Medicinal Chemistry of Aldose Reductase Inhibitors Eric R. Larson, Christopher A. Lipinski, and Reinhard Sarges Pfizer, Central Research, Groton, Coirnecficuf 06340 .......... ................. ..................... toins ...................................... ................................. ................... A. Tolrestat (AY-27,773) ........... IV. Non-Steroidal A ........... ............. ructural Informatio ........... 159 160 160 165 167 167 168 170 170 171 172 174 176 177 178 180 181 183 I. INTRODUCTION This article reviews the medicinal chemistry of aldose reductase (AR) in- hibitors and attempts to focus on those aspects of this area of research that are likely to be of most interest to the medicinal chemist. A review article (in a subsequent issue) by Peter Kador of the NEI reviews the biology of aldose reductase inhibitors. The pharmacology of aldose reductase inhibitors has recently been re- viewed,' as have approaches to the medical treatment of cataract.' The po- tential of aldose reductase inhibitors for control of diabetic complications has been summarized in a perspective article3 and the animal, clinical, and me- dicinal chemistry structure activity relationships (SARs) have been concisely s~mmarized.~ Inhibitors of aldose reductase span a remarkable diversity of structures but, nevertheless, almost all inhibitors are acidic to some degree and can generally be grouped into certain functional classes. Major structural classes are the acidic cyclic amides and the carboxylic acids with members of both classes currently in clinical study. Non-steroidal antiinflammatory agents and an- tiallergy agents also inhibit aldose reductase in vitro, although both classes Medicinal Research Reviews, Vol. 8, No. 2, 159-186 (1988) 0 1988 John Wiley & Sons, Inc. CCC 0198-6325/88/020159-28$O4.00

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Medicinal Chemistry of Aldose Reductase Inhibitors
Eric R. Larson, Christopher A. Lipinski, and Reinhard Sarges Pfizer, Central Research, Groton, Coirnecficuf 06340
. . . . . . . . . . . . . . . . . . . . . . . . . . .
..................... toins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
A. Tolrestat (AY-27,773) . . . . . . . . . . .
IV. Non-Steroidal A . . . . . . . . . . . . . . . . . . . . . . . .
ructural Informatio . . . . . . . . . . .
159 160 160 165 167 167 168 170 170 171 172 174 176 177 178 180 181 183
I. INTRODUCTION
This article reviews the medicinal chemistry of aldose reductase (AR) in- hibitors and attempts to focus on those aspects of this area of research that are likely to be of most interest to the medicinal chemist. A review article (in a subsequent issue) by Peter Kador of the NEI reviews the biology of aldose reductase inhibitors.
The pharmacology of aldose reductase inhibitors has recently been re- viewed,' as have approaches to the medical treatment of cataract.' The po- tential of aldose reductase inhibitors for control of diabetic complications has been summarized in a perspective article3 and the animal, clinical, and me- dicinal chemistry structure activity relationships (SARs) have been concisely s~mmarized.~
Inhibitors of aldose reductase span a remarkable diversity of structures but, nevertheless, almost all inhibitors are acidic to some degree and can generally be grouped into certain functional classes. Major structural classes are the acidic cyclic amides and the carboxylic acids with members of both classes currently in clinical study. Non-steroidal antiinflammatory agents and an- tiallergy agents also inhibit aldose reductase in vitro, although both classes
Medicinal Research Reviews, Vol. 8, No. 2, 159-186 (1988) 0 1988 John Wiley & Sons, Inc. CCC 0198-6325/88/020159-28$O4.00

160 LIPINSKI et al.
lack significant in vivo AR inhibitory activity. Numerous weakly acidic phenols and natural product extracts have also been reported to possess AR inhibitory activity in vitro. In addition, the relatively few, non-acidic AR inhibitors are also described. Sections of this review focus on (agents having both in vitro and in vivo activities and on a model for the AR inhibitor site which was, in part, derived from in vitro SAR obtained with antiallergy agents.
From a medicinal chemistry viewpoint, the translation of in vitro activity to pharmacologically useful in vivo activity is one of the key central challenges in developing a clinically useful AR inhibitor. In vitro activity, as measured by inhibition of isolated bovine lens aldose reductase (BLAR), rat lens aldose reductase (RLAR), or human placental aldose reductase (HPAR), has to trans- late to a reduction of sorbitol levels in specific tissues, which accumulate this polyol, in an animal model following oral dosing. This review describes the structural features influencing in vitro activity and, more importantly, those leading to the relatively few derivatives which have clearly shown significant in vivo activity.
We have also devoted considerable space to a discussion of the history of the discovery of sorbinil, the prototype hydantoin AR inhibitor, in order to exemplify the role of synthetic modification in the medicinal chemistry of this class of agents. The clinical and pharmacological activities of sorbinil have been recently summarized in the publication of a recent ~ymposium.~
Although the tertiary structure of aldose reductase is unknown, a large body of literature has been pubIished on the properties of the enzyme and we have attempted to review the available information from a chemical per- spective.
11. ACIDIC CYCLIC AMIDES
The Discovery of Sorbinil
Sorbinil ( 1 ) is the prototype member of this class of AR inhibitors which came from a drug discovery program initiated at Pfizer about 15 years ago. At that time, only a few simple longer chain aliphatic acids and alpha-keto acids were known to be inhibitors of bovine lens aldose reductase.6 Tetra-
Eric R. Larson received a Ph. D. in organic chemistry from the University of Cambridge in 1981, and after an American Cancer Society postdoctoral fellowship at Yale, joined the Medicinal Chemistry Department of Pfizer Central Research in 1983, initially in the area of diabetic complications. His interests are in bio-organic chemistry and' mechanistic biochemistry. He is presently Manager of Diabetes and Cancer Medicinal Chemistry.
Christopher A. Liyinski is currently a Research Advisor in the Department of Medicinal Chernistry of Pfifizer Central Research. He rereined his Ph.D. degree from the University of California at Berkeley in 1968. Following an NlH postdoctoral fellowship at the California lnstitute of Technology, he joined Pfizer in 1970. Working in the field of general metabolic diseases, he has applied his physical organic and synthetic backgrounds and interest in bioisosteres to the design of enzyme inhibitors for the therapy of gastrointestinal and diabetic diseases.
Reinhard Sarges received a Ph.D. degree in organic chemistry from the Uniziersity of FrankfurtlMain, Germany in 1962. He spent 2; years as a postdoctoral fellow and visiting scientist at the National Institute of Arthritis and Metabolic Diseases at the NlH in Bethesda, M D , elaborating the structure of the linear gramicidin peptide antibiotics. In 1965, he joined the Medicinal Chemistry Department of Pfizer Central Research in Groton, CT, udiere he is presently a Research Advisor. He has been particularly actizje in the discozlery of novel antidiabetic and CNS drugs.

MEDICINAL CHEMISTRY 161
1 R = H Sorbinil 2 R=CHa M-79175
methyleneglutaric acid was the most potent agent known, being active as an aldose reductase inhibitor in a lens culture system at non-toxic doses.7 How- ever, this compound was apparently unable to penetrate plasma membranes and failed to show activity in a diabetic rat rnodeL8 In our hands, these compounds exhibited in vitro activity against bovine lens aldose reductase. Furthermore, using an acute streptozotocin diabetic rat model, we were able to show that tetramethyleneglutaric acid inhibited sorbitol formation in sciatic nerve at a dose of 500 mg/kg tid' (Fig. 1). However, the simple structure of these aldose reductase inhibitors, coupled with their relatively low in vivo activity, did not provide a basis for improving activity by further molecular manipulation. Based on the limited knowledge of the aldose reductase enzyme that was available, we attempted a rational synthesis approach which pre- pared structures capable of mimicking the substrate glucose or the co-factor NADPH. However, these attempts were not successful in generating com- pounds with inhibitory activity.
A screening approach using inhibition of bovine lens aldose reductase6 served as a convenient high throughput screen and did, however, turn up valuable leads. Compounds which inhibited the isolated enzyme by 50% at concentrations of 1 x M or less were further evaluated for in vizio activity after oral administration in the rat model," using suppression of sorbitol formation in sciatic nerve as the endpoint. One of the first interesting leads, which we discovered in this manner, was 1-benzyl-6-azauracil, CP-15,626." As shown in Figure 2, this compound displayed good in vizlo activity, showing
In vivo EDs0. Nerve (strep. rat)
Reported Found
M BLARa MBLARb mglkg, p.0.C In vitro ICs0 In vitro Ic50
octanoic acid v V v \ C O , " 10.4-1w5"
<?-keto-nonanoic acid dc0," 10-4-10'5a
TMG 10'5-1 0 - 6 2 10'5-10'6 - 500
Alrestatin 10-5-1 0-6 - 10- 250-500
aReference 6. blnhibitory concentration in moles per liter required lo Inhibit bovine lens aldose reductase
CReference 10
dReference 12.
by 50% using D-glyceraldehyde as substrate. Data Plizer Central Research.
Figure 1. Early AR inhibitors and their biological activity.

162 LIPINSKI et al.
In vlvo EDeo, Nerve In vitro IC8o (strep. rat) pA4 BLARa mg/kg, p . ~ . ~
CP-15,626 6 25-100
CP-14,772 25 10-25
_--- -
Alrestatln 1.5 250-500
alnhlbltory concentration In prnoles per liter requlred to inhlhlt bovine lens aldose reductase by 50% using D-glyceraldehyde a8 substrate.
bDeterrnlned according to the method of reference 10. EDso required for 50% inhlbl- tion of sorbitol levels in the Sciatic nerve of streptorotminized rats given 3x (dose) of drug In rng/kg of drug In a 27-hour period.
Figure 2. The Pfizer amide AR inhibitor leads.
50% suppression of sorbitol formation in sciatic nerves of diabetic rats at doses between 25 and 100 mg/kg tid. Further exploration of SAR around the uracil lead either by synthetic derivatives or close analogs available from the Pfizer compound file did not lead to greatly improved activity in our in vivo test system.
Another avenue of exploiting this lead did prove more fruitful. This in- volved a computer-aided search for structurally distinct compounds in the Pfizer files, which retained the basic molecular features of the lead, namely a N-CO-N-CO grouping and the aromatic ring. One of the structures found by this search was the 1-tetralone-derived spirohydimtoin, CP-14,772. Testing
In vitro lCS0 In vivo EDSO phl mg./kg, p.0.
BLARa iNerveb
20 5
25 10-25
&
5 5-1 0
>>loo >25
aRelerence a, Figure 2. bReterence b, Figure 2.
Figure 3. SAR of spirohydantoins modified in the hydantoin bearing ring.

