ĐẠI HỌC HUẾ TRƯỜNG ĐẠI HỌC KHOA HỌC NGUYỄN MINH THÔNG NGHIÊN CỨU CẤU TRÚC, KHẢ NĂNG CHỐNG OXY HÓA CỦA MỘT SỐ POLYPHENOL VÀ DẪN XUẤT TRÊN NỀN FULLERENE (C 60 ) BẰNG PHƯƠNG PHÁP HÓA TÍNH TOÁN LUẬN ÁN TIẾN SĨ HÓA LÝ THUYẾT VÀ HÓA LÝ HUẾ, NĂM 2016

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ĐẠI HỌC HUẾ
TRƯỜNG ĐẠI HỌC KHOA HỌC
NGUYỄN MINH THÔNG
NGHIÊN CỨU CẤU TRÚC, KHẢ NĂNG CHỐNG
OXY HÓA CỦA MỘT SỐ POLYPHENOL VÀ DẪN
XUẤT TRÊN NỀN FULLERENE (C60) BẰNG
PHƯƠNG PHÁP HÓA TÍNH TOÁN
LUẬN ÁN TIẾN SĨ HÓA LÝ THUYẾT VÀ HÓA LÝ
HUẾ, NĂM 2016

ĐẠI HỌC HUẾ
TRƯỜNG ĐẠI HỌC KHOA HỌC
NGUYỄN MINH THÔNG
NGHIÊN CỨU CẤU TRÚC, KHẢ NĂNG CHỐNG
OXY HÓA CỦA MỘT SỐ POLYPHENOL VÀ DẪN
XUẤT TRÊN NỀN FULLERENE (C60) BẰNG
PHƯƠNG PHÁP HÓA TÍNH TOÁN
Chuyên ngành: Hóa lý thuyết và Hóa lý
Mã số: 62.44.01.19
LUẬN ÁN TIẾN SĨ HÓA LÝ THUYẾT VÀ HÓA LÝ
Cán bộ hướng dẫn khoa học:
1. PGS. TS. Phạm Cẩm Nam
2. PGS. TS. Trần Dương
Huế, năm 2016

i
LỜI CAM ĐOAN
Tôi cam đoan đây là công trình nghiên cứu của riêng tôi. Các kết quả nghiên
cứu và các kết luận trong luận án này là trung thực, được các đồng tác giả cho phép
sử dụng và chưa từng công bố trong bất kỳ một công trình nào khác. Việc tham khảo
các nguồn tài liệu đã được trích dẫn và ghi nguồn tài liệu tham khảo đúng quy định.
Tác giả
Nguyễn Minh Thông

ii
LỜI CẢM ƠN
Đầu tiên, tôi xin chân thành bày tỏ lòng biết ơn sâu sắc tới PGS.TS. Phạm Cẩm
Nam và PGS. TS. Trần Dương, những người Thầy đã tận tình hướng dẫn, giúp đỡ,
dành nhiều thời gian và công sức hướng dẫn cho tôi hoàn thành luận án.
Lời cảm ơn của tôi cũng xin được gửi đến TS. Đào Duy Quang, TS. Ngô Thị
Chinh, TS. Phạm Lê Minh Thông ở Đại học Duy Tân và các anh chị trong nhóm
nghiên cứu đã dành thời gian thảo luận khoa học và đóng góp các ý kiến quý báu cho
tôi hoàn thành luận án.
Tôi xin gửi lời cảm ơn chân thành tới quý Cô, Thầy trong khoa Hóa, phòng Đào
tạo Sau đại học, Trường Đại học Khoa học - Đại học Huế và Ban Giám đốc Phân
hiệu Đại học Đà Nẵng tại Kon Tum đã tạo mọi điều kiện thuận lợi để tôi thực hiện
luận án này.
Tôi xin chân thành cảm ơn Quỹ Phát triển Khoa học và Công nghệ Việt Nam
(Vietnam National Foundation for Science and Technology Development −
Nafosted) đã hỗ trợ một phần kinh phí thực hiện luận án.
Xin cảm ơn tất cả bạn bè, đồng nghiệp đã động viên và giúp đỡ tôi trong suốt
thời gian thực hiện luận án.
Nhân dịp này, tôi muốn dành những tình cảm sâu sắc nhất, trân trọng nhất đến
những người thân trong gia đình: Bố Mẹ - những người đã hết lòng nuôi dạy tôi khôn
lớn, luôn động viên hỗ trợ tôi về mọi mặt, các anh chị em đã chia sẻ những khó khăn,
thông cảm và giúp đỡ tôi trong cuộc sống.
Huế, tháng năm 2017
Tác giả
Nguyễn Minh Thông

iii
MỤC LỤC
DANH MỤC CÁC CHỮ VIẾT TẮT ...................................................................... vii
DANH MỤC CÁC BẢNG........................................................................................ ix
DANH MỤC CÁC HÌNH ......................................................................................... xi
MỞ ĐẦU ..................................................................................................................... 1
CHƯƠNG 1. TỔNG QUAN VỀ HỆ CHẤT NGHIÊN CỨU .................................... 5
1.1. TỔNG QUAN VỀ HỢP CHẤT POLYPHENOL CÓ TÁC DỤNG CHỐNG
OXY HÓA ............................................................................................................... 5
1.1.1. Hợp chất polyphenol .................................................................................. 5
1.1.2. Một số nguyên liệu giàu hợp chất polyphenol ở Việt Nam ..................... 10
1.1.3. Cơ chế hoạt động của các hợp chất chống oxy hóa polyphenol .............. 12
1.1.4. Tình hình ứng dụng hóa học tính toán trong nghiên cứu khả năng chống
oxy hóa của hợp chất polyphenol ...................................................................... 13
1.2. TỔNG QUAN VỀ HỢP CHẤT FULLERENE VÀ DẪN XUẤT
FULLERENE ........................................................................................................ 15
1.2.1. Lịch sử phát hiện fullerene ...................................................................... 15
1.2.2. Cấu trúc và liên kết của fullerene ............................................................ 15
1.2.3. Tính chất vật lý ........................................................................................ 17
1.2.4. Đặc điểm electron và khả năng phản ứng ................................................ 19
1.2.5. Tính thơm ................................................................................................ 20
1.2.6. Tính chất hóa học ..................................................................................... 20
1.2.7. Tình hình nghiên cứu fullerene và dẫn xuất fulerene .............................. 22
CHƯƠNG 2. NỘI DUNG VÀ PHƯƠNG PHÁP NGHIÊN CỨU ........................... 23
2.1. NỘI DUNG NGHIÊN CỨU .......................................................................... 23
2.2. TỔNG QUAN VỀ PHƯƠNG PHÁP TÍNH TOÁN ...................................... 23
2.2.1. Cơ sở phương pháp tính toán hóa lượng tử ............................................. 23
2.2.2. Phương pháp bán thực nghiệm (semi-empirical methods) ...................... 26
2.2.3. Phương pháp lý thuyết phiếm hàm mật độ (Density Functional Theory -
DFT methods) .................................................................................................... 27

