W. Ribbe, Chem. Rev. 104 (2004) 1159.[17] Y. Hu, M.W. Ribbe, Acc. Chem. Res. 43 (2009) 475.[18] L.C. Seefeldt, B.M. Hoffman, D.R. Dean, Annu. Rev. Biochem. 78 (2009) 701.[19] R.K. Thauer, FEBS Lett. 27 (1972) 111.[20] T. Reda, C.M. Plugge, N.J. Abram, J. Hirst, Proc. Natl. Acad. Sci. U.S.A. 105 (2008)
10654.[21] V. Artero, M. Fontecave, Coord. Chem. Rev. 249 (2005) 1518.[22] M. Winter, R.J. Brodd, Chem. Rev. 104 (2004) 4245.[23] A.K. Jones, E. Sillery, S.P.J. Albracht, F.A. Armstrong, Chem. Commun. (2002)
866.[24] C. Tard, X. Liu, S.K. Ibrahim, M. Bruschi, L.D. Gioia, S.C. Davies, X. Yang, L.-S.
Wang, G. Sawers, C.J. Pickett, Nature 433 (2005) 610.[25] Z. Li, Y. Ohki, K. Tatsumi, J. Am. Chem. Soc. 127 (2005) 8950.[26] S. Ogo, R. Kabe, K. Uehara, B. Kure, T. Nishimura, S.C. Menon, R. Harada, S.
Fukuzumi, Y. Higuchi, T. Ohhara, T. Tamada, R. Kuroki, Science 316 (2007)585.
[27] A.D. Wilson, R.K. Shoemaker, A. Miedaner, J.T. Muckerman, D.L. DuBois, M.R.DuBois, Proc. Natl. Acad. Sci. U.S.A. 104 (2007) 6951.
[28] C. Mealli, T.B. Rauchfuss, Angew. Chem., Int. Ed. 46 (2007) 8942.[29] M.L. Singleton, N. Bhuvanesh, J.H. Reibenspies, M.Y. Darensbourg, Angew.
Chem., Int. Ed. 47 (2008) 9492.[30] C.d. Tard, C.J. Pickett, Chem. Rev. 109 (2009) 2245.[31] Y. Ohki, K. Yasumura, M. Ando, S. Shimokata, K. Tatsumi, Proc. Natl. Acad. Sci.
U.S.A. 107 (2010) 3994.[32] L.H.S. Marjory Stephenson, Biochem. J. 25 (1931) 205.[33] W. Kempner, F. Kubowitz, Biochem. J. 265 (1933) 245.[34] L.H.S. David Ezra Green, Biochem. J. 28 (1934) 898.[35] O. Warburg, Schwermetalle als Wirkungsgruppen von Fermenten (Heavy
Metals as Prosthetic Groups of Enzymes), Verlag Saenger, Berlin, 1946.[36] J.D. Santangelo, P. Dürre, D.R. Woods, Microbiology 141 (1995) 171.[37] A.L.d. Lacey, V.M. Fernandez, M. Rousset, Coord. Chem. Rev. 249 (2005) 1596.[38] F.A. Armstrong, N.A. Belsey, J.A. Cracknell, G. Goldet, A. Parkin, E. Reisner, K.A.
Vincent, A.F. Wait, Chem. Soc. Rev. 38 (2009) 36.
y Reviews 257 (2013) 42– 63
[39] J.C. Fontecilla-Camps, A. Volbeda, C. Cavazza, Y. Nicolet, Chem. Rev. 107 (2007)4273.
[40] A.L. De Lacey, V.c.M. Fernández, M. Rousset, R. Cammack, Chem. Rev. 107(2007) 4304.
[41] W. Lubitz, E. Reijerse, M. van Gastel, Chem. Rev. 107 (2007) 4331.[42] P.M. Vignais, B. Billoud, Chem. Rev. 107 (2007) 4206.[43] S. Canaguier, V. Artero, M. Fontecave, Dalton Trans. (2008) 315.[44] E. Garcin, X. Vernede, E.C. Hatchikian, A. Volbeda, M. Frey, J.C. Fontecilla-
Camps, Structure 7 (1999) 557.[45] M.Y. Darensbourg, E.J. Lyon, X. Zhao, I.P. Georgakaki, Proc. Natl. Acad. Sci.