MEDICINAL CHEMISTRY 163
In vitro lCso In vlvo EDSO FM mg/kg, p.0.
BLARa Nerveb
25 10-25
>>loo >25
-150 >25 8 e.. 20 5
&$n >>loo >25
>>loo >25
>loo >250
aReference a, Figure 2. bReference b, Figure 2.
Figure 4. SAR showing the importance of the aromatic ring.
of this compound revealed modest inhibitory potency against isolated bovine lens aldose reductase, but unexpected potency in the in vivo model with an ED50 between 10 and 25 mg/kg. This activity level was at least 20 times higher than any previously described. Thus, this spirohydantoin was more potent (see Figure 2) than alrestatin, which was at that time the most potent aldose reductase inhibitor described in the literature.I2
This finding prompted us to carry out a broadly based SAR investigation into compounds related to the spirohydantoin. Testing of hydantoins, derived by the Bucherer-Bergs reaction from 5- or 6-membered ketones fused to a benzene ring, showed that the activity of CP-14,772 was shared by a number of congeners. However, expansion to a 7-membered ring led to a dramatic loss of activity (Figure 3). Similarly, shifting the spiro junction, hydrogenating the benzene ring, or eliminating the fusion of the benzene ring to the ring bearing the hydantoin decreased activity (Figure 4). Hydantoins, which were not linked by a spiro junction to another ring but were disubstituted, such as diphenylhydantoin, showed little activity in our test system (Figure 4). These findings suggested that a precise spatial relationship between the ar- omatic benzene ring and the hydantoin ring is required for good aldose re- ductase inhibitory activity in this series.
Interesting results were obtained when we examined the effect of aromatic substituents on activity. Substitution with alkoxy in the tetralin series, or alkoxy and halogen in the indane ring system, had only minimal effects on activity (Figure 5). However, halogen substitution, especially in the 6-position of the chroman or thiochroman ring systems led to dramatic increases of in vitro and in vivo potency (Figure 6).9
We then turned our attention to determining the effect of chirality on ac- tivity, since the spirohydantoins have an asymmetric carbon atom at the spiro

164 LIPINSKI et al.
In vltro lCoo In vivo EDoo f l mg/kg, p.0.
X BLAR a Nerve b
H 25 10-25 5-OMe 10 25
6,7-(OMe)2 50 >5
H 20 5 6-F 25 5 5-OMe 6 >25 6-OMe 9 10 5,6-(OMe)a 2 10
6-0Me 23 >25 7-OMe 75 10-25
'Reference a, Figure 2. bReference b, Figure 2.
Figure 5. Tetralins and indanes with aromatic substituents.
junction. The 6-fluorochroman and the 6-fluorothiochroman derivatives were readily resolved with brucine, and testing of the separated enantiomers showed that the interaction of these compounds with aldose reductase is highly en- antiospecific (Figure 7). We established by x-ray analysis that the active en- antiomer in the chromane series had the S-configuration. l3 This compound, which was later given the USAN designation sorbinil, was selected for clinical evaluation.
It is interesting to look in retrospect at spirohydantoins like sorbinil and at the original empirical lead that led to their discovery. Assuming that the 1- benzyl-6-azauracil lead, CP-15,626, and sorbinil interact with the same binding site at the enzyme aldose reductase, then it is likely that they overlap their
- __. in vitro ICIo In vivo EDso
f l mg/kg, P.O. BLAR a Nerve b x _________.
H 5 5 6-F 0.9 0.75 6-CI 10.1 0.25-0.75 6-Br 0.5 <1.5 8-CI 0.9 >1.5 6,ECIz <0.1 0.25 6,7-C11 <0.1 G1.5 6-OMe 2 >2.5 6-Me 2 5 6-~Bu 30 NT 6-SO*NMe2 70 >5
H 5 5-10 6-F 0.3 1.5 6-CI 0.3 C2.5 6-Br 7-CI 8-CI 6,S-CIz 6,7432 5.8-CI.
0.2 1
50 <0.1 <0.1 >loo
_. (2.5 >1.5 >1.5
2.5 2.5 NT
6-OMe 5 >5
'Reference 8 . Figure 2 bFieference b. Figure 2
Figure 6. Arornotic substitutents in the chroman and thiochrornan series.

MEDICINAL CHEMISTRY 165
In vifro ICso In vivo EDso PM mglkg, p.0.
8LARa Nerveb "U-NM
Sorbinil (5) 0.15 0.25
R-enantiomer 4.4 25
Racemate 0.9 0.75
S-enantlomer (0.1 0.75
R-enantlomer 5 NT
Racemate 0.3 1.5
'Reference a, Figure 2. bReferencc b, Flgure 2.
Figure 7. The effect of chirality on biological activity.
essential molecular features as shown in Figure 8. If this model is correct, their three-dimensional overlap is probably much more complex than the simple connectivity of aromatic ring, carbon spacer, and N-CO-N-CO group, that guided the original computer search.
The precise mechanism by which sorbinil inhibits aldose reductase is still unknown. Kinetic studies have indicated that binding of sorbinil is noncom- petitive with either substrate or ~o-factor.'~ These findings indicate that sor- binil is not an inhibitor either at the substrate or enzyme NADI'H binding site.
With the discovery of sorbinil, spirohydantoins have become a potentially very useful class of aldose reductase inhibitors. They differ generically from carboxylic acid-type inhibitors by their excellent translation of in vitro to in vivu activity, a feature already evident in the first representative, CP-14,772 (Figure 2). A plausible reason for this advantageous behavior in vivo could be the higher pKa values of hydantoins versus those of carboxylic acids. This property seems to translate to lower protein binding and better tissue pen- e t ra t i~n. '~
Spiro Imides
M79,175(2), a 2-methyl analog of sorbinil, is an interesting compound since the 2-substituent creates an additional asymmetric center in the spirohydan-
3

166 LIPINSKI et al.
F133° Sorblnll
CP-15*626
Overlap: Sorblnll.CP-3 5,626
Figure 8. Overlap of the minimal energy confirmations of the original amide lead with the spirohydantoin sorbinil.
toin nucleus. When achiral6-fluoro-2-methyl-chromanone is subjected to the Bucherer-Bergs hydantoin synthesis, four tompounds consisting of two en- antiomeric pairs of diastereomeric hydantoins are formed in an 85:15 ratio. The predominant diastereomer consists of the 2R, 4s and 2S, 4R enantiomeric pair (2 ,3 ) . This has been resolved via the salt with methyl cinchonidium hydroxide and the 2R-CH3-4S enantiomer (2) hatritlg the same chirality at the 4-hydantoin center as sorbinil is the most active (of the four isomers. NMR and x-ray diffraction studies show that the 2-methyl group is equatorial and that the solution conformation is the same as in the crystal form.16 This differs from sorbinil, which in solution exists as two chair conformers with a low- energy barrier for intercon~ersion.’~
M79,175 exhibits enhanced inhibitory activity against rabbit lens AR (IC-50 6.5 X lP), reflecting the beneficial effects of methylation at the 2-position. Replacement of the 6-F in this compound by a 643 group slightly increases potency, while introduction of a nitro group at the 8-position causes a small decrease in potency. l7 Like sorbinil, M79,175 is a non-competitive inhibitor against rabbit LAR when DL-glyceraldehyde is used as substrate.17 M79,175 decreases lens sorbitol levels in streptozotocin-diabetic rats in the range 0.1 to 1.0 mg/kg PO and delays development of cataracts in galactosemic rats.I8
Another class of spiroimides, the racemic oxazolidenediones 4 3 , were found to possess BLAR activity residing in the ( -) enantiomers having the S con- figuration. These compounds were resolved as cinchonidine adducts and the ( - ) enantiomer of 5 was shown to have the 4s configuration at the spiro center by x-ray studies. Activity in this series was enhanced in 6-halo analogs
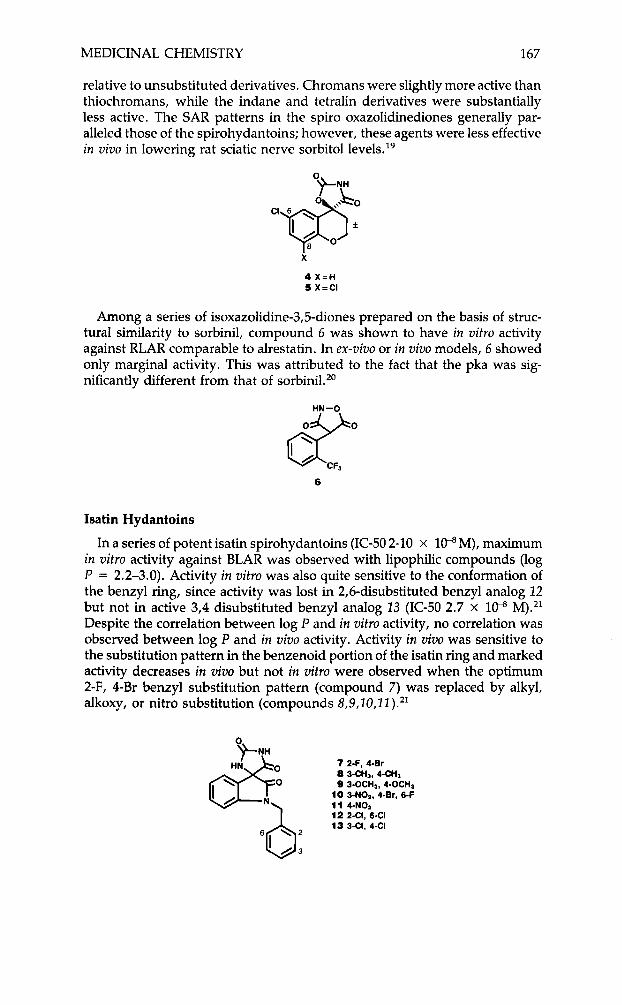
MEDICINAL CHEMISTRY 167
relative to unsubstituted derivatives. Chromans were slightly more active than thiochromans, while the indane and tetralin derivatives were substantially less active. The SAR patterns in the spiro oxazolidinediones generally par- alleled those of the spirohydantoins; however, these agents were less effective in vivo in lowering rat sciatic nerve sorbitol 1e~els . l~
4 X = H 5 X=CI
Among a series of isoxazolidine-3,5-diones prepared on the basis of struc- tural similarity to sorbinil, compound 6 was shown to have in vitro activity against RLAR comparable to alrestatin. In ex-vivo or in vivo models, 6 showed only marginal activity. This was attributed to the fact that the pka was sig- nificantly different from that of sorbinil.20
d"" 6 CFa
Isatin Hydantoins
In a series of potent isatin spirohydantoins (IC-50 2-10 x l0-S M), maximum in vitro activity against BLAR was observed with lipophilic compounds (log P = 2.2-3.0). Activity in vitro was also quite sensitive to the conformation of the benzyl ring, since activity was lost in 2,6-disubstituted benzyl analog 22 but not in active 3,4 disubstituted benzyl analog 13 (IC-50 2.7 x 1P M).21 Despite the correlation between log P and in vitro activity, no correlation was observed between log P and in vivo activity. Activity in vivo was sensitive to the substitution pattern in the benzenoid portion of the isatin ring and marked activity decreases in v im but not in vitro were observed when the optimum 2-F, 4-Br benzyl substitution pattern (compound 7) was replaced by alkyl, alkoxy, or nitro substitution (compounds 8,9,10,22).**
7 2-F, 4-Er
9 3-OCH3, 4-OCHs 10 3-N02, 4-Br, 6-F
8 3-CH3, 4-c&
' N\ 11 4 4 0 , 12 2-CI,6-CI 13 341, 4 4