iv
2.2.4. Phương pháp ONIOM ............................................................................. 28
2.3. CÁC PHẦN MỀM TÍNH TOÁN ................................................................... 31
2.3.1. Phần mềm Gaussian 09W ........................................................................ 31
2.3.2. Phần mềm Gaussview 5 ........................................................................... 31
2.4. HÓA HỌC TÍNH TOÁN ỨNG DỤNG CHO NGHIÊN CỨU CÁC HỢP
CHẤT POLYPHENOL VÀ DẪN XUẤT FULLERENE .................................... 32
2.4.1. Tối ưu hóa cấu trúc (geometry optimization) .......................................... 32
2.4.2. Xác định trạng thái chuyển tiếp và tính hàng rào năng lượng ................. 32
2.4.3. Năng lượng điểm đơn (single point energy) ............................................ 34
2.4.4. Các đại lượng nhiệt động xác định khả năng chống oxy hóa của hợp chất
polyphenol ......................................................................................................... 35
2.4.5. Mô hình hóa các hệ phân tử trong dung môi ........................................... 36
CHƯƠNG 3. KẾT QUẢ VÀ THẢO LUẬN ............................................................ 40
3.1. KHẢO SÁT PHƯƠNG PHÁP TÍNH NĂNG LƯỢNG PHÂN LY LIÊN KẾT
O−H ....................................................................................................................... 40
3.1.1. Cách chọn mô hình ONIOM 2 lớp .......................................................... 40
3.1.2. Áp dụng cho các dẫn xuất của phenol ..................................................... 42
3.1.3. Áp dụng cho các hợp chất flavonoid ....................................................... 45
3.1.4. Nhận xét ................................................................................................... 50
3.2. KHẢO SÁT PHƯƠNG PHÁP TÍNH NĂNG LƯỢNG ION HÓA .............. 50
3.2.1. Giới thiệu ................................................................................................. 50
3.2.2. Đánh giá tính chính xác của phương pháp tính IE ở mức lý thuyết PM6
........................................................................................................................... 51
3.2.3. Nhận xét ................................................................................................... 52
3.3. KHẢ NĂNG CHỐNG OXY HÓA CỦA CÁC HỢP CHẤT POLYPHENOL
CÓ NGUỒN GỐC TỪ CÂY SA KÊ (ARTOCARPUS ALTILIS) ......................... 53
3.3.1. Lựa chọn một số hợp chất polyphenol có chứa trong cây sa kê .............. 53
3.3.2. Cơ chế chuyển nguyên tử hydro (HAT) - Năng lượng phân ly liên kết
(BDE) ................................................................................................................. 54
3.3.3. Cơ chế chuyển electron chuyển proton (SET-PT) ................................... 57

v
3.3.4. Nhận xét ................................................................................................... 60
3.4. KHẢ NĂNG CHỐNG OXY HÓA CỦA CÁC HỢP CHẤT POLYPHENOL
CÓ NGUỒN GỐC TỪ VỎ MĂNG CỤT (GARCINIA MANGOSTANA) ............ 61
3.4.1. Các hợp chất polyphenol có chứa trong vỏ măng cụt ............................. 61
3.4.2. Cơ chế chuyển nguyên tử hydro (HAT) - Năng lượng phân ly liên kết
(BDE) ................................................................................................................. 62
3.4.3. Cơ chế chuyển electron chuyển proton (SET-PT) ................................... 66
3.4.4. Cơ chế chuyển proton chuyển electron (SPLET) .................................... 68
3.4.5. Nhận xét ................................................................................................... 71
3.5. KHẢ NĂNG DẬP TẮT GỐC TỰ DO CỦA MỘT SỐ HỢP CHẤT
POLYPHENOL ..................................................................................................... 71
3.5.1. Giới thiệu ................................................................................................. 71
3.5.2. Năng lượng phân ly liên kết và năng lượng ion hóa của một số hợp chất
polyphenol ......................................................................................................... 72
3.5.3. Phản ứng dập tắt gốc tự do peroxyl (CH3OO•) của một số hợp chất
polyphenol ......................................................................................................... 74
3.5.4. Nhận xét ................................................................................................... 79
3.6. CHỨC NĂNG HÓA FULLERENE BẰNG CÁC DẪN XUẤT MALONATE
............................................................................................................................... 80
3.6.1. Giới thiệu ................................................................................................. 80
3.6.2. Cơ chê phản ứng Bingel - Hirsch ............................................................ 81
3.6.3. Phản ứng đóng vòng giữa fullerene và dẫn xuất flavonoid malonate có
nguồn gốc từ chalcone, flavone và flavanone ................................................... 85
3.6.4. Nhận xét ................................................................................................... 89
3.7. KHẢ NĂNG CHỐNG OXY HÓA CỦA CÁC DẪN CHẤT LAI HÓA
FULLERENE VÀ CHALCONE, FLAVONE, FLAVANONE ........................... 89
3.7.1. Giới thiệu ................................................................................................. 89
3.7.2. Khả năng chống oxy hóa của các dẫn chất lai hóa fullerene và chalcone,
flavone, flavanone .............................................................................................. 90
3.7.3. Nhận xét ................................................................................................... 91