U.S.A. 100 (2003) 3683.[46] D.M. Heinekey, J. Organomet. Chem. 694 (2009) 2671.[47] C. Zirngibl, R. Hedderich, R.K. Thauer, FEBS Lett. 261 (1990) 112.[48] K. Ma, C. Zirngibl, D. Linder, K.O. Stetter, R.K. Thauer, Arch. Microbiol. 156
(1991) 43.[49] R. von Bünau, C. Zirngibl, R.K. Thauer, A. Klein, Eur. J. Biochem. 202 (1991)
1205.[50] G.C. Hartmann, A.R. Klein, M. Linder, R.K. Thauer, Arch. Microbiol. 165 (1996)
187.[51] G. Buurman, S. Shima, R.K. Thauer, FEBS Lett. 485 (2000) 200.[52] S. Shima, U. Ermler, Eur. J. Inorg. Chem. 2011 (2011) 963.[53] M.J. Corr, J.A. Murphy, Chem. Soc. Rev. 40 (2011) 2279.[54] B. Schwörer, R. Thauer, Arch. Microbiol. 155 (1991) 459.[55] C. Zirngibl, W. Van Dongen, B. Schwörer, R. Von Bünau, M. Richter, A. Klein,
R.K. Thauer, Eur. J. Biochem. 208 (1992) 511.[56] J. Schleucher, C. Griesinger, B. Schwoerer, R.K. Thauer, Biochemistry 33 (1994)
3986.[57] E.J. Lyon, S. Shima, G. Buurman, S. Chowdhuri, A. Batschauer, K. Steinbach,
R.K. Thauer, Eur. J. Biochem. 271 (2004) 195.[58] S. Shima, O. Pilak, S. Vogt, M. Schick, M.S. Stagni, W. Meyer-Klaucke, E.
Warkentin, R.K. Thauer, U. Ermler, Science 321 (2008) 572.[59] T. Hiromoto, K. Ataka, O. Pilak, S. Vogt, M.S. Stagni, W. Meyer-Klaucke, E.
Warkentin, R.K. Thauer, S. Shima, U. Ermler, FEBS Lett. 583 (2009) 585.[60] S. Shima, E.J. Lyon, M. Sordel-Klippert, M. Kauss, J. Kahnt, R.K. Thauer, K.
Steinbach, X. Xie, L. Verdier, C. Griesinger, Angew. Chem., Int. Ed. 43 (2004)2547.
[61] O. Pilak, B. Mamat, S. Vogt, C.H. Hagemeier, R.K. Thauer, S. Shima, C. Vonrhein,E. Warkentin, U. Ermler, J. Mol. Biol. 358 (2006) 798.
[62] E.J. Lyon, S. Shima, R. Boecher, R.K. Thauer, F.-W. Grevels, E. Bill, W. Roseboom,S.P.J. Albracht, J. Am. Chem. Soc. 126 (2004) 14239.
[63] S. Shima, E.J. Lyon, R.K. Thauer, B. Mienert, E. Bill, J. Am. Chem. Soc. 127 (2005)10430.
[64] H. Barker, Antonie van Leeuwenhoek 6 (1940) 201.[65] A. Berkessel, Curr. Opin. Chem. Biol. 5 (2001) 486.[66] A.M. Royer, M. Salomone-Stagni, T.B. Rauchfuss, W. Meyer-Klaucke, J. Am.
Chem. Soc. 132 (2010) 16997.[67] M. Korbas, S. Vogt, W. Meyer-Klaucke, E. Bill, E.J. Lyon, R.K. Thauer, S. Shima,
J. Biol. Chem. 281 (2006) 30804.[68] A. Dey, J. Am. Chem. Soc. 132 (2010) 13892.[69] X. Wang, Z. Li, X. Zeng, Q. Luo, D.J. Evans, C.J. Pickett, X. Liu, Chem. Commun.
(Cambridge, UK) (2008) 3555.[70] J.M. Rawson, R.E.P. Winpenny, Coord. Chem. Rev. 139 (1995) 313.[71] J.A. Wright, P.J. Turrell, C.J. Pickett, Organometallics 29 (2010) 6146.[72] A.M. Royer, T.B. Rauchfuss, S.R. Wilson, Inorg. Chem. (Washington, DC, US) 47
(2008) 395.[73] F.A. Cotton, G. Wilkinson, Advanced Inorganic Chemistry, 5th Edn, Wiley, New
York, 1988.[74] A.R. Sadique, W.W. Brennessel, P.L. Holland, Inorg. Chem. (Washington, DC,
US) 47 (2008) 784.[75] D.Y.C. Melgarejo, G. M., S.A. Koch, in: 234th National Meeting of the American
Chemical Society, American Chemical Society, Washington, DC, Boston, MA,United States, 2007.
[76] Y. Guo, H. Wang, Y. Xiao, S. Vogt, R.K. Thauer, S. Shima, P.I. Volkers, T.B. Rauch-fuss, V. Pelmenschikov, D.A. Case, E.E. Alp, W. Sturhahn, Y. Yoda, S.P. Cramer,Inorg. Chem. (Washington, DC, US) 47 (2008) 3969.
[77] M. Salomone-Stagni, F. Stellato, C.M. Whaley, S. Vogt, S. Morante, S. Shima,T.B. Rauchfuss, W. Meyer-Klaucke, Dalton Trans. 39 (2010) 3057.
[78] B.V. Obrist, D. Chen, A. Ahrens, V. Schunemann, R. Scopelliti, X. Hu, Inorg.Chem. (Washington, DC, US) 48 (2009) 3514.
