RESEARCH ARTICLE Modelling brain dopamine-serotonin vesicular transport disease in Caenorhabditis elegans Alexander T. Young 1 , Kien N. Ly 1, *, Callum Wilson 2 , Klaus Lehnert 1 , Russell G. Snell 1 , Suzanne J. Reid 1 and Jessie C. Jacobsen 1 ABSTRACT Brain dopamine-serotonin vesicular transport disease is a rare disease caused by autosomal recessive mutations in the SLC18A2 gene, which encodes the VMAT2 protein. VMAT2 is a membrane protein responsible for vesicular transport of monoamines, and its disruption negatively affects neurotransmission. This results in a severe neurodevelopmental disorder affecting motor skills and development, and causes muscular hypotonia. The condition was initially described in a consanguineous Saudi Arabian family with affected siblings homozygous for a P387L mutation. We subsequently found a second mutation in a New Zealand family (homozygous P237H), which was later also identified in an Iraqi family. Pramipexole has been shown to have some therapeutic benefit. Transgenic Caenorhabditis elegans were developed to model the P237H and P387L mutations. Investigations into dopamine- and serotonin-related C. elegans phenotypes, including pharyngeal pumping and grazing, showed that both mutations cause significant impairment of these processes when compared with a non-transgenic N2 strain and a transgenic containing the wild-type human SLC18A2 gene. Preliminary experiments investigating the therapeutic effects of serotonin and pramipexole demonstrated that serotonin could successfully restore the pharyngeal pumping phenotype. These analyses provide further support for the role of these mutations in this disease. KEY WORDS: VMAT, Cat-1, Dopamine-serotonin transport disease, Caenorhabditis elegans, Animal model INTRODUCTION Brain dopamine-serotonin vesicular transport disease is a rare neurological disease caused by mutations in the vesicular monoamine transporter 2 (VMAT2; encoded by SLC18A2), an H + ATPase antiporter required for transport of neurotransmitters into presynaptic vesicles. This disease was first reported in 2013, in eight members of a consanguineous Saudi Arabian family who carry an autosomal recessive mutation in SLC18A2 (OMIM: 193001, Refseq NM_003054.4; P387L, c.1160C>T) (Rilstone et al., 2013). Two further cases harbouring homozygous recessive mutations have since been reported in our previously described New Zealand family (P237H, c. 710C>A) (Jacobsen et al., 2016) and an Iraqi family with the same mutation (Rath et al., 2017). The two mutations identified for this disorder are in evolutionarily conserved positions and lie within transmembrane domains 5 and 10 of VMAT2, respectively. Affected individuals display truncal hypotonia and have impaired motor skills. Other symptoms include lack of head control, uncontrolled eye movements, abnormal posturing, tremor, dysdiadochokinesia, dysarthria, dystonia and parkinsonism, as well as oropharyngeal and nasal secretions, extreme sweating, fatigue, poor distal circulation, sleep disruptions and hypernasal speech. However, there is some diversity in the phenotypic spectrum of the disease across the three published cases (Jacobsen et al., 2016; Rath et al., 2017; Rilstone et al., 2013). Remarkably, all three reports describe various degrees of improvement after treatment with the dopamine agonist pramipexole [a dopamine D2/D3 receptor preferring agonist (Kvermo et al., 2006)], including improved parkinsonism, gaining control of eye movement, improved fine motor skills, improved learning ability and gaining the ability to walk (Rath et al., 2017; Rilstone et al., 2013). Interestingly, cases treated at an earlier age responded better to pramipexole; for example, the therapeutic improvement in the New Zealand case (at age 14) was not as remarkable as the Iraqi case (at age 7) despite harbouring the same mutation (Jacobsen et al., 2016). Interestingly, patients treated with the dopamine precursor L-DOPA demonstrated worsening of symptoms (Jacobsen et al., 2016; Rilstone et al., 2013). To investigate the effects of the original P387L mutation, Rilstone et al. expressed wild-type (WT) and mutant VMAT2 in COS-7 cells (Rilstone et al., 2013). The mutant VMAT2 demonstrated reduced serotonin signalling, but glycosylation of the protein was not affected, indicative of normal protein processing. Furthermore, serotonin signalling in mutant VMAT2 was slightly higher than WT VMAT2 inhibited with reserpine (a VMAT2 blocker), suggesting that the mutation does not result in complete loss of function (Rilstone et al., 2013). To expand on the functional work by Rilstone et al. and support the role of the P237H mutation in disease, we developed Caenorhabditis elegans models of brain dopamine-serotonin vesicular transport disease. Caenorhabditis elegans is a nematode that is commonly used as a simple multicellular model of human diseases. Caenorhabditis elegans exist predominantly as female hermaphrodites with very few males, and can develop in 3 days from eggs to adults. Self-fertilising adults can produce about 300 progeny, enabling access to very large populations and making them a simple, cost-effective option for studying the biology of neuronal signalling. Caenorhabditis elegans have a VMAT homologue, cat-1, which has 49% amino acid sequence similarity to human VMAT2, with higher regions of conservation in the transmembrane domains where the mutations reside. cat-1 is located on Received 17 May 2018; Accepted 10 September 2018 1 The School of Biological Sciences and Centre for Brain Research, The University of Auckland, Auckland 1010, New Zealand. 2 Adult and Paediatric National Metabolic Service, Auckland City Hospital, Auckland 1023, New Zealand. *Author for correspondence ([email protected]) K.N.L., 0000-0003-0741-6001; C.W., 0000-0003-2034-767X; K.L., 0000-0003- 4598-4966; R.G.S., 0000-0002-8166-4014; S.J.R., 0000-0002-8068-6529 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed. 1 © 2018. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2018) 11, dmm035709. doi:10.1242/dmm.035709 Disease Models & Mechanisms

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

RESEARCH ARTICLE
Modelling brain dopamine-serotonin vesicular transport diseasein Caenorhabditis elegansAlexander T. Young1, Kien N. Ly1,*, Callum Wilson2, Klaus Lehnert1, Russell G. Snell1, Suzanne J. Reid1 andJessie C. Jacobsen1
ABSTRACTBrain dopamine-serotonin vesicular transport disease is a raredisease caused by autosomal recessive mutations in the SLC18A2gene, which encodes the VMAT2 protein. VMAT2 is a membraneprotein responsible for vesicular transport of monoamines, and itsdisruption negatively affects neurotransmission. This results in asevere neurodevelopmental disorder affecting motor skills anddevelopment, and causes muscular hypotonia. The condition wasinitially described in a consanguineous Saudi Arabian family withaffected siblings homozygous for a P387L mutation. Wesubsequently found a second mutation in a New Zealand family(homozygous P237H), which was later also identified in an Iraqifamily. Pramipexole has been shown to have some therapeuticbenefit. Transgenic Caenorhabditis elegans were developed tomodel the P237H and P387L mutations. Investigations intodopamine- and serotonin-related C. elegans phenotypes, includingpharyngeal pumping and grazing, showed that both mutations causesignificant impairment of these processes when compared with anon-transgenic N2 strain and a transgenic containing the wild-typehuman SLC18A2 gene. Preliminary experiments investigating thetherapeutic effects of serotonin and pramipexole demonstrated thatserotonin could successfully restore the pharyngeal pumpingphenotype. These analyses provide further support for the role ofthese mutations in this disease.