vi
3.8. THIẾT KẾ CÁC HỢP CHẤT CHỐNG OXY HÓA CÓ NGUỒN GỐC TỪ
CÂY SA KÊ VÀ MĂNG CỤT TRÊN NỀN FULLERENE ................................ 91
3.8.1. Giới thiệu ................................................................................................. 91
3.8.2. Chức năng hóa fullerene với hợp chất altilisin J và mangostin có nguồn
gốc từ cây sa kê và vỏ măng cụt ........................................................................ 93
3.8.3. Khả năng chống oxy hóa của các dẫn xuất fullerene-altilisin J và fullerene-
mangostin ........................................................................................................... 96
3.8.4. Nhận xét ................................................................................................... 97
NHỮNG KẾT LUẬN CHÍNH CỦA LUẬN ÁN ..................................................... 99
NHỮNG ĐỊNH HƯỚNG NGHIÊN CỨU TIẾP THEO ........................................101
DANH MỤC CÁC CÔNG TRÌNH LIÊN QUAN ĐẾN LUẬN ÁN .....................102
TÀI LIỆU THAM KHẢO .......................................................................................104
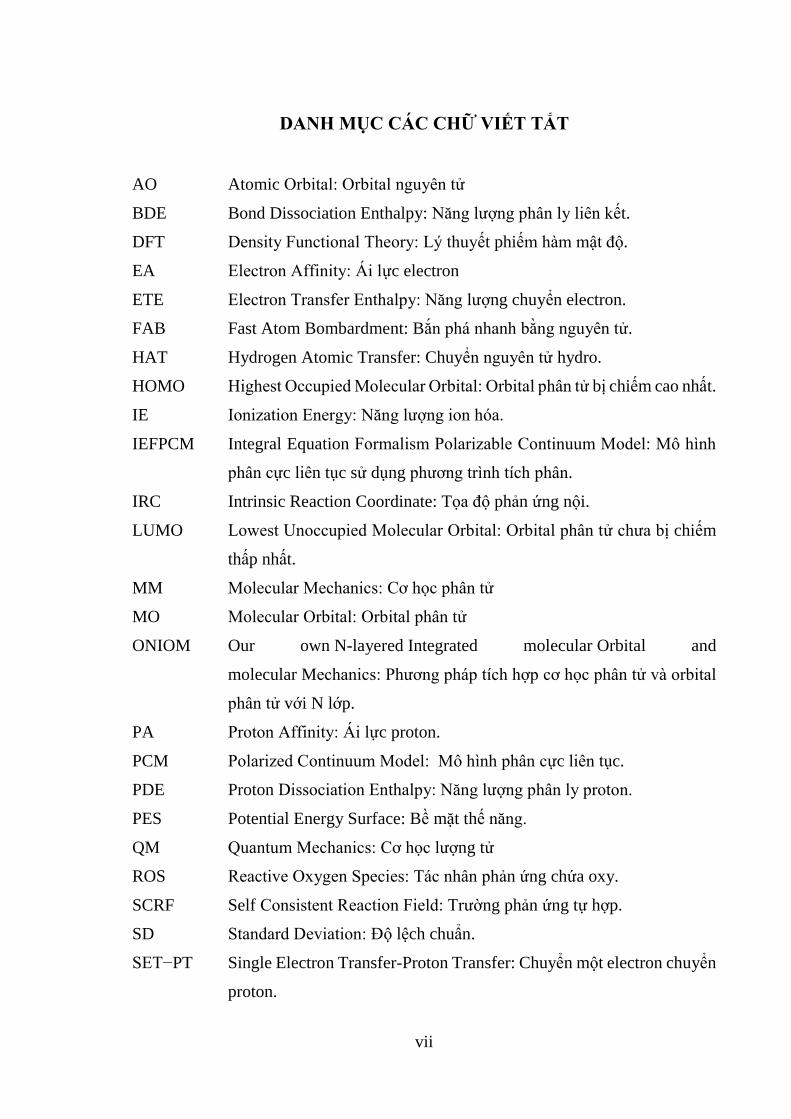
vii
DANH MỤC CÁC CHỮ VIẾT TẮT
AO Atomic Orbital: Orbital nguyên tử
BDE Bond Dissociation Enthalpy: Năng lượng phân ly liên kết.
DFT Density Functional Theory: Lý thuyết phiếm hàm mật độ.
EA Electron Affinity: Ái lực electron
ETE Electron Transfer Enthalpy: Năng lượng chuyển electron.
FAB Fast Atom Bombardment: Bắn phá nhanh bằng nguyên tử.
HAT Hydrogen Atomic Transfer: Chuyển nguyên tử hydro.
HOMO Highest Occupied Molecular Orbital: Orbital phân tử bị chiếm cao nhất.
IE Ionization Energy: Năng lượng ion hóa.
IEFPCM Integral Equation Formalism Polarizable Continuum Model: Mô hình
phân cực liên tục sử dụng phương trình tích phân.
IRC Intrinsic Reaction Coordinate: Tọa độ phản ứng nội.
LUMO Lowest Unoccupied Molecular Orbital: Orbital phân tử chưa bị chiếm
thấp nhất.
MM Molecular Mechanics: Cơ học phân tử
MO Molecular Orbital: Orbital phân tử
ONIOM Our own N-layered Integrated molecular Orbital and
molecular Mechanics: Phương pháp tích hợp cơ học phân tử và orbital
phân tử với N lớp.
PA Proton Affinity: Ái lực proton.
PCM Polarized Continuum Model: Mô hình phân cực liên tục.
PDE Proton Dissociation Enthalpy: Năng lượng phân ly proton.
PES Potential Energy Surface: Bề mặt thế năng.
QM Quantum Mechanics: Cơ học lượng tử
ROS Reactive Oxygen Species: Tác nhân phản ứng chứa oxy.
SCRF Self Consistent Reaction Field: Trường phản ứng tự hợp.
SD Standard Deviation: Độ lệch chuẩn.
SET−PT Single Electron Transfer-Proton Transfer: Chuyển một electron chuyển
proton.

viii
SPLET Sequential Proton Loss Electron Transfer: Chuyển proton mất electron.
TCE Thermal Correction to Enthalpy: Hiệu chỉnh năng lượng dao động nhiệt
đối với enthalpy.
TCGFE Thermal Correction to Gibbs Free Energy: Hiệu chỉnh năng lượng dao
động nhiệt đối với năng lượng tự do Gibbs.
ZPE Zero Point Energy: Năng lượng điểm không.

ix
DANH MỤC CÁC BẢNG
Bảng 1.1. Độ tan của C60 trong các dung môi phổ biến ........................................... 18
Bảng 3.1. Các giá trị ∆S trong phương pháp ONIOM(ROB3LYP/6-311++G
(2df,2p):PM6) đối với model 1A .............................................................................. 44
Bảng 3.2. Giá trị BDE(O−H) của các ubiquinol (kcal/mol) ..................................... 45
Bảng 3.3. BDE(O−H) của các dẫn xuất phenol - kcal/mol ...................................... 47
Bảng 3.4. BDE(O−H) của các hợp chất flavonoid trong Hình 3.4 (kcal/mol) ......... 48
Bảng 3.5. Giá trị năng lượng ion hóa (eV) tính bằng phương pháp PM6 của các dẫn
xuất phenol và giá trị thực nghiệm ............................................................................ 51
Bảng 3.6. Năng lượng phân ly liên kết O–H và năng lượng phân ly proton tính bằng
phương pháp PM6 (kcal/mol) ................................................................................... 55
Bảng 3.7. BDE(O–H) của 12 hợp chất polyphenol có nguồn gốc từ cây sa kê tính
bằng phương pháp ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) .......................... 57
Bảng 3.8. Giá trị năng lượng ion hóa (eV) tính bằng phương pháp PM6 của 12 hợp
chất nghiên cứu ......................................................................................................... 58
Bảng 3.9. Năng lượng phản ứng của 12 hợp chất có nguồn gốc từ cây sa kê tính bằng
phương pháp PM6 (kcal/mol) ................................................................................... 59
Bảng 3.10. Giá trị BDE (OH) của các hợp chất nghiên cứu tính bằng phương pháp
PM6 ........................................................................................................................... 64
Bảng 3.11. Giá trị BDE(OH) của 14 hợp chất xanthone có nguồn gốc tử vỏ măng
cụt tính bằng phương pháp ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) ............. 65
Bảng 3.12. Năng lượng ion hóa của 14 hợp chất xanhthone có nguồn gốc từ vỏ măng
cụt tính bằng phương pháp PM6 ............................................................................... 66
Bảng 3.13. Các giá trị PA, ETE và PDE của 14 hợp chất xanhthone có nguồn gốc từ
vỏ măng cụt tính ở mức lý thuyết B3LYP/6-31+G(d,p)//PM6 trong pha khí và nước
................................................................................................................................... 68
Bảng 3.14. Giá trị BDE(O−H) và IE trong pha khí của một số hợp chất polyphenol
................................................................................................................................... 73