168 LIPINSKI et al.
Fluorene Hydantoins
Spirofluorenyl hydantoins have been reported to possess potent in vitro and in vivo activity. The monofluoro derivative, alconil (AL-1567, 24), and difluoroderivative (AL-1576, 25), have respective IC-50’s of 4.3 and 2.4 x against RLAR and 2.4 x and 8.9 x lo-’ against HPAR.22 In a series of mono-substituted compounds (e.g., 24) tested as racemates, the order of de- creasing potency for 2-substituents was 2-F > 2SCH3 > 2SOCH3 > 2S02CH3.23 When the fluoro substituent was moved from the 2 to the 1 or 4 positions, an activity decrease was observed. The racemic AL-1567 ( 2 4 ) is very similar in potency to sorbinil in zritro when tested against RLAR24 or BLAR.25
4
14 X = H , Z = N H 16 Z=CHZ 17 Z = S 18 Z = O 15 X = F , Z = N H 2 3 X = F , Z = S
Both AL-1567 ( 1 4 ) and the symmetric, achiral AL-1576 ( 2 5 ) prevented lens cataract formation when p e n to rats in single doses of 5-16 and 0.5-4 mg/kg/day for 15 weeks. Both compounds also decreased sciatic nerve sorbitol levels in a dose-dependent manner. AL-1576, the more potent agent, completely pre- vented sciatic nerve conduction velocity deficits in the streptozotocin diabetic rat at 1.5-4 mg/kg/day. A daily dose of 1.5 mg/kg/day halted the progression of cataracts which had been allowed to form for 2 months before the start of drug treatment. 22
When the spirohydantoin ring in AL-1567 ( 2 4 ) is replaced by related spi- roimide systems, the relative potency of the two series depends on whether HPAR or RLAR is used as the enzyme. Against HPAR, the order of potency is succinimide (26) > thiazolidinedione (1 7) > hydantoin ( 2 4 ) > oxazolidi- nedione (28), whereas against RLAR, the order of potency is 24 > 17 > 26 > 18.26 In a series of spiroimides related to AL-1567, the racemic thiazolidi- nedione (19) lacking a fluoro substituent is surprisingly potent against HPAR (IC-50 1.6 x On the other hand, corresponding hydantoins, succinim- ides, and oxazolidinediones are all about 10 times less potent than 29.
Very good activity has been reported for the 4-<aza analogs of 29. The thia- zolidinedione (20, IC-50 3.8 x is less active against RLAR than related
0 +NH
19 x = s , Y=CH, Z=CH 20 X = S , Y=CH, Z = N &” 22 21 X=NH, X=NH, ‘V=CH, Y = N , Z = C H Z = N ‘ A
hydantoin 22, (IC-50 9.9 x lo-’). Against HPAR, 21 (IC-50 9.4 x is more active than the 2-aza analog (22, IC-50, 2.6 x Replacement of the hy- dantoin NH in AL-1576 ( 2 5 ) by sulfur leads to the slightly more potent 23, (IC-50, 6.2 x M), which appears to be the most active agent in the series against HPAR. 26

MEDICINAL CHEMISTRY 169
Cyclic Imides
Among a series of 5-benzylthiazolidinediones developed originally as hy- poglycemic agents, only weak activity against HPAR was detected in deriv- atives such as 24. An effort to improve activity led to an exploration of the
H
24
SAR about phenyl analogs lacking the methylene bridge.27 Moderate activity was observed with the thiazolidinedione analog of diphenyl hydantoin (25),
R ' dNH '
which is substantially more potent against RLAR (IC-50 1 x 1CP M) than diphenylhydantoin itself (IC-50 >> M).28 Most of the derivatives studied were thiazolidinediones containing a single substituted phenyl ring appended to the thiazolidinedione moiety. The best in vitro activity among mono-sub- stituted phenyl derivatives was observed with 26. However, in contrast to the good in vitro activity, very little activity was observed in inhibiting lens swelling in a rat lens culture assay. The in vitro activity of 26 and related analogs led to a focus on derivatives incorporating benzene ring alkoxy sub- stituents. More than 25 analogs with 3,4-dialkoxy substitution were prepared, and although many of these were no more potent in vitro than the best 4 mono-alkoxy derivatives, some members showed good activity in the lens swelling assay. Among the best derivatives in this assay were the 3-ethoxy, 4-butoxy and 3-ethoxy, 4-pentyloxy derivative^.^^ The latter derivative (CT- 112, 27) was selected for further study.
CT-112 (27) was tested at 25 mg/kg p.0. in streptozotocin-induced diabetic rats, and significantly reduced sciatic nerve sorbitol levels and improved tail
0
27 R = CSH, 28 R = (CH2)zCHOHCHOHCHa
RO
nerve motor conduction velo~ities.'~ This compound is extensively metabo- lized in rat and dog, and in these species has plasma half-lives of 4.3 h and 9.0 h, respectively. One of the metabolites (28) bearing a hydroxylated 4- pentyloxy substituent exhibited slightly better activity in the lens swelling

170 LIPINSKI et al.
assay than the parent drug, while other metabolites had activity comparable to that of the parent."f3'
In an extensive study, over 50 hydantoin and thiohydantoin derivatives were examined as inhibitors of RLAR and BLAR." In general, inhibitory activity against the two enzymes was very similar and did not differ by more than a factor of 3. Among the compounds studied, the best activity was observed in a series of 1-phenylsulfonyl hydantoins. The para-bromo analog, 29, was the most active agent with activity nearly 5 times that of the phenyl
29
unsubstituted derivative. Diphenylhydantoin with a pka of 8.02 and 29 with a pka of 6.04 were studied as RLAR inhibitors over the pH range 6.0 to 7.5. The inhibitory activity of diphenylhydantoin increased, while that of 29 de- creased as the pH was raised. This was interpreted as indicating that di- phenylhydantoin inhibits the enzyme in its ionized form, while 29 inhibits in its non-ionized form.28
Derivative 29, in addition to AR inhibitory activity, is also a weak hypo- glycemic agent, an effect that was discovered when 29 was tested for hypo- glycemic activity in rabbits on the basis of its resemblance to sulfonylureas. However, doses of 50 and 100 mglkg p.0. were less hypoglycemic than a 50 mg/kg p.0. dose of t o l b ~ t a m i d e . ~ ~ A 50 mg/kg dose of 29 reduced galactitol levels in galactose fed rats by 50% in sciatic nerve and 25% in the retina. At this dose, lens galactitol levels declined only slightly; however, a marked decrease in cataract formation was observed and this was interpreted as sug- gesting an additional effect of 29 beyond the small decrease in osmotic pres- sure that might result from weak AR i n h i b i t i ~ n . ~ ~
111. CARBOXYLIC ACIDS
Tolrestat (AY-27,773)
(30) is a potent inhibitor of BLAR (IC50 3.5 x 10-8)34 and is reported to exist in the crystalline state as the thermodynamically more stable rotamer (30) in which the two bulky carboxymethyl and naphthyl substituents are trans.35
CF, 30

MEDICINAL CHEMISTRY 171
In solution, the equilibrium ratio between the trans rotamer 30 and cis rotamer 31 is about 3 to 1 in the dark, while the ratio is about 1 to 1 in the presence
of light. The rates of rotamer interconversion are both solvent and pH de- pendent. NMR studies show that the thioamide side chain lies out of the plane of the naphthyl ring due to a peri interaction with the naphthalene ring 8-H. Data on the pH-rate profile interconversion of the two rotamers suggest that there may be a hydrogen bond between the acetic acid carboxyl hydrogen and the thioamide sulfur in the trans tolrestat rotamer (30).35
Tolrestat inhibits formation of sorbitol in human red blood cells incubated with glucose (IC50 3 x lo4 M) and effectively inhibits galactitol or sorbitol formation in galactosemic or diabetic rat sciatic nerve and lens. The effec- tiveness varies according to experimental conditions and ED,,'s are reported to be in the range of 5 to 15 mg/kg ~ 0 . ~ ~ Tolrestat in a dose-dependent manner is also effective in preventing cataracts in galactose fed rats37 and retinal capillary basement membrane thickening3'
In contrast to the similar i iz uitro AR inhibitory potency across animal spe- cies, tolrestat differs markedly in metabolic profile between rat, on the one hand, and dog, monkey, and man on the other hand. In the rat, tolrestat has a short half-life (3.5 h) and is extensively metabolized. In dog, monkey, and man, half-lives are 9 to 12 h and the drug is largely unmetabolized. In all species, the drug is well absorbed and highly protein b o ~ n d . ~ ~ , ~ ~ In diabetics, the elimination half-life was higher and the bioavailability was slightly reduced relative to normal subjects.41
In clinical trials, tolrestat (50 to 200 mg p.0.) has been reported to reduce elevated red blood cell sorbitol levels in diabetic patient~,~' and in an open trial was regarded as beneficial in the treatment of diabetic n e ~ r o p a t h y . ~ ~
Alrestatin (32, AY-22,284), the lead compound which preceded tolrestat, is 100 times less active than tolrestat, both in uitro against BLAR and in vivo
32 R = H 33 R=CH,
in its ability to reduce galactitol formation in the sciatic nerve of rats on a high galactose diet.34 In clinical trials, alrestatin produced significant differ- ences over placebo only at high oral dose^,^,^^ while after intravenous dosing