KEY WORDS: VMAT, Cat-1, Dopamine-serotonin transport disease,Caenorhabditis elegans, Animal model
INTRODUCTIONBrain dopamine-serotonin vesicular transport disease is a rareneurological disease caused by mutations in the vesicularmonoamine transporter 2 (VMAT2; encoded by SLC18A2), an H+
ATPase antiporter required for transport of neurotransmitters intopresynaptic vesicles. This disease was first reported in 2013, in eightmembers of a consanguineous Saudi Arabian family who carry anautosomal recessive mutation in SLC18A2 (OMIM: 193001, RefseqNM_003054.4; P387L, c.1160C>T) (Rilstone et al., 2013).Two further cases harbouring homozygous recessive mutations
have since been reported in our previously described New Zealandfamily (P237H, c. 710C>A) (Jacobsen et al., 2016) and an Iraqifamily with the same mutation (Rath et al., 2017). The twomutations identified for this disorder are in evolutionarily conservedpositions and lie within transmembrane domains 5 and 10 ofVMAT2, respectively.
Affected individuals display truncal hypotonia and have impairedmotor skills. Other symptoms include lack of head control,uncontrolled eye movements, abnormal posturing, tremor,dysdiadochokinesia, dysarthria, dystonia and parkinsonism, as wellas oropharyngeal and nasal secretions, extreme sweating, fatigue,poor distal circulation, sleep disruptions and hypernasal speech.However, there is some diversity in the phenotypic spectrum ofthe disease across the three published cases (Jacobsen et al., 2016;Rath et al., 2017; Rilstone et al., 2013). Remarkably, all threereports describe various degrees of improvement after treatment withthe dopamine agonist pramipexole [a dopamine D2/D3 receptorpreferring agonist (Kvermo et al., 2006)], including improvedparkinsonism, gaining control of eye movement, improved finemotor skills, improved learning ability and gaining the ability to walk(Rath et al., 2017; Rilstone et al., 2013). Interestingly, cases treated atan earlier age responded better to pramipexole; for example, thetherapeutic improvement in the New Zealand case (at age 14) was notas remarkable as the Iraqi case (at age 7) despite harbouring the samemutation (Jacobsen et al., 2016). Interestingly, patients treated withthe dopamine precursor L-DOPA demonstrated worsening ofsymptoms (Jacobsen et al., 2016; Rilstone et al., 2013).
To investigate the effects of the original P387L mutation,Rilstone et al. expressed wild-type (WT) and mutant VMAT2 inCOS-7 cells (Rilstone et al., 2013). The mutant VMAT2demonstrated reduced serotonin signalling, but glycosylation ofthe protein was not affected, indicative of normal proteinprocessing. Furthermore, serotonin signalling in mutant VMAT2was slightly higher than WT VMAT2 inhibited with reserpine(a VMAT2 blocker), suggesting that the mutation does not result incomplete loss of function (Rilstone et al., 2013).
To expand on the functional work by Rilstone et al. and supportthe role of the P237H mutation in disease, we developedCaenorhabditis elegans models of brain dopamine-serotoninvesicular transport disease. Caenorhabditis elegans is a nematodethat is commonly used as a simple multicellular model of humandiseases. Caenorhabditis elegans exist predominantly as femalehermaphrodites with very few males, and can develop in 3 daysfrom eggs to adults. Self-fertilising adults can produce about 300progeny, enabling access to very large populations andmaking thema simple, cost-effective option for studying the biology of neuronalsignalling. Caenorhabditis elegans have a VMAT homologue,cat-1, which has 49% amino acid sequence similarity to humanVMAT2, with higher regions of conservation in the transmembranedomains where the mutations reside. cat-1 is located onReceived 17 May 2018; Accepted 10 September 2018
1The School of Biological Sciences and Centre for Brain Research, The Universityof Auckland, Auckland 1010, New Zealand. 2Adult and Paediatric NationalMetabolic Service, Auckland City Hospital, Auckland 1023, New Zealand.
*Author for correspondence ([email protected])
K.N.L., 0000-0003-0741-6001; C.W., 0000-0003-2034-767X; K.L., 0000-0003-4598-4966; R.G.S., 0000-0002-8166-4014; S.J.R., 0000-0002-8068-6529
This is an Open Access article distributed under the terms of the Creative Commons AttributionLicense (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use,distribution and reproduction in any medium provided that the original work is properly attributed.