x
Bảng 3.15. Giá trị năng lượng của các phần tử tham gia phản ứng giữa một số hợp
chất polyphenol và gốc tự do CH3OO• tính ở mức lý thuyết ONIOM (B3LYP/6-
311G++(2df,2p):PM6) .............................................................................................. 75
Bảng 3.16. Giá trị năng lượng của các phần tử tham gia phản ứng giữa ion âm
dimethyl bromomalonate và fullerene tính ở mức lý thuyết B3LYP/6-31G(d)//PM6
trong pha khí (Đơn vị: hartree) ................................................................................. 81
Bảng 3.17. Giá trị năng lượng của các phần tử tham gia phản ứng giữa ion âm
flavonoid malonate và fullerene tính ở mức lý thuyết B3LYP/6-31G(d)//PM6 trong
pha khí ....................................................................................................................... 86
Bảng 3.18. Giá trị BDE(O−H) và IE của dẫn xuất C60-chalcone, C60-flavone và C60-
flavanone ................................................................................................................... 90
Bảng 3.19. Giá trị năng lượng của các phần tử tham gia phản ứng giữa dẫn xuất
malonate có nguồn gốc từ altilisin J, mangostin và fullerene tính ở mức lý thuyết
B3LYP/6-31G(d)//PM6 trong pha khí ...................................................................... 93
Bảng 3.20. Giá trị BDE(O−H), EA và IE của mangostin, altilisin J, C60-mangostin -
1, C60-mangostin-2 và C60-altilisin J ......................................................................... 97

xi
DANH MỤC CÁC HÌNH
Hình 1.1. Cấu trúc của các hợp chất polyphenol đơn giản ......................................... 5
Hình 1.2. Cấu trúc tổng quát của hợp chất flavonoid ................................................. 6
Hình 1.3. Cấu trúc của hợp chất flavone .................................................................... 6
Hình 1.4. Cấu trúc của hợp chất flavonol ................................................................... 7
Hình 1.5. Cấu trúc của hợp chất flavanone ................................................................ 7
Hình 1.6. Cấu trúc của hợp chất dihydroflavonol ...................................................... 8
Hình 1.7. Cấu trúc của hợp chất flavanol ................................................................... 8
Hình 1.8. Cấu trúc của hợp chất chalcone .................................................................. 9
Hình 1.9. Cấu trúc của hợp chất isoflavone ............................................................... 9
Hình 1.10. Cấu trúc của hợp chất anthocyanidin ..................................................... 10
Hình 1.11. Hình ảnh cây sa kê .................................................................................. 10
Hình 1.12. Hình ảnh về cây và vỏ quả măng cụt...................................................... 11
Hình 1.13. Sơ đồ các cơ chế chống oxy hóa ............................................................ 13
Hình 1.14. a) Icosahedron b) Truncated Icosahedron c) Fullerenes, C60 ................ 16
Hình 1.15. Cấu trúc của C60: a) Liên kết ở cầu nối vòng (6,6); b) Liên kết ở cầu nối
vòng (5,6) .................................................................................................................. 16
Hình 1.16. Góc hình chóp (p = (π - 900) của: a) C lai hóa sp2 phẳng; b) C lai hóa
sp3 tứ diện và c) C lai hóa sp2 không phẳng .............................................................. 17
Hình 1.17. Giản đồ orbital phân tử Huckel (HMO) của C60 và benzene ................. 19
Hình 1.18. Một số phản ứng của C60 ........................................................................ 21
Hình 1.19. Cơ chế phản ứng Bingel ......................................................................... 22
Hình 2.1. Sơ đô phân chia hê phân tư băng phương phap ONIOM: (a) ONIOM hai
lơp, (b) ONIOM ba lơp, và (c) ONIOM n-lơp ......................................................... 30
Hình 2.2. Giản đồ năng lượng .................................................................................. 33
Hình 2.3. Mô hình Onsager (SCRF = Dipole) ......................................................... 37
Hình 2.4. Mô hình IEFPCM (SCRF = IEFPCM) ..................................................... 37
Hình 2.5. Mô hình IPCM (SCRF = IPCM) .............................................................. 38
Hình 2.6. Mô hình SCIPCM (SCRF = SCIPCM) .................................................... 38

xii
Hình 3.1. Cách chọn các mô hình ONIOM 2 lớp ..................................................... 41
Hình 3.2. So sánh giá trị BDE(O-H) của dẫn xuất phenol được tính bằng 7 mô hình
ONIOM với dữ liệu thực nghiệm .............................................................................. 43
Hình 3.3. Cấu trúc và mô hình ONIOM 2 lớp của ubiquinol-n (n = 2, 6, 10) ......... 45
Hình 3.4. Các hợp chất flavonoid dùng để khảo sát mô hình ONIOM .................... 46
Hình 3.5. Sự tương quan giữa IE adiabatic và giá trị thực nghiệm .......................... 52
Hình 3.6. Cấu trúc của 12 hợp chất nghiên cứu có nguồn gốc từ cây sa kê ............ 54
Hình 3.7. Cấu trúc của các hợp chất có nguồn gốc từ vỏ măng cụt và hợp chất liên
quan ........................................................................................................................... 62
Hình 3.8. Một số hợp chất chống oxy hóa polyphenol ............................................ 72
Hình 3.9. Cấu trúc của trạng thái trung gian Int-1, Int-2 và trạng thái chuyển tiếp
(TS) của phản ứng giữa quecertin và gốc tự do CH3OO• ......................................... 74
Hình 3.10. Giản đồ năng lượng phản ứng giữa một số hợp chất polyphenol và gốc tự
do CH3OO• ................................................................................................................ 77
Hình 3.11. Các tọa độ phản ứng nội (IRC) của phản ứng giữa hợp chất polyphenol
và gốc tự do CH3OO• ................................................................................................ 78
Hình 3.12. Sự tương quan giữa BDE và hàng rào năng lượng (Er) của phản ứng giữa
gốc tự do (CH3OO•) và một số hợp chất polyphenol ................................................ 79
Hình 3.13. Phản ứng Bingel - Hirsch giữa các dẫn xuất của malonate và fullerene 80
Hình 3.14. Giản đồ năng lượng của phản ứng giữa ion âm dimethyl bromomalonate
và fullerene ................................................................................................................ 83
Hình 3.15. Cấu trúc hình học của trạng thái trung gian, trạng thái chuyển tiếp, phức
sản phẩm và sản phẩm của phản ứng giữa ion âm [CBr(COOCH3)2]− và C60. Độ dài
liên kết được tính bằng Angstroms (Å) ..................................................................... 84
Hình 3.16. Các tọa độ phản ứng nội (IRC) của phản ứng giữa ion âm
[CBr(COOCH3)2]− và C60 .......................................................................................... 85
Hình 3.17. Giản đồ năng lượng của phản ứng flavonoid malonate với fullerene .... 87
Hình 3.18. Cấu trúc hình học của trạng thái trung gian, trạng thái chuyển tiếp, phức
sản phẩm và sản phẩm của phản ứng giữa flavonoid malonate và fullerene. Độ dài
liên kết được tính bằng Angstroms (Å) ..................................................................... 88

xiii
Hình 3.19. Các tọa độ phản ứng nội (IRC) của phản ứng giữa flavonoid malonate và
fullerene ..................................................................................................................... 88
Hình 3.20. Phản ứng Bingel - Hirsch giữa các dẫn xuất của malonate và fullerene 92
Hình 3.21. Giản đồ năng lượng của phản ứng giữa ion âm dimethyl bromomalonate
có nguồn gốc từ cây sa kê và măng cụt với fullerene ............................................... 94
Hình 3.22. Cấu trúc tối ưu của trạng thái trung gian, trạng thái chuyển tiếp, phức sản
phẩm và sản phẩm của phản ứng giữa polyphenol malonate và fullerene ................ 95
Hình 3.23. Tọa độ phản ứng (IRC) của phản ứng giữa dimethyl bromomalonate có
nguồn gốc từ cây sa kê và măng cụt với fullerene .................................................... 96