172 LIPINSKI et al.
some subjective improvement of signs of neurclpathy in the lower limbs as well as slightly improved sensory nerve conduction velocity was reported.46 Alrestatin itself is achiral. However, a potential chiral environment in the vicinity of the receptor site binding the alrestatin acetic acid side chain has been searched for by testing the R and S enantiomers of 33 a chiral alrestatin propionic acid analog. Both enantiomers were similar in activity; the R en- antiomer being slightly the more active (IC50 4 )< against RLAR).47 This lack of receptor enantiospecificity differs markedly from the marked receptor enantiospecificity observed with the chiral sorbinil.
Statil (ICI-128,436)
(34) , which is currently in clinical studies, is the key member of a series of phthalazinone carboxylic acids, and like alrestatin and tolrestat is an acetic
2-F, 4-Br 3-Br, 4-Rr, 3-CI, 4-CI 2-CI, 6-CI 2-F, 5-Br
5-Br
acid derivative. Like tolrestat, it is also a potent inhibitor of AR from various tissues including HPAR (IC50 2-15 X 10-8 M). bz vitro activity of the parent phthalazinone acetic acid is increased by appending a benzyl group to the phthalazinone amide nitrogen. Replacing benzyl by methyl or propyl leads to inactive compounds. Halogenation of the benzyl group greatly enhances activity. The pattern of halogenation is critical. For example, the small increase in activity which was observed on introduction of a benzyl 2-fluoro group was later incorporated into the design of the optimum 2-fluoro-4-bromobenzyl moiety in ICI-128,436.
In the phthalazinone carboxylic acids, in vitro activity increases with in- creasing lipophilicity up to a log P value of about 3. Activity in vivo but not in vitro drops off with very lipophilic derivatives such as the 3,4,5-tribromo- benzyl derivative 35. Substituents other than halogen maintain moderate in vitro potency; however, compounds with non-halogen substituents lose the ability to lower sorbitol levels in vivo in the diabetic rat nerve. The marked effect of benzyl substitution pattern is clear from the excellent in vitro activity of the 3,4-dichloro derivative 36 and inactivity of the 2,6-dichloro derivative 37. As in the case of the structurally related N-benzyl isatin hydantoins (7- 13), the 2,6-dichlorobenzyl phthalazinone acetic derivative ( 3 7 ) was inactive due to its inability to adopt a required conformation. Moving the bromo substituent in the benzyl group of ICI-128,436 from the 4 to 5 benzyl position (e.g., 3 8 ) results in a 50-fold decrease in in vitro activity.
Among several series of phthalazinone and quinolone acetic acids, plots of the log P vs. IC50 showed that the better compounds in vivo occurred at the point where the curve began to flatten, suggesting a balance between a lower optimum log P for tissue penetration and a higher log P for enzyme inhibition.

MEDICINAL CHEMISTRY 173
In maximizing in vivo potency, the ICI workers reported that no group was as effective as the N-4-bromo-2-fluorobenzyl substituent in ICI-128,436.21*48
In diabetic rats models, ICI-128,436 lowers nerve sorbitol levels (EDSO, 5 mgkg p.0.) and reverses motor nerve conduction (MNCV) deficits and my- oinositol depletion. Tolerance to the ability of ICI-128,436 to lower nerve sorbitol levels did not occur even after 74 days of dosing. Cataracts in diabetic rats were prevented by chronic doses as low as 25 mgkg.49,50 Plasma half- life in the rat was 10 h,51 while that in man was 13 h following single doses up to 1000 mg5’
Other Acids
ICI-105,552 (39), a 3-methyl quinolone acetic acid, is about one-half as potent (ICSO 5 x lo4 M) against BLAR as ICI-128,436 and corresponding half
39 R = CHI 40 R = H 41 R = CH(CH&
as potent in lowering sciatic nerve sorbitol levels in diabetic rats.’* Removal of the 3-methyl group leads to derivatives such as 40 with reduced potency both in vitro and in z ~ i v o . ’ ~ ~ ~ ~ As in the phthalazinone series, the best in vitro activity in these series is seen with compounds which have halogen substi- tuents on the 1-benzyl group. Activity in vitro is improved in the desmethyl series by addition of a fluorine at the 6-position as in 42 (ICSO 6 X 1W M against BLAR).
Increasing the bulk of the 3-methyl to 3-isoprop)tl as in 42 greatly decreases in vitro activity (ICS0 2.2 x 10-5),21 an effect that was attributed to a change in the steric relationship between the carboxyl group and the planar aromatic backbone.
The animal pharmacology of 39 in lens and nerve is generally similar to ICI-128,436, except for the somewhat lower potency. Sorbitol levels are re- duced in lens, sciatic nerve, and seminal vesicles, but not kidney in diabetic rats.53 In vitro, 39 prevented sorbitol accumulation in monkey kidney cells cultured in high glucose medium; however, this compound did not inhibit rat kidney basement membrane thickening after 1 year of oral the rap^.^^,^^

174 LIPINSKI et al.
ICI-105,552 (39) did not proceed to clinical studies, apparently due to crys- tallization of the compound in the kidneys of animak4*
ONO-2235 (43), an acetic acid in clinical studies in Japan, is a potent in- hibitor of RLAR and HPAR (IC50 1 and 2.6 x lo4 M, respectively) and is the
43
major E,E geometric isomer produced in the condensation of a-methyl-cin- namaldehyde with rhodanine-N-acetic acid. Enzyme kinetic studies indicate that this compound, like many AR inhibitors, is an uncompetitive inhibitor against RLAR.56 When given in food, 43 lowers diabetic rat sciatic nerye sorbitol levels and improves tail nerve MNCV.57-59 Studies assessed a variety of parameters related to retinal microangiopathy in streptozotocin diabetic rats fed a fructose rich diet containing 43. Even after 8 months of experimental diabetes, retinal capillary basement membrane thickening could be prevented by the addition of 43 to the diet.60
A single 200 mg oral dose of 43 in diabetic subjects decreased the rise in red cell sorbitol levels during a glucose tolerance test.61 When given orally at 600 mg for 8 weeks, 43 relieved a variety of symptomatology related to diabetic neuropathy in a small open study.62
Santen 110-790 (44) and Santen 118-79 (45) are members of a class of thia- zolidine carboxylic acids related to series of angiotensin covering enzyme (ACE) inhibitors. In studies examining in vitro activity, both 44 and 45 were extremely potent against RLAR (IC50 3.7 and 4.3 x l O - ' O M, r e spe~ t ive ly ) .~~ ,~
The best of a series of benzopyran-Zone acetic acids such as 46 (ICm 2.0 x 10-8 M) were equivalent in vitro to sorbinil (ICs0 1.7 x 1C8 M, in this assay) as
b NO2
44 L J P
45
inhibitors of crude RLAR. The acetic acid moiety at C-4 gave optimum activity in contrast to 4-carboxylic, acrylic, or propionic acid derivative^.^^ No in vivo activities were reported for these compounds.
46

MEDICINAL CHEMISTRY 175
Cyclandelic acid (47), a human metabolite of Cyclospasmol, a drug used for treatment of peripheral and cerebral vascular disorder, weakly inhibits rat
%o&co*H OH H CH,
47
nerve aldose reductase (ICs0 1 x uivo are not described.
M).66 The effects of this derivative in
IV. NON-STEROIDAL ANTIINFLAMMATORY AGENTS
Since most ARIs are acidic, it is not surprising that there should be some degree of overlap between ARI activity in uitro and non-steroidal antiinflam- matory (NSAI) activity. However, in uivo effects attributable to AR inhibition have not been impressive, and dose escalation to increase AR efficacy is problematical because of the side effects observed at high doses with NSAI agents. Among NSAI agents, sulindac (48) appears to be the most active as
F
CHs-X
an in uitro inhibitor of cataractous HLAR and normal BLAR approaching the in uitro activity of ~ o r b i n i l . ~ ~ The rank order of NSAIs in in uitro AR inhibitory potency is sulindac > sulindac sulfide > indomethacin > oxyphenbutazone > sodium salicylate.
Sulindac and its antiinflammatory active sulfide metabolite (49) have been extensively studied. The sulfide metabolite applied topically in three doses of 100 mg during a 24-h period penetrates rapidly into diabetic rat eye and decreases sorbitol buildup by 38% .68 As assessed by chemical analysis, sulin- dac and sorbinil are equi-active in decreasing sorbitol buildup in rat lenses or rabbit sciatic nerve incubated in high glucose medium.69 Sorbitol levels can also be monitored in intact lenses incubated with drug in l-13C glucose media by NMR spectroscopy. Using this method, both sulindac and sorbinil were equiactive in lowering sorbitol levels in rabbit lenses. Ibuprofen, acetamino- phen, and aspirin were significantly less active.69
In a double-blind study in seven human volunteers receiving 300 mg su- lindac bid, in contrast to plasma levels, only very low levels of the parent sulfoxide and sulfide and sulfone metabolites were found in red cells, and these levels were too Iow to influence polyol pathway metabolite levels. The conclusion was drawn that, despite good in uitro AR activity, sulindac is likely to have little effect on the oxidative pathways implicated in diabetic compli- cations unless high levels of the drug are used.” Either sulindac does not