1
© 2018. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2018) 11, dmm035709. doi:10.1242/dmm.035709
Disea
seModels&Mechan
isms

the X chromosome, and is expressed in all dopamine- andserotonin-containing cells in C. elegans. Like human VMAT2,C. elegans VMAT is responsible for the transport of dopamine andserotonin into synaptic vesicles (Duerr et al., 1999). Caenorhabditiselegans lacking VMAT are viable and grow well, but are deficientfor dopamine- and serotonin-associated behaviours, suggesting thatneurotransmitter function is dependent on synaptic vesicle loading(Duerr et al., 1999). We subsequently generated three transgenic
lines – cat-1(−)-huVMAT2(+), cat-1(−)-huVMAT2(P387L) andcat-1(−)-huVMAT2(P237H) – to assess the impact of thesemutations on VMAT2 function.
RESULTSTransgenic strain developmentThree transgenic C. elegans lines were successfully generated usingthe MosSCI method: cat-1(−)-huVMAT2(+), which is homozygous
Fig. 1. Confirmation of transgenics. (A) Sanger traces confirming that the mutations are present in the transgenes integrated into the C. elegans strains.Mutation sites are indicated by red boxes. (B) Confirmation of gene expression by RT-PCR (primers seqhVMATf1 and seqhVMATr1). RT negative controlscontained no reverse transcriptase. P237H (NZ), P387L (SA),WT. (C) Prior to Sanger sequencing,C. eleganswere genotyped by PCR. EachC. elegans required2 wells for assessment: the first well contains a positive control (SSU18A+SSU26R ∼950 bp) and a test for the transgene (primers unc5402 and seqSNB01,2657 bp). Primers NM3884 and NM3880, which flank the integration site, were used to determine insertion of the construct. The second well for each sample onlyshow a product (2945 bp) if one chromosome does not have the construct inserted, and no band if the C. elegans is homozygous. The top series of bands are allfrom C. elegans of a single parent, deemed heterozygous, as all possible genotype combinations are represented. The lower bands are from a homozygousparent, as all C. elegans show presence of the transgene, and a copy on each chromosome. Primer sequences are available in Table S3. TG, transgene.
2
RESEARCH ARTICLE Disease Models & Mechanisms (2018) 11, dmm035709. doi:10.1242/dmm.035709
Disea
seModels&Mechan
isms
for the WT human SLC18A2 gene; cat-1(−)-huVMAT2(P237H),carrying the P237H (c.710C>A) human mutation; and cat-1(−)-huVMAT2(P387L), carrying the P387L (c.1160C>T) humanmutation. The genotype was confirmed using polymerase chainreaction (PCR; Fig. 1C), and the transgenic status of the final strainswas confirmed by Sanger sequencing (Fig. 1A). Successfultranscription of the transgene was verified using reversetranscription PCR (RT-PCR; Fig. 1B). The crossing method onlyselected for cat-1 knockouts, which was confirmed by Sangersequencing (Fig. S2).The transgenic C. elegans lines were subsequently assessed for
dopamine- and serotonin-dependent behaviours.
PhenotypingGrazingGrazing behaviour is a well-established phenotype in C. elegans,and is known to be dependent on both dopamine and serotonin
(Chase and Koelle, 2007). It was therefore used to assess the effectof both mutations on dopamine and serotonin signalling.
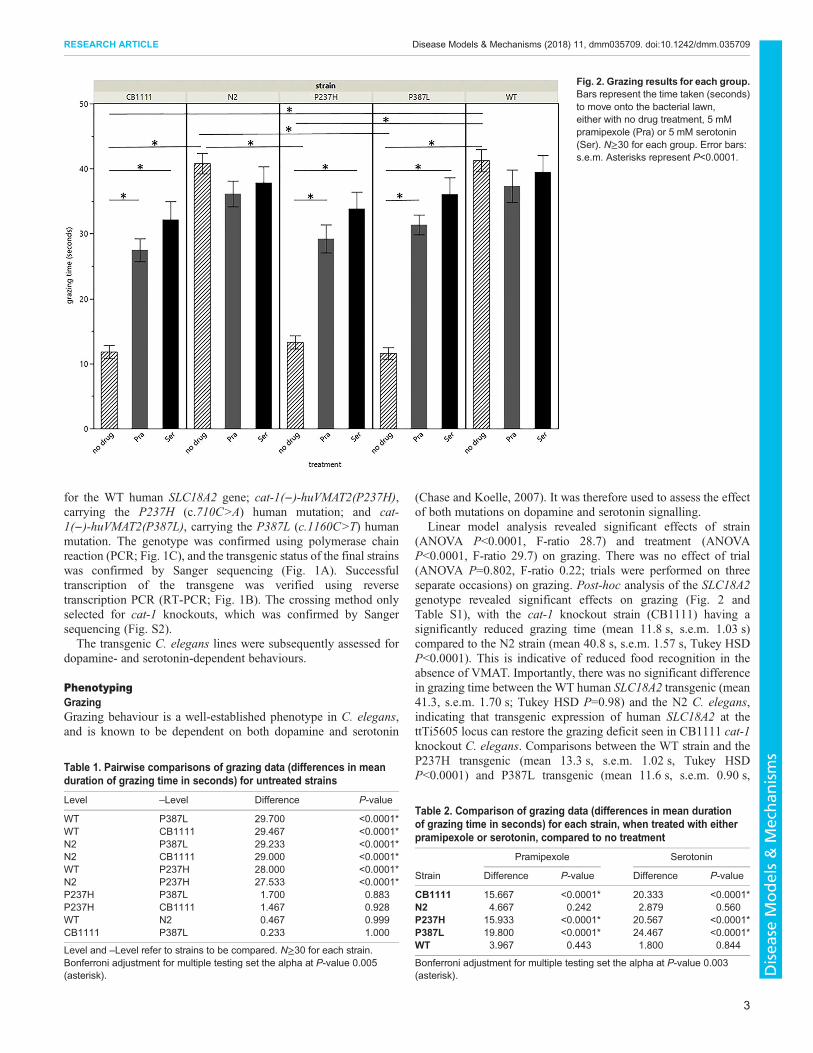
Linear model analysis revealed significant effects of strain(ANOVA P<0.0001, F-ratio 28.7) and treatment (ANOVAP<0.0001, F-ratio 29.7) on grazing. There was no effect of trial(ANOVA P=0.802, F-ratio 0.22; trials were performed on threeseparate occasions) on grazing. Post-hoc analysis of the SLC18A2genotype revealed significant effects on grazing (Fig. 2 andTable S1), with the cat-1 knockout strain (CB1111) having asignificantly reduced grazing time (mean 11.8 s, s.e.m. 1.03 s)compared to the N2 strain (mean 40.8 s, s.e.m. 1.57 s, Tukey HSDP<0.0001). This is indicative of reduced food recognition in theabsence of VMAT. Importantly, there was no significant differencein grazing time between the WT human SLC18A2 transgenic (mean41.3, s.e.m. 1.70 s; Tukey HSD P=0.98) and the N2 C. elegans,indicating that transgenic expression of human SLC18A2 at thettTi5605 locus can restore the grazing deficit seen in CB1111 cat-1knockout C. elegans. Comparisons between the WT strain and theP237H transgenic (mean 13.3 s, s.e.m. 1.02 s, Tukey HSDP<0.0001) and P387L transgenic (mean 11.6 s, s.e.m. 0.90 s,