[79] P. Halder, A. Dey, T.K. Paine, Inorg. Chem. 48 (2009) 11501.[80] W.-F. Liaw, C.-H. Chen, G.-H. Lee, S.-M. Peng, Organometallics 17 (1998) 2370.[81] B. Li, T. Liu, C.V. Popescu, A. Bilko, M.Y. Darensbourg, Inorg. Chem. (Washing-
ton, DC, US) 48 (2009) 11283.[82] W.-F. Liaw, N.-H. Lee, C.-H. Chen, C.-M. Lee, G.-H. Lee, S.-M. Peng, J. Am. Chem.
Soc. 122 (2000) 488.[83] T. Liu, B. Li, C.V. Popescu, A. Bilko, L.M. Perez, M.B. Hall, M.Y. Darensbourg,
Chem. Eur. J. 16 (2010) 3083.[84] A.M. Royer, T.B. Rauchfuss, D.L. Gray, Organometallics 28 (2009) 3618.[85] D. Chen, A. Ahrens-Botzong, V. Schunemann, R. Scopelliti, X. Hu, Inorg. Chem.
(Washington, DC, US) 50 (2010) 5249.[86] P.J. Turrell, J.A. Wright, J.N.T. Peck, V.S. Oganesyan, C.J. Pickett, Angew. Chem.,
Int. Ed. 49 (2010) 7508.[87] D. Chen, R. Scopelliti, X. Hu, Angew. Chem., Int. Ed. 49 (2010) 7512.[88] D. Chen, A. Ahrens-Botzong, V. Schünemann, R. Scopelliti, X. Hu, Inorg. Chem.
50 (2011) 5249.[89] D. Chen, R. Scopelliti, X. Hu, Angew. Chem., Int. Ed. 50 (2011) 5671.

emistr
[108] M.T. Stiebritz, A.R. Finkelmann, M. Reiher, Eur. J. Inorg. Chem. (2010) 1163.
S. Dey et al. / Coordination Ch
[90] D. Chen, R. Scopelliti, X.L. Hu, Angew. Chem., Int. Ed., http://dx.doi.org/10.1002/anie.201107634, in press.
[91] S.P.J. Albracht, in: G. Hauska, R. Thauer (Eds.), The Molecular Basis of BacterialMetabolism, Springer, Berlin, 1990, pp. 40–51.
[92] J. Schleucher, B. Schwoerer, R.K. Thauer, C. Griesinger, J. Am. Chem. Soc. 117(1995) 2941.
[93] B. Schwörer, V.M. Fernandez, C. Zirngibl, R.K. Thauer, Eur. J. Biochem. 212(1993) 255.
[94] A.R. Klein, V.M. Fernández, R.K. Thauer, FEBS Lett. 368 (1995) 203.[95] R. Breslow, J. Am. Chem. Soc. 79 (1957) 1762.
[96] A. Berkessel, R.K. Thauer, Angew. Chem., Int. Ed. 34 (1995) 2247.[97] G.A. Olah, A. Germain, H.C. Lin, D.A. Forsyth, J. Am. Chem. Soc. 97 (1975) 2928.[98] G.A.S.P. Olah, G. K., J. Sommer, Wiley, New York, 1985.[99] G.A.a.K. Olah, D. A., Wiley-Interscience, 2007.[100] A.P. Scott, B.T. Golding, L. Radom, New J. Chem. 22 (1998) 1171.
y Reviews 257 (2013) 42– 63 63
[101] S. Bartoschek, G. Buurman, R.K. Thauer, B.H. Geierstanger, J.P. Weyrauch, C.Griesinger, M. Nilges, M.C. Hutter, V. Helms, ChemBioChem 2 (2001) 530.
[102] J. Cioslowski, G. Boche, Angew. Chem., Int. Ed. 36 (1997) 107.[103] J.H. Teles, S. Brode, A. Berkessel, J. Am. Chem. Soc. 120 (1998) 1345.[104] B.H. Geierstanger, T. Prasch, C. Griesinger, G. Hartmann, G. Buurman, R.K.
Thauer, Angew. Chem., Int. Ed. 37 (1998) 3300.[105] A.R. Klein, G.C. Hartmann, R.K. Thauer, Eur. J. Biochem. 233 (1995) 372.[106] N. Nakatani, Y. Nakao, H. Sato, S. Sakaki, Chem. Lett. 38 (2009) 958.[107] M.T. Stiebritz, M. Reiher, Inorg. Chem. (Washington, DC, US) 49 (2010) 5818.
[109] X. Yang, M.B. Hall, J. Am. Chem. Soc. 130 (2008) 14036.[110] X. Yang, M.B. Hall, J. Am. Chem. Soc. 131 (2009) 10901.[111] T. Hiromoto, E. Warkentin, J. Moll, U. Ermler, S. Shima, Angew. Chem., Int. Ed.
48 (2009) 6457.
Related Documents






![Exploration of H2 binding to the [NiFe]-hydrogenase active ...](https://static.cupdf.com/doc/110x72/61f93edcd3f9ea74a822ec1c/exploration-of-h2-binding-to-the-nife-hydrogenase-active-.jpg)