176 LIPINSKI et al.
penetrate the red cell membrane or, if it does, it is tightly bound to cell membrane proteins or to soluble proteins such as haemoglobin in the red cell.
Leakage of intravenously administered fluorescein into the eye is associated with vascular retinopathy and higher hemoglobin Alc levels in diabetic pa- tients. In a randomized double-blind trial, 200 mg sulindac bid was given for 6 months to 24 insulin-dependent patients with little or no retinopathy. This treatment decreased fluorescein leakage into the eye and reduced the increase in blood-retinal barrier ~ermeability.~' In an extension of this work, sorbinil showed a similar effect to sulindac in reducing blood-retinal barrier perme- ability. 72
Aspirin has been proposed for the treatment of human senile cataracts based in part on epidemiological evidence, suggesting a decreased incidence of cataracts in rheumatoid arthritis patients taking high doses of aspirin for prolonged periods of Aspirin's action is based in part on the very weak effects of the aspirin metabolite-sodium salicylate as an inhibitor of cataractous HLAR and BLAR74 or aspirin itself as an inhibitor of rabbit LAR.69 Aspirin also competes with the tryptophan binding site in human serum albumin and decreases levels of bound and total tryptophan. This might be a desirable effect, since plasma tryptophan levels are reported to be increased in senile cataract patients and the amino acid has been suspected to be ca- tara~togenic .~~
The putative anticataract effects of aspirin have also been linked to possible decreases in lens protein gluc~sylat ion.~~ In the rat, aspirin acetylates a low pka albumin lysine, which may be the major site of gluc~sylation.~~ Blocking glucosylation could be expected to block protein aggregate formation due to a variety of mechanisms. Glucosylation may induce conformational changes in the protein, rendering sulfhydryl groups more susceptible to oxidative c r~ss l ink ing .~~ Also, glucosylated proteins may cross-link via non-reducible fluorescent cross-links (the browning reaction) as is observed in collagen-rich tissues7* The optical density increase that occurs when rabbit lens protein is incubated with tryptophan in the presence of high glucose-6 phosphate levels is a model for the browning reaction. In this model, sodium salicylate partially blocks the density increase,74 an effect that cannot be due to acetylation of a lysine residue since sodium salicylate is not an acetylating agent.
The lysine salt of the clinically used NSAI carboxylic acid bendazac (50)
50 '0 has been reported to improve visual acuity in cataractous patients in a small double-blind study. 79,80 In vitro, bendazac is more effective than indometh- acin in inhibiting thermal denaturation of rat lens proteins. In vivo following oral dosing, bendazac slowly accumulates in rat lens and inhibits thermal

MEDICINAL CHEMISTRY 177
lens protein denaturatiom81 Following x-ray treatment, lens water content increased, insoluble protein increased, glutathione levels decreased and sol- uble protein sulfhydryl content decreased in rabbit lenses over a 6 to 12 week period. These effects were all partially blocked by daily 30-day treatment with bendazac (100 mg/kg ip) terminating on the day of X-ray treatment.s2
V. ANTIALLERGY AGENTS
Substituted xanthone-2 carboxylic acids, which are well known antiallergy agents, inhibit RLAR in vitro. Among 7-sulfamoyl derivatives, best activity (83% inhibition at Za"-MJ. was observed with 52, a more water soluble deriv- ative of the active N,N-dimethyl derivative 52. Activity was decreased when
CH, \
51 R = (CH~),OH 52 R = C n J
N-On.S. 7 h
the substituent at C-7 contained an acidic hydrogen. Based on previously published antiallergy SAR, the authors concluded that there was no structural correlation between AR inhibition and antiallergy SAR.83 When compound 52 was administered into the fused eyelid pouch of galactose maintained rats, intra-cellular lens fluid accumulation and damage to lens fiber protein com- ponents and cataract formation was partially prevented by dosing with a lo" M solution from birth to 12 days.84
A partial overlap in SAR has been observed among antiallery agents, xan- thine oxidase inhibitors, and AR inhibitory agents. The speculative proposal was advanced that a common feature leading to the three activities might be the ability of the inhibitors to interfere with univalent electron transfer^.'^ Compounds such as the antiallergy agent lodoxamide (53) inhibit xanthine oxidase (48% at 8 x lo-* M), while the related oxamic acid (54) inhibits both
i N
53
.. CHJ 0
54
milk xanthine oxidase (39% at 8 x M) and RLAR and HPAR (IC-50 0.5 and 8.3 x 10-6 M, re~pectively).~~, '~
A quantitative relationship has been observed between decreasing energy of the lowest unoccupied molecular orbital (LUMO) of the carbonyl moiety present ip antiallergy agents and increasing antiallergy activity as measured by the pet passive cutaneous anaphylaxis assay.87 A low LUMO value indicates that a carbonyl group is easily polarized and can act as a charge transfer bridge between an electron rich nudeaphilic group approaching the carbonyl carbon an4 an electron acceptor group close to the carbonyl oxygen. A correlation was also observed between LUMO energies and inhibition of RLAR in a series of closely related 4-oxo-4H-chromens but not with the multiple series

178 LIPINSKI et al.
R
C02H H
57 R2
55 56
of structurally divergent antiallergy agents exemplified by 55, 56, and 57.86 Among these, the fragment 59 was identified as common to several series.
58
X=O. NH
59
VI. AR INHIBITOR SITE MODEL
A model for the AR inhibitory site has been proposed based on a combi- nation of AR enzyme inactivation experiments and AR inhibitory antiallergy agent SAR.I4 The observation that AR can be inactivated by 2-bromo-4’-ni- troacetophenone, a reagent which reacts well with guanidine groups, led to the suggestion that an arginine might be located at the AR active site. Aldose reductase is also inactivated by 4-nitrobenzenesulfonyl fluoride, a reagent known to inactivate tyrosine residues, suggesting the presence of a tyrosine residue. The kinetics of inhibition by both 2-bromo-4’-nitroacetophenone and 4-nitrobenzenesulfonyl fluoride were interpreted as consistent with a tyrosine and a guanidine being located at the enzyme site occupied by inhibitor mol- ecu le~ . ’~
A wide range of antiallergy agents such as 55, 56, and 57 inhibit ARE6 and also contain a polarizable carbonyl group. This, combined with the inacti- vation experiments, led to the suggestion that the carbonyl moiety functions as a charge transfer bridge between a basic oxygen nucleophileas in a tyrosine hydroxyl and a protonated arginine acting as the proton d o n ~ r . ’ ~ , ~ ~ Features that were noted as common in the AR active antiallergy agents were the polarizable carbonyl, a generally planar molecular structure preferably con- taining an aromatic hydroxyl, and an acidic moiety.86
Key features of the active site model are two parallel lipophilic regions and a charge transfer pocket accommodating a reactive carbonyl moiety. The model defines broad ranges for aromatic ring to carbonyl distances and distances from the aromatic ring plane to the carbonyl group as well as distances from the carbonyl moiety to a presumably optimally positioned receptor tyrosine hydroxyl and acidic protonated guanidine as in arginine.I4
The model serves a useful purpose in focusing attention on the lipophilic aromatic backbone of many inhibitors and in particular on the role that struc- tural modifications might make on carbonyl group polarizability . Limitations of the model are the lack of precision relating the position of a carbonyl moiety with respect to the aromatic plane and the failure to focus on the position of

MEDICINAL CHEMISTRY 1 79
an inhibitor acidic moiety with respect to the aromatic plane. The problem with carbonyl moiety orientation is well illustrated by the fact that the sorbinil hydantoin carbonyls lie in a plane orthogonal to the aromatic plane, whereas the carbonyl in antiallergy AR inhibitors lie in the aromatic plane. Because an acidic moiety is found in virtually all AR inhibitors, its location relative to other important structural features is likely to be an important determinant of activity but is not accounted for by the model. ICI workers, in particular, have drawn attention to an overlapping relationship between the carboxylic acid moiety in ICI-128,436 and the acidic imidic N-H proton in sorbinil.''
VII. PHENOLS
Interest in flavonoids as aldose reductase inhibitors stems from the broad in vitro aldose reductase inhibitory activity observed with this class of com- pounds. The major limitations of flavonoids as potential drugs are poor oral activity, which is likely related to the poor aqueous solubility of many mem- bers, and the erratic absorption and poor half-life found with many phenolic medicinal agents. Thus, while a large volume of literature exists on phenols as in vitro AR inhibitors, this likely reflects the ease of discovery of AR activity in this class rather than the potential of this class as useful medicinal agents.
Compounds in the flavonoid class that have been studied include the fla- vones, with the chromone ring 2,3 double bond (e.g., 60), the flavanols which contain a hydroxyl or substituted oxygen moiety at C-3 (60, R = OH or OR), and the flavanones in which the 2,3 double bond is reduced (62, R = H).
The aromatic ring at C-2 may contain a variety of hydroxyl or modified hy- droxyl groups (X) while the fused benzene ring of the flavonoid nucleus can also contain a variable number of free or modified hydroxyl groups ( Y ) . The oxygen at C-3 may be either a free hydroxyl (aglvcwe, 60, R = OH') or covalently linked to a sugar residue (glycone, 60, R = OR').
Systematic SAR studies within defined series of compounds have not been conducted. Thus, meaningful SAR data are not available and much confu- sion exists in the area. For example, one group has concluded that addition of sugar residues to the flavonoid appears to cause a major decrease in po- t e n ~ y , * ~ while the opposite conclusion, namely that monoglycosides have a higher activity than the corresponding aglycones, has been reached by an- other group studying a different set of compounds.90 Furthermore, given the poor solubility of many flavonoids, changes in relative activity occa- sioned by use of solubilizing cosolvents serves further to confound at- tempts to determine precise SAR since it is frequently necessary to use sol-