Fig. 2. Grazing results for each group.Bars represent the time taken (seconds)to move onto the bacterial lawn,either with no drug treatment, 5 mMpramipexole (Pra) or 5 mM serotonin(Ser). N≥30 for each group. Error bars:s.e.m. Asterisks represent P<0.0001.
Table 1. Pairwise comparisons of grazing data (differences in meanduration of grazing time in seconds) for untreated strains
Level –Level Difference P-value
WT P387L 29.700 <0.0001*WT CB1111 29.467 <0.0001*N2 P387L 29.233 <0.0001*N2 CB1111 29.000 <0.0001*WT P237H 28.000 <0.0001*N2 P237H 27.533 <0.0001*P237H P387L 1.700 0.883P237H CB1111 1.467 0.928WT N2 0.467 0.999CB1111 P387L 0.233 1.000
Level and –Level refer to strains to be compared. N≥30 for each strain.Bonferroni adjustment for multiple testing set the alpha at P-value 0.005(asterisk).
Table 2. Comparison of grazing data (differences in mean durationof grazing time in seconds) for each strain, when treated with eitherpramipexole or serotonin, compared to no treatment
Strain
Pramipexole Serotonin
Difference P-value Difference P-value
CB1111 15.667 <0.0001* 20.333 <0.0001*N2 4.667 0.242 2.879 0.560P237H 15.933 <0.0001* 20.567 <0.0001*P387L 19.800 <0.0001* 24.467 <0.0001*WT 3.967 0.443 1.800 0.844
Bonferroni adjustment for multiple testing set the alpha at P-value 0.003(asterisk).
3
RESEARCH ARTICLE Disease Models & Mechanisms (2018) 11, dmm035709. doi:10.1242/dmm.035709
Disea
seModels&Mechan
isms

Tukey HSD P<0.0001) demonstrated that both mutations causesignificant grazing impairment, reducing it to levels comparablewith the cat-1 knockout. Pairwise comparisons between untreatedstrains are shown in Table 1.To provide some preliminary insight into the effects of
exogenous serotonin and pramipexole on C. elegans VMATfunction, the grazing phenotype was evaluated in the presence ofeither 5 mM serotonin or 5 mM pramipexole. Treatment with eithercompound significantly improved grazing in the P237H, P387L andCB1111 strains (Tukey HSD P<0.0001 for each treatment acrossall three strains, Table 2). While serotonin consistently showed agreater effect than pramipexole (Fig. 2), this difference was notsignificant (Tukey HSD P>0.1) for any strain. There was nosignificant treatment effect for either drug on either of thecontrol strains.
Pharyngeal pumpingPharyngeal pumping in C. elegans is another well-definedphenotype, dependent on serotonin signalling, and was used toinvestigate serotonin signalling in these models (Fig. 3 andTable S2).Linear model analysis revealed significant effects of strain
(ANOVA P<0.0001, F-ratio 132.2) and treatment (ANOVAP<0.0001, F-ratio 26.1) on pharyngeal pumping. There was noeffect of trial (ANOVA P=0.757, F-ratio 0.27; trials were performedon three separate occasions) on grazing. Post-hoc analysis of theSLC18A2 genotype revealed that all pairwise comparisons betweenstrains were significantly altered (Tukey HSD P<0.005, Table 3).N2 had the greatest number of pumps per minute (mean 269,s.e.m. 2.33), and the CB1111 strain had the least (mean 177,
s.e.m. 3.04). Transgenic expression of WT human SLC18A2(mean 240, s.e.m. 2.34) partially restored the deficit seen inCB1111 (Tukey HSD P<0.0001), but the value remainedsignificantly lower (P<0.0001) than levels observed in the N2strain. The mutant VMAT2 variants also displayed significantlymore pumps (P237H mean 198, s.e.m. 3.2; P387L mean 209,s.e.m. 2.48), when compared to the CB1111 knockout strain(Tukey HSD P<0.0001 for both comparisons, Table 3). However,pumping in both mutation-carrying strains was significantly lessthan that observed in the WT C. elegans, where 69% of thedifference between CB1111 and N2 levels was restored (TukeyHSD P<0.0001), while only a 23.1% and 34.4% rescue wasachieved for the P237H and P387L strains, respectively.