1
MỞ ĐẦU
Sự thoái hóa của tế bào là nguyên nhân chính gây nên các bệnh tật trong cơ
thể con người. Bệnh ung thư cũng có liên quan đến sự thoái hóa của tế bào, các tế
bào ác tính nói chung hoạt động hơn tế bào bình thường trong việc tạo ra superoxide
(các hợp chất chứa liên kết đơn O−O). Các bệnh tật này là kết quả của sự tạo ra quá
nhiều tác nhân phản ứng chứa oxy (Reactive Oxygen Species−ROS) trong các hoạt
động trao đổi chất của tế bào, dẫn đến sự hư tổn tế bào bao gồm peroxy hóa lipid,
hình thành sản phẩm cộng DNA, quá trình oxy hóa protein, làm mất hoạt tính enzyme
và cuối cùng có thể dẫn đến chết tế bào [40]. Cơ thể động vật hay con người thường
lưu giữ các hợp chất có tính chống oxy hóa cao như gluthathione, vitamin E, vitamin
C... Khi hàm lượng các chất chống oxy hóa trong cơ thể giảm xuống sẽ làm tăng nguy
cơ hủy hoại các tế bào. Tuy nhiên, những ảnh hưởng bất lợi của ROS có thể được
ngăn ngừa bằng cách bổ sung chế độ ăn giàu thực phẩm có chứa chất chống oxy hóa
(các loại đậu, rau và trái cây tươi…) với tác dụng có lợi cho sức khỏe con người [17,
105]. Các chất chống oxy hóa tự nhiên như các hợp chất polyphenol có khả năng loại
bỏ gốc tự do, ngăn chặn quá trình oxy hóa rất hiệu quả. Trong tự nhiên, cây sa kê và
vỏ măng cụt được biết đến là một nguồn hợp chất polyphenol rất dồi dào. Vấn đề
nghiên cứu thực nghiệm về hoạt tính chống oxy hóa của các hợp chất chiết xuất từ
cây sa kê và vỏ măng cụt yêu cầu trải qua nhiều giai đoạn phức tạp: Sàn lọc từ các
hợp chất tự nhiên, phân lập, xác định cấu trúc, thử nghiệm hoạt tính sinh học... đòi
hỏi một khối lượng công việc rất lớn. Thực tế cho thấy trên thế giới xu hướng kết hợp
nghiên cứu giữa các nhóm tính toán lý thuyết và thực nghiệm phát triển rất mạnh mẽ.
Các tính toán lý thuyết có thể cung cấp các thông tin nền tảng như các thông số cấu
trúc, năng lượng và một số tính chất quan trọng khác góp phần định hướng làm thực
nghiệm và ngược lại kết quả thực nghiệm sẽ làm sáng tỏ và chứng minh tính đúng
đắn của tính toán lý thuyết. Do đó, việc bố trí nghiên cứu thực nghiệm và nghiên cứu
hóa tính toán đan xen nhau một cách hợp lý sẽ giúp giảm thiểu được khối lượng thực
nghiệm và kết quả thu được cũng được lý giải một cách logic và khoa học hơn. Mặc
dù, các kết quả thực nghiệm về hoạt tính sinh học của các hợp chất chiết xuất từ cây

2
sa kê và vỏ măng cụt đã được báo cáo [49, 57, 72, 126], nhưng việc mô phỏng đánh
giá khả năng chống oxy hóa bằng hóa học tính toán là một bước mới, chưa có nghiên
cứu lý thuyết nào phân tích khả năng chống oxy hóa của các hợp chất này cũng như
xác định cơ chế của phản ứng dập tắt gốc tự do.
Ngoài hợp chất polyphenol thì gần đây nhiều nghiên cứu đã chứng minh rằng
fullerene (C60) phản ứng với các gốc tự do và hoạt động như một chất dập tắt gốc tự
do hiệu quả. Điều này đã được thử nghiệm cả in vitro và in vivo [56, 78]. Hiệu quả
chống oxy hóa của chúng được chứng tỏ cao hơn so với vitamin E trong việc ngăn
cản sự oxy hóa của lipid bởi các tác nhân superoxide và gốc tự do hydroxyl [125].
Tuy nhiên, việc ứng dụng fullerene trong lĩnh vực làm chất chống oxy hóa còn hạn
chế bởi vì nó chỉ đóng vai trò như chiếc lồng thu nhận các gốc tự do có trung tâm
phản ứng ở nguyên tử carbon chứ không phải là chất chống oxy hóa xảy ra theo cơ
chế ngắt mạch. Do vậy, fullerene không có hiệu quả trong việc dập tắt phản ứng của
các gốc tự do peroxyl trong giai đoạn phát triển mạch của phản ứng dây chuyền [25,
119]. Để cải thiện điều này, một số phương pháp chức năng hóa fullerene đã được đề
xuất tạo ra một loạt các dẫn xuất C60 với những tính chất vật lý và hóa học khác nhau
[50, 90]. Cho đến nay đã có nhiều chất được sử dụng làm chất chống oxy hóa trong
nhiều lĩnh vực khác nhau. Một vấn đề đang được quan tâm hiện nay là làm sao tạo ra
các chất chống oxy hóa hiệu quả cao không độc hại đến môi trường và con người để
có thể sử dụng trong y học, thực phẩm, dược phẩm… Một trong những cách tốt nhất
để lợi dụng cấu trúc độc đáo của fullerene là dùng nó để làm nền cho các phân tử
thuốc đính tử vào [2, 30, 46, 110]. Trong những năm qua, một số lượng lớn các nghiên
cứu thực nghiệm đã tập trung vào tính chất chống oxy hóa của dẫn xuất fullerene-
polyphenol [20, 21, 24]. Tuy nhiên, một nghiên cứu lý thuyết có tính hệ thống về mối
quan hệ giữa đặc điểm cấu trúc và khả năng chống oxy hóa của dẫn xuất fullerene-
polyphenol, cũng như nghiên cứu định hướng để thiết kế và hỗ trợ cho quá trình bán
tổng hợp các phân tử có hoạt tính sinh học cao hơn so với hợp chất tự nhiên chưa
được thực hiện.