180 LIPINSKI et al.
vents such as DMSO or propylene glycol. Thus, it is not surprising that, in a series of 73 flavonoids tested against RLAR, AR potencies were found to be solvent de~endent . ’~
In comparing flavonoids to other ARI inhibitors, quercetin serves as a useful benchmark. In vitro, IC-50’s for quercetin, alrestatin, and sorbinil against human lens aldose reductase are 1.0, 6.0, and 0.2 x 1CP M, respectively.” In vim activity is only modest, however. In galactosemic rats, sciatic nerve galactitol levels are only reduced following very high oral doses (700 mg/kg, q u e r ~ e t i n , ~ ~ 1100 mg/kg, alrestatin12). This contrasts with sorbinil, which low- ers sorbitol levels at less than 1 mgkg in streptozotocinized rats.”
Among a series of 27 3’,4’-dihydroflavonoids, the best activity against RLAR and BLAR (10 x quercetin) was observed in a 3’,4’-di-hydroxy-5,6,7,8-tetra- metho~yflavone.~~ A related 5,7,8-trimethoxy 6-hydroxyflavone exhibited similar activity, while isoflavones (phenyl ring at C-3 instead of C-2) and coumarins (carbonyl transposed from C-4 to C-2) were inactive.94
In another series, best activity against HLAR (10 x quercetin) was found in a 3-0,acetyl-7,3’,4’,-trisulfate derivative of quercetin, the aglycone of quer- citrin.” Inhibition of RLAR, BLAR, and HLAR by quercetin has been reported to be noncompetitive with respect to substrate and c o f a ~ t o r . ~ ~ , ~ ~ In a study with the water soluble 7-O-(P-hydroxyethyl)-quercetin and 5,7,3‘,4‘-tetra-O-@- hydroxyethy1)-rutin, which are established RLAR inhibitor^,^^ no inhibitory activity was observed on sorbitol accumulation in cultured monkey kidney epithelial cells,% in contrast to the good activity observed with the carboxylic acid inhibitor ICI-105,552.
Not all phenolic AR inhibitors are as structurally complex as the flavonoids; thus a synthetic relatively simple tetrahydroxy benzophenone, 70A196 (62),
62
with RLAR inhibitory activity 10 x quercetin inhibited cataract formation in diabetic rats fed a xylose diet containing 2% of drug.96
Among natural products with folk medical uses, the flavonoid silybin in- hibits HPAR.97 Brazilin and haematoxylin (631, plant pigments with some
no R = H Brazilin &: / 0 R =OH Haemmatoxylin
no n
63
structural resemblance to 62, were found to be very weak inhibitors (IC-50 .01 x 1P) of BLAR. It is interesting to note that brazilin is the main phenolic constituent of the aqueous extract of sappan wood, which is used for treatment of diabetic complications in Southern Korean folk medicine.98
MEDICINAL CHEMISTRY 181
VIII. NON-ACIDIC AND COMPETITIVE INHIBITORS
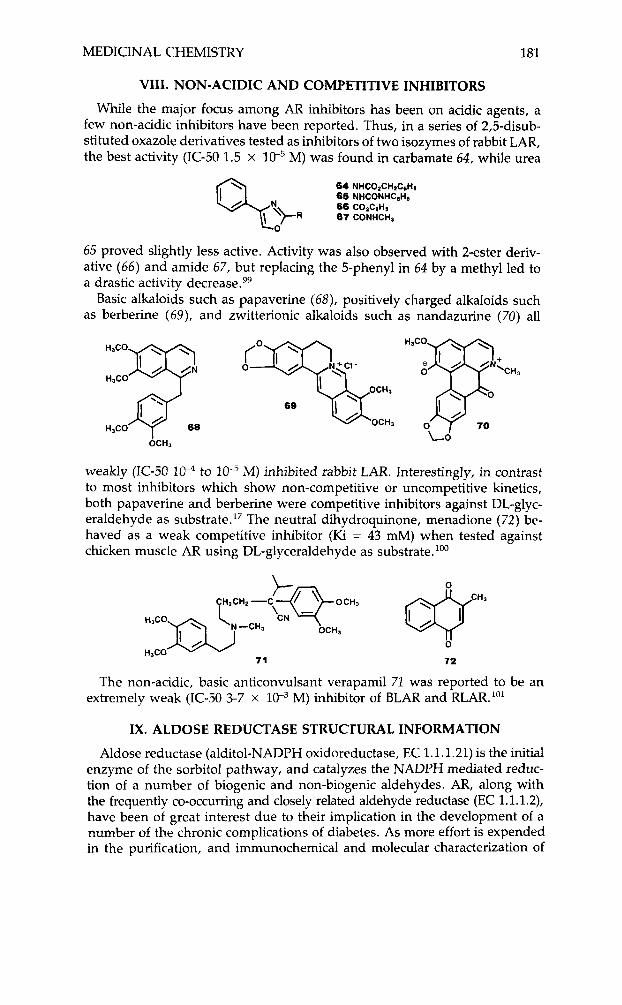
While the major focus among AR inhibitors has been on acidic agents, a few non-acidic inhibitors have been reported. Thus, in a series of 2,5-disub- stituted oxazole derivatives tested as inhibitors of two isozymes of rabbit LAR, the best activity (IC-50 1.5 x l W 5 M) was found in carbamate 64, while urea
64 NHCO&H&eHa 65 NHCONHCllHs
q ' > R 66 87 COiCiHS CONHCH,
0
65 proved slightly less active. Activity was also observed with 2-ester deriv- ative ( 6 6 ) and amide 67, but replacing the 5-phenyl in 64 by a methyl led to a drastic activity decrease.99
Basic alkaloids such as papaverine (68), positively charged alkaloids such as berberine (69), and zwitterionic alkaloids such as nandazurine (70) all
'CH, H,CO
69
OCHi 0 70 L O
H,CO
OCH,
weakly (IC-50 1 P to M) inhibited rabbit LAR. Interestingly, in contrast to most inhibitors which show non-competitive or uncompetitive kinetics, both papaverine and berberine were competitive inhibitors against DL-glyc- eraldehyde as ~ubstrate. '~ The neutral dihydroquinone, menadione (72) be- haved as a weak competitive inhibitor (Ki = 43 mM) when tested against chicken muscle AR using DL-glyceraldehyde as substrate. loo
0 HJCO
71 72
The non-acidic, basic anticonvulsant verapamil 71 was reported to be an extremely weak (IC-50 3-7 x M) inhibitor of BLAR and RLAR."'
IX. ALDOSE REDUCTASE STRUCTURAL INFORMATION
Aldose reductase (alditol-NADPH oxidoreductase, EC 1.1.1.21) is the initial enzyme of the sorbitol pathway, and catalyzes the NADPH mediated reduc- tion of a number of biogenic and non-biogenic aldehydes. AR, along with the frequently co-occurring and closely related aldehyde redudase (EC 1.1.1 .Z), have been of great interest due to their implication in the development of a number of the chronic complications of diabetes. As more effort is expended in the purification, and immunochemical and molecular characterization of

182 LIPINSKI et al.
these proteins, it is becoming apparent that a large number of other reductases previously described in the older literature are identical to these enzymes. Excellent reviews of this subject have been p u b l i ~ h e d , ' ~ ~ , ~ ~ ~ and the reader is referred to these for more detailed discussion. This section will focus instead on the characterization of aldose reductase from a molecular perspective.
Aldose reductase is a monomeric NADPH-dependent oxidoreductase with a fairly broad substrate specificity and commonly co-occurs with the closely related, but immunochemically distinct aldehyde reductase. lo4-lo6 Although it does exhibit substantial substrate overlap with aldehyde reductase, aldose reductase generally shows a higher substrate affinity (lower Km) and a higher catalytic efficiency in processing simple aldoses (glucose, xylose, glyceral- dehyde). The broad substrate recognition of these enzymes implies a large substrate binding domain.
Aldose reductase also shows a high affinity for biogenic aldehydes other than aldoses (some arylacetaldehydes and isocorticosteroids); however, the physiological significance of these observations has not yet been conclusively demonstrated and caution should be exercised in applying teleological inter- pretations. lo7-'09
The estimates of affinity of pure aldose reductase for its endogenous sub- strate glucose vary, and may represent variations in the purity of enzyme utilized for those determinations. A careful analysis of kinetic parameters for the reduction of both anomeric forms of D-glucose by homogeneous bovine lens aldose reductase, shows that the enzyme has an affinity (Km = 0.66 pM) for the chemically relevant aldehydo form, commensurate with the phys- iological levels of that substrate.'1° No mutarotase activity has been demon- strated for the enzyme, although the initial step of hemi-acetal opening might be a property of the protein, which could be masked by subsequent catalytic turnover to polyol. The high affinity of aldose reductase for sugar substrates which show reduced propensity to form cyclic hemi-acetals provides addi- tional evidence for the more favored processing of the aldehydo form. This may give some insight into the character of the substrate binding domain.
NADPH affinities of the two enzymes are similar. Aldose reductase can also function with NADH as a hydride source, although with two to three orders of magnitude lower affinity relative to NADPH. Aldehyde reductase has a strict requirement for NADPH.11'-113
Kinetic analyses of the turnover of sugar aldehydes to polyols by aldose reductase have shown biphasic behavior, and it is possible that these results may have been due to the presence of multiple forms of carbonyl reductases. Early preparations of the enzyme may have been complicated by protease modification of the protein during purification114 and/or oxidative sulfhydryl modification. 'I5 Other explanations have been advanced for the non-linear kinetics observed for the enzyme, including cooperative effects116 and inter- fering non-enzymatic substrate reaction^."^ The biphasic behavior of the en- zyme has also been attributed to activation by the presence of glucose, glucose- 6-phosphate, and NADI'H.'18,119 These authors have also proposed that al- dose reductase is a monomeric protein (alpha subunit), and one of two human aldehyde reductases is a heterodimer of aldose reductase and another protein (beta subunit), which itself is devoid of enzyme activity. A third monomeric protein represents the second form of human aldehyde reductase.'*O
Highly purified aldose reductase shows apparent ordered bi-bi kinetics, al-