When the pharyngeal pumping phenotype was re-assessed inthe presence of either 5 mM pramipexole or serotonin (Table 4),we found that serotonin restored the mean number of pumps perminute in the CB1111 (mean 238, s.e.m. 3.4), P387L (mean 234,s.e.m. 3.01) and P237H (mean 233, s.e.m. 3.03) strains (P<0.0001for all strains when compared to no drug treatment) to levels closeto those seen in the WT transgenic (with or without serotonin)(Tukey HSD P<0.003). In addition, a more rhythmic, consistentpumping action (when compared to the untreated knockout ormutation-carrying strains) was observed with serotonin. This wasnot quantified, and requires further validation. Only the P387Lstrain significantly increased in pumping rate after additionof pramipexole (Tukey HSD P=0.0027), demonstrating adifferentiated effect from the P237H mutation (Tukey HSDP=0.4712). Interestingly, pumping rate was adversely affectedfollowing treatment with both pramipexole and serotonin in the N2strain (Table 4).
Fig. 3. Pharyngeal pumping resultsfrom each group. N≥90 for each testwithout drugs. N=30 for each drugtreatment group. Bars represent theaverage number of pharyngeal pumps,either with no drug treatment, 5 mMpramipexole (pra) or 5 mM serotonin(ser). Error bars: s.e.m. Asterisksrepresent significant differences(P<0.0001). For simplicity, significancefor all untreated strain comparisonsare not shown; all were significantlydifferent (Tukey HSD P-value lessthan 0.003).
4
RESEARCH ARTICLE Disease Models & Mechanisms (2018) 11, dmm035709. doi:10.1242/dmm.035709
Disea
seModels&Mechan
isms

DISCUSSIONWe have utilised C. elegans as a model to validate the role of twoknown SLC18A2mutations (P237H and P387L) in brain dopamine-serotonin vesicular transport disease. While previous functionalinvestigations of the P387L mutation revealed a severe reduction,but not complete loss, in VMAT2 transport ability (Rilstone et al.,2013), there has been no research into the consequence of theP237H mutation that has been observed in both the New Zealandand Iraqi families. We present here the first animal models of thedisease. Three full-length cDNA transgenic models were developed(one for each mutation and WT human SLC18A2 gene), and theimpact of both mutations on monoamine neurotransmitter transportwas assessed and compared to a previously established C. eleganshomologue (cat-1) knockout model and a WT N2 strain.It is known that VMAT2 plays an important role in dopamine and
serotonin signalling; therefore, phenotypes dependent on theseneurotransmitters [grazing and pharyngeal pumping (Loer andKenyon, 1993; Sawin et al., 2000)] were evaluated in the models toprovide insight into the relative function of the mutations. Resultsfrom both assays demonstrated a significant impairment in bothstrains carrying the human mutations, which was comparable to thatof the cat-1 knockout (CB1111). Interestingly, comparisons of thephenotypic function of the strains carrying the P237H and P387Lvariants suggest that the P237H mutation may have a moresignificant impact on protein function, which aligns with thecomparative phenotypic description of the cases (Jacobsen et al.,2016; Rath et al., 2017; Rilstone et al., 2013). Further functionalwork assessing the impact of these mutations on protein function isrequired to confirm their relative effects.The grazing assay, designed to assess food recognition by
measuring the time taken for the length of the C. elegans to moveonto the bacterial lawn (and indicative of dopamine and serotonin
signalling), demonstrated a deficit in the ability to recognise food inthe P237H (mean 13.3 s) and P387L (mean 11.6 s) C. elegansstrains, despite theWT SLC18A2 transgenic displaying normal foodrecognition (mean 41.3 s).
The pharyngeal pumping experiments revealed the meanpumping rate of the P237H and P387L strain to be 32% and48% of the WT strain, respectively. It is important to consider that,although the CB1111 strain has no functional VMAT gene, thepharynx in these animals still pumps, suggesting that this is notentirely dependent on VMAT pathways. Cell culture experimentsby Rilstone et al. evaluating serotonin uptake of vesiclescontaining mutant VMAT2 (P387L mutation) showed that thelevel of uptake was less than 5 pmol/mg over 10 min, compared toan uptake level of 25∼30 pmol/mg in WT VMAT2-containingvesicles (Rilstone et al., 2013). This demonstrated the level ofneurotransmitter transport in the P387L mutant protein to beapproximately 16-20% of normal function (compared to the 48%rescue of the pharyngeal pumping phenotype in the assaypresented here). Utilising both data sets, one may postulate thata small percent of transport can provide a relatively greater level ofrescue (i.e. 20% of normal transport results in a 48% rescue ofphenotype). However, any conclusions from the phenotypingassays should only point to relative and not absolute effectson neurotransmitter transport. The data from the pharyngealpumping assays presented here provide evidence that bothmutations result in a very low level of protein function whencompared to theWTVMAT2 protein, which is consistent with thatreported by Rilstone et al.
Interestingly, grazing and phenotypic pumping were not fullyrestored to levels seen in N2 C. elegans, despite showing significantimprovement relative to the CB1111 knockout strain. This isperhaps not unexpected, given that the proteins are only 49% similar(Duerr et al., 1999). It would be interesting to assess the effects ofthe mutations at the equivalent positions in cat-1.