3
Với những lý do nêu trên, chúng tôi chọn đề tài luận án tiến sĩ: “Nghiên cứu
cấu trúc, khả năng chống oxy hóa của một số polyphenol và dẫn xuất trên nền
fullerene (C60) bằng phương pháp hóa tính toán“.
Mục tiêu của luận án:
- Nghiên cứu khả năng chống oxy hóa của các hợp chất polyphenol có nguồn gốc
từ cây sa kê, vỏ măng cụt và một số dẫn xuất fullerene-polyphenol bằng phương pháp
hóa tính toán.
- Thiết kế các hợp chất có khả năng chống oxy hóa từ dẫn xuất malonate có nguồn
gốc từ cây sa kê và vỏ măng cụt trên nền fullerene thông qua phản ứng Bingel-Hirsch.
Ý nghĩa khoa học của luận án:
Triển khai một hướng nghiên cứu mới, phù hợp với xu thế chung trên thế giới
cũng như các điều kiện của Việt Nam: Tìm kiếm chất chống oxy hóa xanh thân thiện
môi trường. Bên cạnh đó, luận án còn có vai trò đóng góp vào việc khẳng định khả
năng tổng hợp các chất chống oxy hóa trên nền fullerene đáp ứng được yêu cầu nghiên
cứu và hướng tới việc ứng dụng trong nước.
Những đóng góp mới của luận án:
- Đã tiến hành nghiên cứu một cách có hệ thống khả năng chống oxy hóa của một
số hợp chất polyphenol có nguồn gốc từ cây sa kê và vỏ măng cụt thông qua 3 cơ chế
HAT, SET−PT và SPLET. Trong đó, hợp chất S12, M10 và M11 được xem là những
chất chống oxy hóa tiềm năng với giá trị BDE(O−H) trong pha khí lần lượt là 77,3;
82,3; 82,8 kcal/mol.
- Đã làm rõ được cơ chế phản ứng Bingel − Hirsch giữa ion âm dimethyl
bromomalonate và fullerene (C60). Trong bốn đường phản ứng thì đường phản ứng
đi qua trạng thái chuyển tiếp TS(6,6)-1 để hình thành sản phẩm ở vị trí liên kết (6,6) là
thuận lợi về mặt nhiệt động, điều này cũng phù hợp với kết quả thực nghiệm.
- Đã thiết kế các hợp chất chống oxy từ một số dẫn xuất malonate của altilisin J
và mangostin trên nền fullerene bằng phương pháp hóa tính toán. Các hợp dẫn xuất
fullerene mới có khả năng chống oxy hóa cao hơn so với hợp chất ban đầu, được đánh
giá thông qua các thông số đặc trưng như: BDE, IE và EA.

4
Những đóng góp mới của luận án đã được công bố trên các tạp chí Quốc tế uy
tín trong danh mục ISI cũng như trên các tạp chí Quốc gia uy tín:
- Journal of Molecular Modeling, 2016, 22, pp.113-121.
- Chemical Physics Letters, 2016, 652, pp.56–61.
- Chemical Physics Letters, 2015, 625, pp. 30–35.
- Springer International Publishing, 2015, 46, pp. 464-469.
- Chemical Physics Letters, 2014, 613, pp. 139–145.
- Tạp chí KHCN - ĐHĐN, 2016, Số 5(102), trang 95-98.
- Tạp chí Khoa học Đại học Huế, 2016, Số 3(117).
- Tạp chí Khoa học và Công nghệ (Viện Hàn lâm KH&CN Việt Nam), 2016
Số 54(2B), trang 149-155.
Cấu trúc của luận án gồm các phần sau:
- Mở đầu
- Chương 1: Tổng quan về hệ chất nghiên cứu
- Chương 2: Nội dung và phương pháp nghiên cứu
- Chương 3: Kết quả và thảo luận
- Những kết luận chính của luận án
- Định hướng nghiên cứu tiếp theo
- Danh mục các công trình liên quan đến luận án
- Tài liệu tham khảo

5