MEDICINAL CHEMISTRY 183
though this may vary with the source of the protein and ~ubstrate.”~The mech- anism of substrate turnover is still debated, but it is likely that initial binding of co-factor precedes substrate binding, followed by reduction, product release, and expulsion of spent co-factor. Hydride transfer from the co-factor has been cor- related with isocitrate dehydrogenase (EC 1.1.1.42) and glucose-6-phosphate dehydrogenase (EC 1.1.1.49) and has been shown to involve transfer of the pro- 4R hydrogen, leading to the classification of aldose reductase as an A-type en- zyme.”’ A study of the reduction of D-glyceraldehyde by glycerol dehydrogen- ase (aldose reductase) derived from rabbit muscle was correlated by elegant dou- ble labelling experiments with aldolase and triosephosphate isomerase. The reduction was shown to proceed by delivery of the pro-4R hydride of NADPH to the re-face of the carbonyl.’22 The presentation of the aldehyde and orientation of the substrate are, therefore, well-defined.
Covalent modification/protection studies have yielded a crude picture of the substrate binding domain, which can be summarized as two anion-binding sites, and a hydrophobic region.lZ3 The co-factor binding domain appears to contain basic amino acid side chains (lysine, arginine, and a histidine)lo3 and the presence of a free protein cysteine residue.lz4 Until more detailed struc- tural information is forthcoming (modelling from primary sequence or ideally, single crystal x-ray analysis), one may only speculate as to the gross topology of the protein.
Physicochemical characterization of bovine lens aldose reductase (sedi- mentation, circular dichroism behavior) points to a near spherical shape, with a fairly low alpha-helical content (ca. 5%).ll6 The enzyme used in this study showed biphasic, double reciprocal plot kinetics, which may be due to the presence of isoenzymes of aldose reductase as is seen in bovine liver.lZ5 Nevertheless, the low alpha-helical content and spherical nature is consistent with the relatively high proline content found in the aldose/aldehyde reduc- tases. 108,1267127 One may speculate that the nucleotide binding domain is as- sociated with a “dinucleotide binding helix,” a compact region of beta-alpha- beta secondary structure conserved in a number of dinucleotide binding en- zymes.128 When primary sequence information is available for the protein, the presence of this structural feature will be readily discerned.
A great deal of interest has been generated around the characterization of inhibitor binding to aldose reductase. Given the uncertainities mentioned above with regard to the enzyme kinetics observed in the processing of sub- strate and co-factor, it is even more problematic to consider the characteri- zation of individual inhibitor kinetics as a straightforward undertaking. Recent attempts at defining ”the” aldose reductase inhibitor binding site are seriously compromised by the lack of evidence that the structurally unrelated inhibitors considered in the analysis bind in a similar fashion to the enzyme.14 Minor variations in close structurally related inhibitors can lead to pronounced al- terations in the sense of binding, as shown by Matthews in high-resolution x-ray crystallographic studies of the binding of the inhibitors of therm~lysin.”~
REFERENCES 1. P. F. Kador, W. G. Robison, Jr., and J. H. Kinoshita, Ann. Rev. Pharmacol. Toxicol., 25, 691
2. P. F. Kador, Ophthalmol., 90, 352 (1983). 3. P. F. Kador, J. H. Kinoshita, and N. E. Sharpless, 1. Med. Chem., 28, 841 (1985).
(1985).

184 LIPINSKI et al.
4. C. A. Lipinski and N. 1. Hutson, Ann. Rep. Med. Chem.. 19, 169 (1984). 5. Metabolism Clinical and Experimental, 35, No. 4, Suppl. 1, April 1986. 6. S. Hayrnan and J. H. Kinoshita, 1. Biol. Chem., 240, 877 (1965). 7. J. H. Kinoshita, D. Dvornik, M. Kraml, and K. H. Gabbay, Biochim. Biophys. Acta, 158, 472
8. J. C. Hutton, P. J. Schofield, J. F. Williams, and F. C. Hollows, Biochem. Pharmacol., 23,
9. R. Sarges and M. J. Peterson, Metabolism, 35, No. 4, Suppl. 1, 101 (1986).
(1968).
2991 (1974).
10. M. J. Peterson, R. Sarges, C. E. Aldinger, and D. l’. MacDonald, Metabolism, 28, Suppl. 1,
11. B. L. Mylari, M. W. Miller, H. L. Howes, S. K. Figdor, J . E. Lynch, and R. C. Koch, 1. Med.
12. D. Dvornik, N. Sirnard-Duquesne, M. Kraml, K. Sestanj, K. H. Gabbay, J. H. Kinoshita,
13. R. Sarges, J. Bordner, 8 . W. Dominy, M. J. Peterson, and E. B. Whipple, 1. Med. Chem.,
14. P. F. Kador and N. E. Sharpless, Mol. Pharmacol., 24, 521 (1983). 15. R. A. Ronfeld, Pfizer Central Research, Groton, CT, unpublished observations. 16. Y. Kawakarni, Y. Nagai, H. Ono, K. Ueda, T. Sato, and S. Tanaka, Heterocycles, 21, 583
17. N. Nakai, Y. Fujii, K. Kobashi, and K. Nomura, Arch. 13iochem. Biophys., 239, 491 (1985). 18. H. Ono, Y. Nozawa, and S. Hayano, Nippon Ganku Gakkui Zasshi, 86, (9), 253 (1982). 19. R. C. Schnur, R. Sarges, and M. J. Peterson, I. Med. Chem., 25, 1451 (1982). 20. A. B. Richon, M. E. Maragoudakis, and J. S. Wasvary, J. Med. Chem., 25, 745 (1982). 21. D. R. Brittain, Spec. Pub1.-R. SOC. Chem. 1986,55 (SCI-RSC Med. Chem. Syrnp., 3rd.’ 1985),
22. 8 . W. Griffin, M. L. Chandler, and L. DeSantis, ARVO Abstracts. Invest. Ophthalmol. Vis.
23. B. M. York, Jr., European Patent 0 092 385, April 14, 1983. 24. 8 . M. York, Jr., United States Patent 4,438,272, March 20, 1984. 25. P. Miiller, 0. Hockwin, and C. Ohrloff, Ophthalmic Res., 17, 115 (1985). 26. B. M. York, Jr., European Patent 0 137 333, Dec. 9, 1984. 27. T. Sohda, K. Mizuno, E. Irnamiya, H. Tawada, K. Meguro, Y. Kawamatsu and Y. Yamamoto,
28. K. Inagaki, I. Miwa, Y. Yashiro, and J. Okuda, Chem. Pharm. Bull., 30, 3244 (1982). 29. R. Kikkawa, I. Hatanaka, and Y. Shigeta, Frontiers in Diabetes, Diabetic Microangiopathy, H.
30. K. Mizuno, T. Sohda, K. Meguro, and Y. Kawamatsu, I . Takeda Res. Lab., 42, 227 (1983). 31. T. Doi, T. Tsukarnoto, H. Torii, K. Yoshida, and S. Tanayama, 1. Takeda Res. Lab., 43, 19
32. I. Miwa, K. Inagaki, T. Yashiro, and J. Okuda, Chem. Pharm. B u L , 32, 2030 (1984). 33. J. Okuda, K. Yashima, K. Inagaki, and I. Miwa, Chem. Pharm. Bull., 33, 2990 (1985). 34. K. Sestanj, F. Bellini, S. Fung, N. Abraham, A. Treasurywala, L. Humber, N. Simard-
35. H-K Lee and G. Querijero, 1. Pharm. Sci., 74, 273 (1985). 36. N. Simard-Duquesne, E. Greselin, J. Dubuc, and D. Dvornik, Metab. Clin. Exp., 34,885 (1985). 37. N. Simard-Duquesne, E. Greselin, R. Gonzalez, and D. Dvornik, Proc. Soc. E x p . Biol. Med.,
178, 599 (1985). 38. W. G. Robison, P. F. Kador, Y. Akagi, J. H. Kinoshita, R. Gonzalez, and D. Dvornik,
Diabetes, 35, 295 (1986). 39. D. R. Hicks, M. Kraml, M. N. Cayen, J. Dubuc, S. Ryder, and D. Dvornik, Clin. Pharmacol.
Ther., 36, 493 (1984). 40. M. N. Cayen, D. R. Hicks, E. S. Ferdinandi, M. Krarnl, E. Greselin, and D. Dvornik, Drug
Metab. Disp., 13, 412 (1985). 41. D. R. Hicks, M. Kraml, M. N. Cayen, J. Dubuc, S. Ryder, and D. Dvornik, Clin. Pharmacol.
Ther., 36, 493 (1984). 42. D. Dvornik, R. Gonzalez, D. Hicks, T. Smith, S. Ryder, J. F. Muilane, P. Raskin, J. Rosen-
stock, P. Challis, J. Caro, and W. Fore, Diabetes Research and Clinical Practice, Suppl. 1,5146 (1985).
456 (1979).
Chem., 20, 475 (1977).
S. D. Varma, and L. 0. Meroia, Science, 182, 1146 (1973).
28, 1716 (1985).
(1984).
210-240.
Sci. 26 (Suppl.), 136 (1986).
Chem. Pharm. Bull. , 30, 3601 (1982).
Abe and M. Hoshi, Eds., Karger, Basel, New York, 1983, Vol. 3, p. 195.
(1984).
Duqeusne, and D. Dvornik, 1. Med. Chem., 27, 255 (1984).