Preliminary analysis of the therapeutic benefits of serotonin andpramipexole were assessed in these models using the establishedgrazing and pharyngeal pumping phenotypes. Serotoninadministration was successful in restoring pumping rates inthe P237H, P387L and CB1111 strains, improving both thefrequency (number per minute) and function (rhythmic cadence).Furthermore, serotonin administration successfully restored grazingdeficits seen in the P237H, P387L and CB1111 strains. Previouswork has identified successful recovery of serotonin signalling inCB1111 C. elegans (Loer and Kenyon, 1993), supporting thisobservation. The addition of pramipexole appeared to partiallyrestore the pharyngeal pumping phenotype observed in the P387LC. elegans but not in the P237H or CB1111 strains, reflecting adifference in mutant protein function and suggesting thattherapeutic success may be dependent on the location of themutation. Additionally, the addition of pramipexole successfullyrestored grazing deficits in the P237H, P387L and CB1111 strains,
Table 3. Pairwise Tukey HSD tests of pharyngeal pumping data(differences in mean pumping rate per minute) for untreated strains
Level –Level Difference P-value
N2 CB1111 69.153 <0.0001*N2 P237H 53.293 <0.0001*WT CB1111 49.867 <0.0001*N2 P387L 41.332 <0.0001*WT P237H 34.007 <0.0001*P387L CB1111 27.821 <0.0001*WT P387L 22.046 <0.0001*N2 WT 19.287 <0.0001*P237H CB1111 15.860 <0.0001*P387L P237H 11.961 0.002*
Level and –Level refer to strains to be compared. N≥30 for each strain.Bonferroni adjustment for multiple testing set the alpha at P-value 0.005(asterisk).
Table 4. Comparison of pharyngeal pumping data (differences in meanpumping rate) for each strain, when treated with either pramipexoleor serotonin, compared to no treatment
Strain
Pramipexole Serotonin
Difference P-value Difference P-value
CB1111 5.600 0.588 61.800 <0.0001*N2 38.444 <0.0001* 22.811 <0.0001*P237H 6.800 0.471 35.467 <0.0001*P387L 17.158 0.003* 25.558 <0.0001*WT 12.333 0.024 0.6333 0.990
Bonferroni adjustment for multiple testing set the alpha at P-value 0.003(asterisk).
Table 5. Caenorhabditis elegans strains used for phenotypingexperiments
Strain name Genotype
P237H cat-1(−)-huVMAT2(P237H)P387L cat-1(−)-huVMAT2(P387L)WT cat-1(−)-huVMAT2(+)N2 Bristol Wild typeCB1111 (Duerr et al., 1999) cat-1(−)
5
RESEARCH ARTICLE Disease Models & Mechanisms (2018) 11, dmm035709. doi:10.1242/dmm.035709
Disea
seModels&Mechan
isms

providing evidence that pramipexole can rescue dopamine-dependent phenotypes in the C. elegans models.Interestingly, the N2 strain displayed hyperactive movement
when treated with either serotonin or pramipexole (fast backwardmovement, rapid twitches and increases in sporadic pumping), andwere never observed to be stationary and eating. In comparison, theWT strain exhibited no adverse behaviours following drugtreatment. Therefore, the improved pumping rate observed in thetransgenic animals may not be caused directly by pramipexole, butindirectly via mechanisms that impact the grazing ability ofC. elegans. Excess pramipexole may also result in over-stimulation of the C. elegans. Further experiments are required todetermine dose effects of both serotonin and pramipexole acrossthese strains, as well as the basal levels of these neurotransmittersand the affinity of C. elegans receptors for pramipexole.A significant consideration from the case reports is that the
response to treatment appears to have a lower impact in olderchildren, as demonstrated by the greater improvement in youngerpatients following pramipexole treatment (Rilstone et al., 2013).Given that C. elegans has a life span of only a few weeks, it is hardto assess age-related effects of drug treatment and follow-up ina longer-living organism will be required. Interestingly,administration of L-DOPA to both cohorts of patients causedworsening of symptoms (Jacobsen et al., 2016; Rilstone et al.,2013). This may be due to L-DOPA-induced cytotoxicity, orapoptosis-induced effects of increased signalling on dopaminergicneurons (Jeon et al., 2010). It would be interesting to assess theeffects of L-DOPA in these models.Both the case reports and the C. elegans transgenics described
here demonstrate that bypassing the need for vesiculartransportation and directly targeting neurotransmitter receptorssuccessfully alleviates some of the symptoms caused by thedisease. Considering the potential therapeutic impacts forchildren harbouring mutations in the SLC18A2 gene, the
importance of an early diagnosis to optimise treatment,prevent cell damage or death and increase quality of lifeis clear. Based on these C. elegans studies, it is possiblethat dual administration of pramipexole and serotonin (as5-hydroxytryptophan) may be of benefit.
ConclusionThe C. elegans models described here are the first animal modelsdeveloped for brain dopamine-serotonin vesicular transport disease,and provide insight into the relative biological impact of thesemutations, which may ultimately help guide management andtreatment options for this disease.
MATERIALS AND METHODSCaenorhabditis elegans maintenanceSynchronised C. elegans populations were maintained on NematodeGrowth Medium (NGM) on 35×10 mm agar Petri dishes in 15 or 20°Cincubators, and were inspected daily to ensure that there was noovercrowding or contamination. Escherichia coli HB101 was used as afood source.
Development of transgenic linesTransgenics were generated following the MosSCI method using theC. elegans strain EG6699 (locus ttTi5605, chromosome 2: 0.78, genomicposition: II:8420158..8420158) (Frøkjaer-Jensen et al., 2012, 2014). Theplasmid consisted of a Mos1 transposon containing SLC18A2 (full-lengthhuman cDNA) under a synaptobrevin (snb-1) promoter (expressed inC. elegans neurons and localised to synaptic regions, and used in the cat-1knockout transgenic generated by Duerr et al., 1999), unc-119(+) andhomologous sites (L and R) as shown in Fig. 4. Vectors were alsoconstructed carrying both the P237H and P387L mutations within thehuman cDNA (Table 5). It is unclear whether all C. elegans neurons expresscat-1; therefore, there could be a potential confounding effect if the humanprotein is expressed in neurons in which endogenous cat-1 is not expressedin the normal physiological state.