CHƯƠNG 1. TỔNG QUAN VỀ HỆ CHẤT NGHIÊN CỨU
1.1. TỔNG QUAN VỀ HỢP CHẤT POLYPHENOL CÓ TÁC DỤNG CHỐNG
OXY HÓA
Polyphenol là các hợp chất có nguồn gốc tự nhiên và tồn tại trong thực vật
được chứng minh là có khả năng chống oxy hóa vô cùng hiệu quả. Polyphenol có thể
bảo vệ cơ thể, giúp cơ thể chống lại nhiều loại bệnh khác nhau do gốc tự do gây ra.
Đặc điểm chung của chúng là trong phân tử có vòng thơm (vòng benzen) chứa một
hay hai, ba... hoặc nhiều nhóm hydroxyl (OH) gắn trực tiếp vào vòng benzen. Tùy
thuộc vào số lượng và vị trí tương hỗ của các nhóm OH với bộ khung hóa học mà các
tính chất lý hoá học hoặc hoạt tính sinh học thay đổi.
1.1.1. Hợp chất polyphenol
- Những hợp chất polyphenol đơn giản
Chỉ có một số ít hợp chất phenolic đơn giản (hợp chất chỉ chứa 1 vòng benzen)
tồn tại trong tự nhiên. Chẳng hạn, resorcinol (1,3-dihydroxybenzen) và
phloroglucinol (1,3,5-trihydroxybenzen) là những hợp chất được hình thành từ nhựa
cây và vỏ cây ăn trái (Hình 1.1).
OHO
HO
OH
OH
Acid shikimic
OHO
OH
O
OH
O
CH2
Acid chorismic
O OH
Acid cinamic
HO OH HO OH
OHO
HO
OH
OH
OH
Resorcinol Phloroglucinol Acid gallic
OHO
OH
OCH3
Acid vanillic
HO
OH
OCH3
Aldehyd vanillin
O OH O OH
OH
OHOH
O OH
OCH3
OH
O OH
OCH3
OH
HO
O OH
OCH3
OH
H3CO
OHO O
Acid p-coumaricAcid caf f eic Acid ferulic Acid 5-hydroxyf erulic Acid sinapic Umbellif erone
Hình 1.1. Cấu trúc của các hợp chất polyphenol đơn giản
Acid phenolic là những hợp chất có đặc điểm của nhóm chức caboxylic (ví
dụ: acid gallic, acid vanillic...) và aldehyd hydroxybenzoic là những hợp chất có đặc
6
điểm của nhóm chức aldehyd (ví dụ: aldehyd vanillin). Các acid cinnamic phổ biến
là acid cinnamic, acid p-coumaric, acid caffeic, acid ferulic, acid 5-hydroxyferulic và
acid sinapic (Hình 1.1).
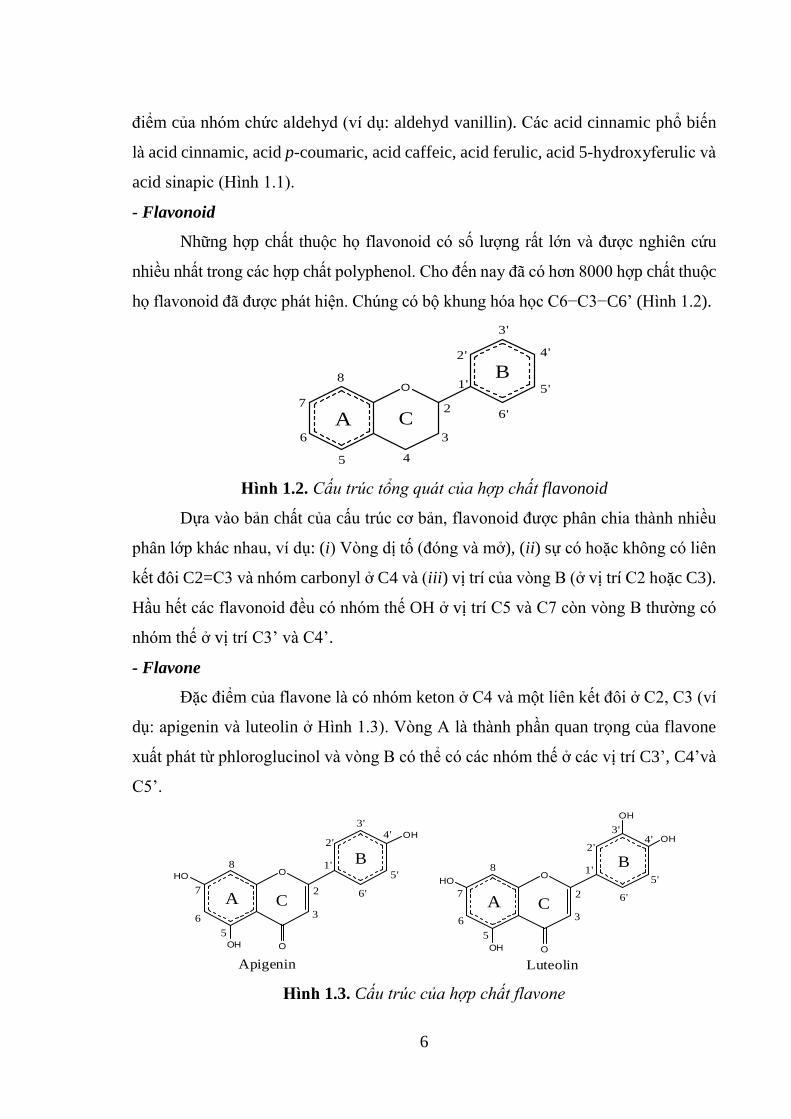
- Flavonoid
Những hợp chất thuộc họ flavonoid có số lượng rất lớn và được nghiên cứu
nhiều nhất trong các hợp chất polyphenol. Cho đến nay đã có hơn 8000 hợp chất thuộc
họ flavonoid đã được phát hiện. Chúng có bộ khung hóa học C6−C3−C6’ (Hình 1.2).
O
A
B
C
1'
2'
3'
4'
5'
6'2
3
45
6
7
8
Hình 1.2. Cấu trúc tổng quát của hợp chất flavonoid
Dựa vào bản chất của cấu trúc cơ bản, flavonoid được phân chia thành nhiều
phân lớp khác nhau, ví dụ: (i) Vòng dị tố (đóng và mở), (ii) sự có hoặc không có liên
kết đôi C2=C3 và nhóm carbonyl ở C4 và (iii) vị trí của vòng B (ở vị trí C2 hoặc C3).
Hầu hết các flavonoid đều có nhóm thế OH ở vị trí C5 và C7 còn vòng B thường có
nhóm thế ở vị trí C3’ và C4’.
- Flavone
Đặc điểm của flavone là có nhóm keton ở C4 và một liên kết đôi ở C2, C3 (ví
dụ: apigenin và luteolin ở Hình 1.3). Vòng A là thành phần quan trọng của flavone
xuất phát từ phloroglucinol và vòng B có thể có các nhóm thế ở các vị trí C3’, C4’và
C5’.
O
O
A
B
C
OH
O
O
A
B
C
OH
HOHO
OH OH
OH
Apigenin Luteolin
1'
2'
3'4'
6'
5'1'
2'
3'4'
6'
5'2
3
5
6
7
8
2
3
5
6
7
8
Hình 1.3. Cấu trúc của hợp chất flavone