MEDICINAL CHEMISTRY 185
43. L. Koglin, C. Clark, S. Ryder, and J. F. Mullane, Diabetes, 34, Suppl. 1,202A (1985). 44. J. Fagius and S. Jameson, 1. Neurol. Neurosurg. Psychiatry, 44, 941 (1981). 45. D. J. Handelsman and J. R. Turtle, Diabetes, 30, 459 (1981). 46. A. Culebras, J. Alio, J. L. Herrara, and M. I. Lopez-Fraile, Arch. Neurol., 38,133 (1981). 47. P. F. Kador, J. D. Goosey, N. E. Sharpless, J. Kolish, and D. Miller, Eur. 1. Med. Chem.,
48. D. Stribling, and D. R. Brittain, Znnovative Approaches in Drug Research, A. F. Harms, Ed.,
49. D. R. Tomlinson, J. Townsend, and P. Fretten, Diabetes, 34, 970 (1985). 50. D. Stribling, D. J. Mirrlees, H. E. Harrison, and D. C. N. Earl, Metabolism, 34, 336
51. D. Stribling, D. J. Mirrlees, and D. C. N. Earl, Diabetologia, 25, 196 (1983). 52. S. C. Norris, C. M. Perkins, W. Bastian, D. Mirrlees, and R. A. Yates, Clin. Sci., 69, Suppl.
53. R. Poulsom and H. Heath, Biochem. Pharmacol., 32, 1495 (1983). 54. R. Boot-Hanford and H. Heath, Biochem. Pharmacol., 30, 3065 (1981). 55. R. Poulsom, R. P. Boot-Hanford, and H. Heath, Exp. Eye. Res., 37, 507 (1983). 56. H. Terashima, K. Hama, R. Yamamoto, M. Tsuboshima, R. Kikkawa, I. Hatanaka, and Y.
57. R. Kikkawa, I. Hatanaka, H. Yasuda, N. Kobayashi, Y. Shigeta, H. Terashima, T. Morimura,
58. R. Kikkawa, I. Hatanaka, H. Yasuda, N. Kobayashi, and Y. Shigeta, Metabolism, 33, 212
59. N. Hotta, H. Kakuta, H. Fukasawa, M. Kimura, N. Koh, M. Iida, H. Terashima, T. Mor-
60. K. Kojima, H. Matsubara, T. Harada, K. Mizuno, J. Suzuki, N. Hotta, H. Kakuta, and N.
61. N. Kaman, H. Mabuchi, and R. Takeda, Diabetes Res. Clin. Pract., 1 (Suppl.), 251 (1985). 62. G. Okuno, A. Oki, T. Yamazaki, S. Shibamoto, H. Fukuda, T. Kashiwara, K. Sakuyama,
63. E. Kato, K. Yamamoto, T. Baba, T. Watanabe, Y. Kawashima, H. Masuda, M. Horiuchi,
64. E. Kato, K. Yamamoto, T. Baba, T. Watanabe, Y. Kawashima, M. Horiuch, M. Oya, T. Iso,
65. A. N. Brubaker, J. DeRuiter, and W. L. Whitmer, J . Med. Chem., 29, 1094 (1986). 66. W. W. Van Den Hoven, D. W. R. Hall, and J:W. Burns, Br. 1. Clin. Practice, (Symp. Suppl.),
67. Y. R. Sharma and E. Cotlier, Exp. Eye. Res., 35, 21 (1982). 68. M. Jacobson, Y. R. Sharma, E. Cotlier, and J . D. Hollander, Invest. Ophthalmol. Vis. Sci.,
69. W. F. Williams and J. D. Odom, Science, 233, 223 (1986). 75. M. J. C. Crabbe, G. Freeman, A. 8. HaIder, and A. J. Bron, O ~ h t h a ~ m ~ c Res., 17, 85 (1985). 71. J. G. Cunha Vaz, C. C. Mota, E. C. Leite, J. 8. Abreu, and M. A. Ruas, Arch. Ophthalmol.,
72. J. G. Cunha Vaz, C. Mota, E. Leite, J. R. Faria Abreu, and M. A. Ruas, Diabetes, 34, Suppl.
73. E. Cotlier and Y. R. Sharma, Lancet, 1, 358 (1981). 74. E. Cotlier, Y. R. Sharma, T. Niven, and M. Brescia, Am. j . Med., 74(6A), 83 (1983). 75. E. Cotlier and Y. R. Sharma, Lancet, 1, 607 (1980). 76. J. F. Day, R. W. Thornberg, S. R. Thorpe, and J. W. Baynes, J . B i d . Chem., 254,9394 (1979). 77. G. N. Rao and E. Cotlier, Invest. Ophthalmol. Vis. Sci., 27, 98 (1986). 78. V. M. Monnier, V. Vishwanath, K. E. Frank, C. A: Elmets, P. Dauchot, and R. R. Kohn,
N. Engl. J . Med., 314, 403 (1986); Editorial, Lancet, 1, 1192 (1986). 79. M. Testa, G. Iuliano, and B. Silvestrini, Lancet, 1, 849 (1982). 80. L. Bonomi, G. Marchini, I. DeFranco, S. Perfetti, and M. DeGregoyio, Curr. Ther. Res., 33,
81. B. Silvestrini, B. Catanese, G. Barrillari, E. Iorio, and P. Valeri, Int. 1. Tiss. Reac., V, 217
82. L. Pandolfo, M. A. Livrea, and A. Bono, Exp. Eye. Res., 42, 167 (1986).
16, 293 (1981).
Elsevier, Amsterdam, 1986, p. 297.
(1985).
12, 22P, 1985.
Shigeta, 1. Pharrn. Exp. Therapeut., 229, 226 (1984).
and M. Tsuboshima, Diubetologia, 24, 290 (1983).
(1984).
imura, and N. Sakamoto, Diabetologiu, 28, 176 (1985).
Sakamoto, Jpn. 1. Ophthalmol., 29, 99 (1985).
F. Kawakami, and H. Tako, Diabetes Res. Clin. Pract., 1 (Suppl.), 420 (1985).
M. Oya, T. Iso, and J.4. Iwao, Chem. Pharrn. Bull., 33, 74 (1985).
and J.-I. Iwao, Chem. Pharrn. Bull., 33, 5341 (1985).
34, 31 (1984).
24, 1426 (1983).
103, 1307 (1985).
1, 109A (1985).
727 (1983).
(1983).

186 LIPINSKI et al.
83. J . R. Pfister, W. E. Wymann, J. M. Mahoney, and L. D. Waterbury, 1. Med. Chem., 23,1264
84. A. Beyer-Mears, E. Cruz, J , Nicolas-Alexandre, and E. Yaragiannis, Arch. lnt. Pharmacodyn.
85. G. J. White, Agents Actions, 11, 503 (1981). 86. P. F. Kador, N. E. Sharpless, and J , D. Goosey, Prog. Clin. Biol. Res., 114, 243 (1982). 87. B. V. Cheney, J. B. Wright, C. M. Hall, and R. E. Christofferson, 1. Med. Chem., 21,980 (1978). 88. P. F. Kador and N. E. Sharpless, Biophys. Chem., 8, 81 (1978). 89. S. R. Whittle and A. J. Turner, Biochem. Pharmacol., 11, 1191 (1981). 90. M. Shimizu, T. Ito, S. Terashima, T. Hayashi, M. Arisawa, N. Morita, S. Kurokawa, K. Ito,
91. P. S. Chaudhry, J. Cabrera, H. R. Juliani, and S. D. Varma, Biochem. Pharmacol., 32, 1995
92. S. D. Varma, A. Mizuno, and J. H. Kinoshita, Science, 195, 205 (1977). 93. J . Okuda, I. Miwa, K. Inagaki, T. Horie, and M. Nakayama, Chem. Pharm. Bull., 32, 764
94. J. Okuda, I. Miwa, K. Inagaki, T. Horie, and M. Nakayama, Biochem. Pharmacol., 31, 3807
95. S. D. Varma and J . H. Kinoshita, Biochem. Pharmacol., 25, 2505 (1976). 96. H. Ono and S. Hayano, Nippon Ganka, Gakkui Zasshi, 86,(4), 353 (1982). 97. J. Santos, L. 8 . A. Mira, A. M. Freire, M. Azevedo, and C. Manso, Acta. Med. Portuguesa,
98. C. K. Moon, Y. P. Yun, J. H. Lee, H. Wagner, and Y. S. Shin, Planta. Med., 1, 66 (1985). 99. T. Tanimoto, H. Fukuda, J. Kawamura, M. Nakao, U. Shimada, A. Yamada, and C. Tanaka,
Chem. Pharm. Bull., 32, 1032 (1984). 100. W. S. Davidson and D. G. Murphy, Enzymology of Carbonyl Metabolism 2: Aldehyde Dehydro-
genase, Aldo-Keto Reductase, and Alcohol Dehydrogenase, Alan R. Liss, New York, 1985, p. 251. 101. P. Muller, 0. Hockwin, and C. Ohrloff, Ophthalmic Res., 17, 115 (1985). 102. G. Flynn, Biochem. Pharmacol., 31, 2705 (1982). 103. B. Wermuth, Prog. Clin. B i d . Res., 174, 209 (1985). 104. H. B. Markus, M. Raducha, and H. Harris, Biochem. Med., 29, 31 (1983). 105. H.-P. Wirth and B. Wernuth, Prog. Clin. B i d . Res., 174, 231 (1985). 106. H.-P. Wirth and B. Wernuth, FEBS Lett., 187, 280 (1985). 107. 8 . Wermuth and C. Monder, Eur. 1. Biochem., 131, 423 (1983).
(1980).
Ther., 259, 166 (1982).
and Y. Hashimoto, Phytochemistry, 23, 1885 (1984).
(1983).
(1984).
(1982).
5, 115 (1984).
108. 8 . Wermuth, H.-P. Burgisser, K. Bohrn, and J. P. von Wartburg, Eur. 1. Biochem., 127, 279 (1982).
109. J. A. Cromlish and T. G. Flynn, Ophthalmic Res., 17, 185 (1985). 110. K. Inagaki, I. Miwa, and J. Okuda, Arch. Biochem. Biophys., 216, 337 (1982). 111. J. A. Cromlish and T. G. Flynn, 1. Biol. Chem., 258, 3583 (1983). 112. P. R. Hoffman, B. Wermuth, and J. P. vonwartburg, J. Neurochem., 35, 354 (1980). 113. A. K. Daly and T. J . Mantle, Biochem. j . , 205, 373 (1982). 114. J. A. Cromlish and T. G. Flynn, Biochem. J . , 209, 597 (.L983). 115. B. Wermuth, J. D. B. Munch, and J. P. von Wartburg, 1. B i d . Chem., 352, 3821 (1977). 116. C. M. Sheaff and C. C. Doughty, J . B id . Chem., 251, 2696 (1976). 117. A. B. Halder and M. J. C. Crabbe, Biochern. j . , 219,33 (1984). 118. B. Das and S . K. Srivastava, Diabetes, 34, 1145 (1985). 119. S. K. Srivastava, G. A. Hair, and 8. Das, Proc. Natl. Acad. Sci. U S A , 82, 7222 (1985). 120. S. K. Srivastava, B. Das, G. A. Hair, R. W. Gracy, S. Awasthi, N. H. Ansarai, and M. J.
Petrash, Biochim. Biophys. Acta., 840, 334 (1985). 121. H. 8. Feldman, P. A. Szczepanik, P. Havre, R. J. M. Corrall, L. C. Yu, H. M. Rodman,
8. A. Rosner, P. D. Klein, and 8. R. Landau, Biochem. Biophys. Acta., 480, 14 (1977). 122. D. J. Walton, Biochemistry, 12, 3472 (1973). 123. G. Branlant, E u r . /. Biochem., 121, 407 (1982). 124. A. 8. Halder and M. J. C. Crabbe, Ophthalmic Res., 17, 185 (1985). 125. K. H. Gabbay and E. S. Cathcart, Diabetes, 23, 460 (1974). 126. T. G. Flynn, J. Shires, and D. J. Walton, 1. B i d . Chem.. 250, 2933 (1975). 127. J. A. Cromlish and T. G. Flynn, 1. Biol. Chem., 258, 3416 (1983). 128. R. K. Wierenga, M. C. H. DeMaeyer, and W. G. J. Hal, Biochemistry, 24, 1346 (1985). 129. W. R. Kester, and B. W. Matthews, Biochemistry, 16, 2506 (1977).
Related Documents









![HMG CoA reductase inhibitors [statins] for dialysis patients178940/UQ178940_OA.pdf · HMG CoA reductase inhibitors (statins) for dialysis patients Sankar D Navaneethan1, Rakesh Shrivastava2](https://static.cupdf.com/doc/110x72/5f0740f07e708231d41c12a3/hmg-coa-reductase-inhibitors-statins-for-dialysis-patients-178940uq178940oapdf.jpg)