Microinjection was performed using an Injectman NI 2 micromanipulator(Eppendorf ) attached to a Leica inverted compound microscope.Caenorhabditis elegans were immobilised to slides using a 2% agarosesolution. The plasmid DNA was purified using an Axygen maxiprep kit(50 ng/μl) and injected with pCFJ601 (100 ng/μl, transposase), pAM122(10 ng/μl, peel-1 toxin), pGH8 (10 ng/μl, mCherry in neurons), pCFJ90(2.5 ng/μl, mCherry in the pharynx) and pCFJ104 (5 ng/μl, mCherry in thebody muscle). The injection cocktail was delivered at approximately 1000psi until the mixture appeared to fill the gonad of the C. elegans.
Following microinjection, C. elegans were placed in recovery buffer(66 mM NaCl, 5 mM HEPES pH 7.2, 3 mM CaCl2, 3 mMMgCl2, 2.4 mMKCl and 4% glucose), and transferred to a seeded NGM plate and incubatedat 25°C. After approximately 10 days, the plates containing the injectedC. elegans were transferred to a 34°C incubator for 2 h, to activateexpression of the toxic peel-1 gene.
Caenorhabditis elegans that survived the heat-shock treatment anddisplayed normal movement (successful incorporation of unc-119) wereexamined under a fluorescence microscope to identify any still carrying theextrachromosomal mCherry coding plasmids. Caenorhabditis elegans
Table 6. Primer pairs used to confirm successful integration of theplasmid into chromosome 2 of EG6699 C. elegans
Forwardprimer
Reverseprimer
Product size(base pairs) Targeted region
unc5401 NM3880 3809 Across the left integrationsite (left)
unc5401 NM3887 2836 Inside the left integrationsite (left inner)
unc5402 seqSNB01 2657 Flanking the transgene(middle)
pSNB01 NM3888 2312 Inside the rightintegration site (rightinner)
pSNB01 NM3884 3601 Across the rightintegration site (right)
Primers mapped to the plasmid are available in Fig. S1.
Fig. 4. Linear schematic showing the PCR design to verify transgenic integration in C. elegans chromosome 2. PCR primer pairs used: left(unc5401+NM3880), left inner (unc5401+NM3887), middle (unc5402+seqSNB01), right inner (pSNB01+NM3888) and right (pSNB01+NM3884). R and Lare homologous sites used for insertion into site ttTi5605 on chromosome 2. For a full plasmid map showing genes and primer locations, see Fig. S1.
6
RESEARCH ARTICLE Disease Models & Mechanisms (2018) 11, dmm035709. doi:10.1242/dmm.035709
Disea
seModels&Mechan
isms

showing no signs of mCherry fluorescence were transferred to fresh NGMplates, as they had evidently lost the extrachromosomal arrays and thedesired chromosomal integration had likely occurred. The selected C.elegans were propagated for several generations and bred to homozygosity(demonstrated by loss of the uncoordinated phenotype due to rescue of unc-119), and confirmed by DNA analysis using PCR to identify the presence ofthe transgene using primer pairs specified in Table 6 (primer sequencesavailable in Table S3).
The three resulting strains containing the transgene [huVMAT2(+),huVMAT2(P387L) and huVMAT2(P237H)] were crossbred with theCB1111 cat-1 knockout strain to establish lines homozygous for thetransgenes. These final strains are designated as WT, P387L and P237H(Table 5). Homozygosity was confirmed by PCR and Sanger sequencing.The males used for crossing were created by exposure to ethanol, aspreviously described (Lyons and Hecht, 1997). Primers used for allgenotyping assays are described in Table 6, Table S3 and graphicallyillustrated in Fig. 4.
The N2 and CB1111 strains were provided by the CaenorhabditisGenetics Center (CGC), which is funded by NIH Office of ResearchInfrastructure Programs (P40 OD010440).
Reverse transcriptionRNA was extracted using a previously published single-worm method (Lyet al., 2015), and RT-PCR was used to determine successful transcription ofthe transgenes using the Maxima Reverse Transcription kit (ThermoFisherScientific).
Phenotyping assaysSynchronised adult C. elegans were used for all experiments, and all strainswere assessed in parallel across 3 days. Synchronised populations wereachieved by placing adults on plates for several hours to lay eggs. Onceseveral eggs had been laid, the adults were removed. The C. elegans werehoused at 20°C prior to phenotyping, and assessed in a temperature-controlled room also at 20°C. Bacterial lawn thickness and dryness of theagar was visually inspected in all assays to ensure consistency. Preliminaryphenotyping involved assessing pharyngeal pumping for serotoninsignalling, and grazing behaviour for dopamine and serotonin signalling,as these phenotypes have been shown to be affected by theseneurotransmitters (Chase and Koelle, 2007).
Grazing activity was measured by assessing the behaviour of C. eleganssearching for food (bacterial lawn). To perform this assay, groups of C.elegans were transferred to a fresh agar plate containing many smallbacterial lawns (<2 μl). After encountering the bacterial lawn, the time takenfor a C. elegans to move fully onto the bacterial lawn (1 body length) wasrecorded. To control for increased movement due to transfer ofC. elegans toa new plate, any recordings of individuals encountering the lawn within 1min of plate transfer were excluded. This is a similar method to that appliedby Duerr et al. in their investigation of monoamine-dependent behaviours(Duerr et al., 1999). To measure pharyngeal pumping rate, C. elegans weretransferred to an NGM plate with a bacterial lawn and left for 10 min toacclimate. The rate was determined by focusing on the pharyngeal muscle inindividual worms under a dissecting microscope and counting eachindividual pump over a 60 s period, and tracking the C. elegans as itmoved freely about the plate, as described previously (Raizen et al., 2012).
Drug treatmentTo assess treatment responses, serotonin (Sigma) or pramipexole (Sigma)were spread on top of agarose plates to a final concentration of 5 mM andgiven time to be absorbed. As soon as the solutions were absorbed into theagar (determined by visual inspection), the phenotyping assays wereconducted.
Statistical analysisStatistical analysis was performed using the JMP statistical package (JMP®
11.2.0), in which the individual worms within each trial were treated as
biological replicates. The effect of strain, treatment and trial were assessedusing an ANOVA model because data from grazing and pharyngealpumping trials approximated a normal distribution (determined by visualinspection and Shapiro-Wilks tests). Subsequently, post-hoc (all pairs TukeyHSD) analysis were performed and Bonferroni adjustment for multipletesting was made.
AcknowledgementsWe would like to thank Kristine Boxen at the Auckland Science Analytical Servicesfor Sanger sequencing services.
Competing interestsThe authors declare no competing or financial interests.
Author contributionsConceptualization: K.L., R.G.S., S.J.R., J.C.J.; Methodology: A.T.Y., K.N.L.,S.J.R.; Validation: K.L., S.J.R., J.C.J.; Formal analysis: A.T.Y., K.N.L., S.J.R.;Resources: C.W., J.C.J.; Writing - original draft: A.T.Y., K.N.L.; Writing - review &editing: A.T.Y., K.N.L., C.W., K.L., R.G.S., S.J.R., J.C.J.; Supervision: S.J.R.,J.C.J.; Project administration: S.J.R., J.C.J.; Funding acquisition: J.C.J.
FundingJ.C.J. is supported by a Rutherford Discovery Fellowship from the New ZealandGovernment and administered by the Royal Society of New Zealand. The researchwas also funded by the School of Biological Sciences (The University of Auckland).
Supplementary informationSupplementary information available online athttp://dmm.biologists.org/lookup/doi/10.1242/dmm.035709.supplemental
ReferencesChase, D. L. and Koelle, M. R. (2007). Biogenic amine neurotransmitters in
C. elegans. WormBook 1-15.Duerr, J. S., Frisby, D. L., Gaskin, J., Duke, A., Asermely, K., Huddleston, D.,
Eiden, L. E. and Rand, J. B. (1999). The cat-1 gene of Caenorhabditis elegansencodes a vesicular monoamine transporter required for specific monoaminedependent behaviours. J. Neurosci. 19, 72-84.
Frøkjaer-Jensen, C., Davis, M. W., Ailion, M. and Jorgensen, E. M.(2012). Improved Mos1-mediated transgenesis in C. elegans. Nat. Methods9, 117-118.
Frøkjaer-Jensen, C., Davis, M. W., Sarov, M., Taylor, J., Flibotte, S., LaBella, M.,Pozniakovsky, A., Moerman, D. G. and Jorgensen, E. M. (2014). Random andtargeted transgene insertion in Caenorhabditis elegans using a modified Mos1transposon. Nat. Methods 11, 529-534.
Jacobsen, J. C., Wilson, C., Cunningham, V., Glamuzina, E., Prosser, D. O.,Love, D. R., Burgess, T., Taylor, J., Swan, B., Hill, R. et al. (2016). Braindopamine-serotonin vesicular transport disease presenting as a severe infantilehypotonic parkinsonian disorder. J. Inherit. Metab. Dis. 39, 305-308.
Jeon, S.-M., Cheon, S.-M., Bae, H.-R., Kim, J. W. and Kim, S. U. (2010). Selectivesusceptibility of human dopaminergic neural stem cells to dopamine-inducedapoptosis. Exp. Neurobiol. 19, 155-164.
Kvermo, T., Hartter, S. and Burger, E. (2006). A review of the receptor-binding andpharmacokinetic properties of dopamine agonists. Clin. Ther. 28, 1065-1078.
Loer, M. C. and Kenyon, C. J. (1993). Serotonin-deficient mutants and malemating behavior in the nematode Caenorhabditis elegans. J. Neurosci. 13,5407-5417.
Ly, K., Reid, S. J. and Snell, R. G. (2015). Rapid RNA analysis of individualCaenorhabditis elegans. Methods X 2, 59-63.
Lyons, L. C. and Hecht, R. M. (1997). Acute ethanol exposure inducesnondisjunction of the X chromosome during spermatogenesis. Worm BreedersGazette 14, 52.
Raizen, D., Song, B. M., Trojanowski, N. and You, Y. J. (2012). Methods formeasuring pharyngeal behaviors. WormBook 1-13.
Rath, M., Korenke, G. C., Najm, J., Hoffman, G. F., Hagendorff, A., Strom, T. M.and Felbor, U. (2017). Exome sequencing results in identification and treatmentof brain dopamine-serotonin vesicular transport disease. J. Neurol. Sci.379, 296-297.
Rilstone, J. J., Alkhater, R. A. and Minassian, B. A. (2013). Brain dopamine-serotonin vesicular transport disease and its treatment. N. Engl. J. Med.368, 543-550.
Sawin, E. R., Ranganathan, R. and Horvitz, H. R. (2000). C. elegans locomotoryrate is modulated by the environment through a dopaminergic pathway and byexperience through a serotonergic pathway. Neuron 26, 619-631.
7
RESEARCH ARTICLE Disease Models & Mechanisms (2018) 11, dmm035709. doi:10.1242/dmm.035709
Disea
seModels&Mechan
isms
Related Documents


![TM wetenschappelijk onderzoek v2 - Transcendente Meditatie · [4] BUJATTI, M., and RIEDERER, P. Serotonin, noradrenaline, dopamine metabolites in Transcendental Meditation technique.](https://static.cupdf.com/doc/110x72/5f45826fe5dcf175f017fdcd/tm-wetenschappelijk-onderzoek-v2-transcendente-meditatie-4-bujatti-m-and.jpg)