7
- Flavonol
Hợp chất flavonol là những hợp chất flavone có một nhóm OH ở C3 (Hình
1.4). Flavonol có rất nhiều trong các loài thực vật và phổ biến là: quercetin,
kaempferol và myricetin (Hình 1.4).
O
O
A
B
C
OH
O
O
A
B
C
OH
HOHO
OH OH
OH
Quercetin
O
O
A
B
C
OH
HO
OH
Myricetin
OH OH
OH
OH
Kaempferol
OH
1'
2'
3'4'
5'
6'
1'
2'
3'4'
5'
6'
1'
2'
3'4'
5'
6'
2
35
6
7
8
2
35
6
7
82
35
6
7
8
Hình 1.4. Cấu trúc của hợp chất flavonol
- Flavanone
Đặc điểm của flavanone là có một nhóm keton ở C4 và không có liên kết đôi
ở C2, C3 (2-phenyl-2,3-dihydropyran-4-one). Flavanone là đồng phân của chalcone
và chúng được tổng hợp bằng phản ứng nhân tạo và phản ứng sinh hóa. Flavanone
có một trung tâm bất đối ở C2 tạo nên hai đồng phân quang học rất quan trọng có một
số hoạt tính sinh học. Một số flavanone phổ biến là naringenin, eriodictyol và
hespertin (Hình 1.5).
O
O
A
B
C
OH
HO
OH
O
O
A
B
C
NaringeninFlavanone
1'
2'
3'
4'
5'
6'
1'
2'
3'4'
5'
6'2
3
5
6
7
8 2
3
5
6
7
8
Hình 1.5. Cấu trúc của hợp chất flavanone
- Dihydroflavonol
Hợp chất dihydroflavonol là flavanone được gắn nhóm thế OH ở C3 (2-
phenyl-3-hydroxy-2,3-dihydropyran-4-ones) (Hình 1.6). Chúng có hai carbon bất đối
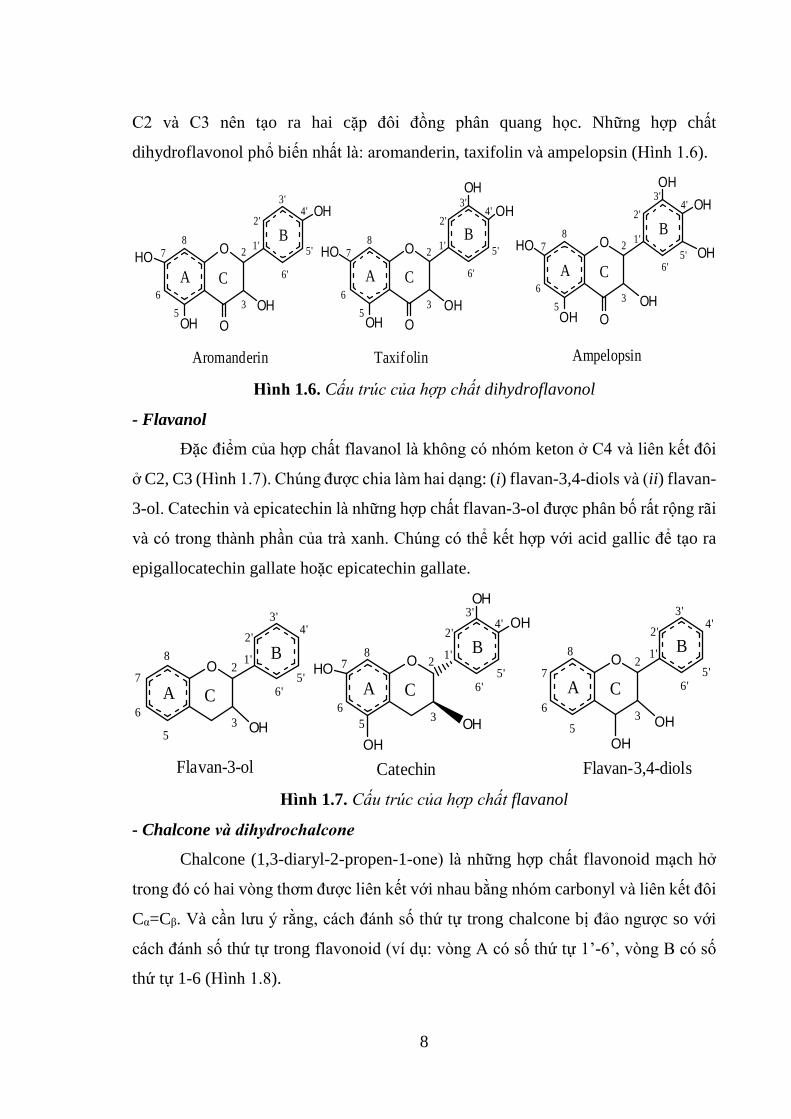
8
C2 và C3 nên tạo ra hai cặp đôi đồng phân quang học. Những hợp chất
dihydroflavonol phổ biến nhất là: aromanderin, taxifolin và ampelopsin (Hình 1.6).
O
O
A
B
C
OH
O
O
A
B
C
OH
HOHO
OH OH
OH
Taxifolin
O
O
A
B
C
OH
HO
OH
Ampelopsin
OH
OH
Aromanderin
OHOH OH
2'
1'
3'4'
5'
6'
2'
1'
3'4'
5'
6'
2'
1'
3'4'
5'6'
2
35
6
78
2
35
6
78
2
35
6
78
Hình 1.6. Cấu trúc của hợp chất dihydroflavonol
- Flavanol
Đặc điểm của hợp chất flavanol là không có nhóm keton ở C4 và liên kết đôi
ở C2, C3 (Hình 1.7). Chúng được chia làm hai dạng: (i) flavan-3,4-diols và (ii) flavan-
3-ol. Catechin và epicatechin là những hợp chất flavan-3-ol được phân bố rất rộng rãi
và có trong thành phần của trà xanh. Chúng có thể kết hợp với acid gallic để tạo ra
epigallocatechin gallate hoặc epicatechin gallate.
O
OH
A
B
C
O
A
B
C
OH
HO
OH
O
A
B
C
Catechin
OH OH OH
OH
Flavan-3-ol Flavan-3,4-diols
1'
2'
3'4'
5'6'
1'
2'
3'4'
5'6'
1'
2'
3'4'
5'6'
2
35
6
7
82
35
6
78
2
35
6
7
8
Hình 1.7. Cấu trúc của hợp chất flavanol
- Chalcone và dihydrochalcone
Chalcone (1,3-diaryl-2-propen-1-one) là những hợp chất flavonoid mạch hở
trong đó có hai vòng thơm được liên kết với nhau bằng nhóm carbonyl và liên kết đôi
Cα=Cβ. Và cần lưu ý rằng, cách đánh số thứ tự trong chalcone bị đảo ngược so với
cách đánh số thứ tự trong flavonoid (ví dụ: vòng A có số thứ tự 1’-6’, vòng B có số
thứ tự 1-6 (Hình 1.8).
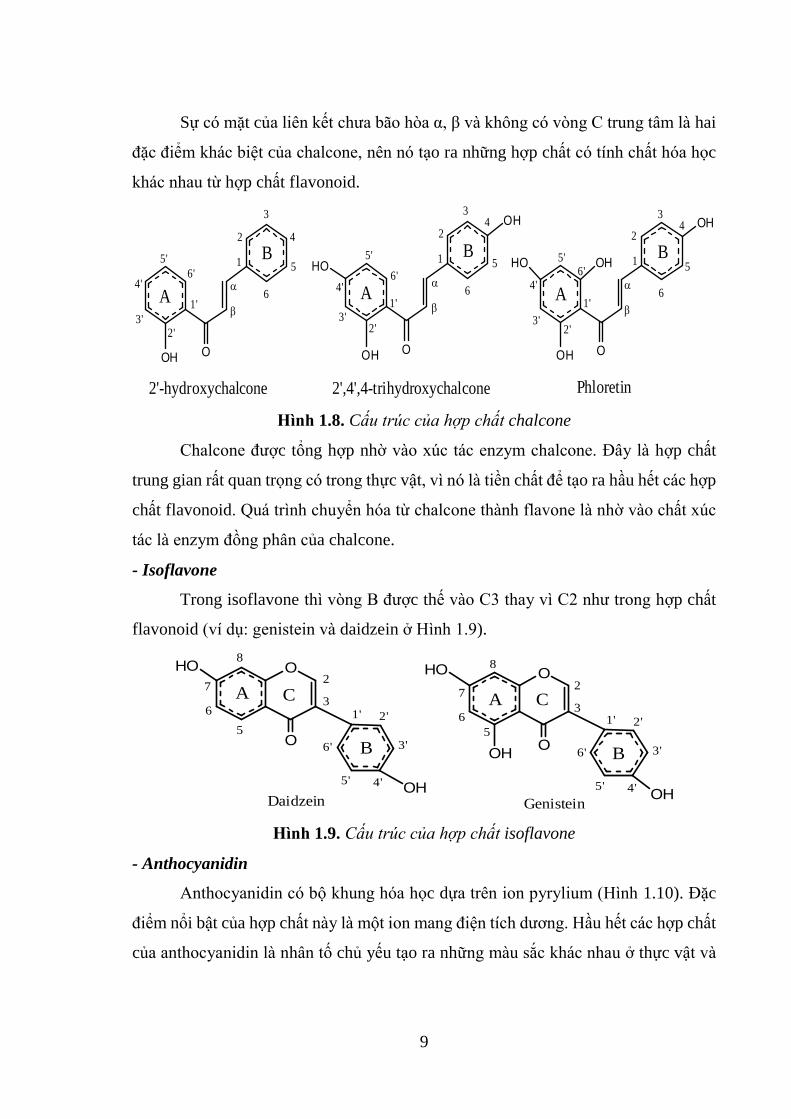
9
Sự có mặt của liên kết chưa bão hòa α, β và không có vòng C trung tâm là hai
đặc điểm khác biệt của chalcone, nên nó tạo ra những hợp chất có tính chất hóa học
khác nhau từ hợp chất flavonoid.
O O OOH OH
HO
OH
HO OH
OH
2'-hydroxychalcone 2',4',4-trihydroxychalcone Phloretin
OH
A A A
B B B
1'
2'3'
4'
5'
6'
1'
2'3'
4'
5'
6'
1'
2'3'
4'
5'6'
1
2
3
4
5
6
1
2
34
5
6
1
2
34
5
6
Hình 1.8. Cấu trúc của hợp chất chalcone
Chalcone được tổng hợp nhờ vào xúc tác enzym chalcone. Đây là hợp chất
trung gian rất quan trọng có trong thực vật, vì nó là tiền chất để tạo ra hầu hết các hợp
chất flavonoid. Quá trình chuyển hóa từ chalcone thành flavone là nhờ vào chất xúc
tác là enzym đồng phân của chalcone.
- Isoflavone
Trong isoflavone thì vòng B được thế vào C3 thay vì C2 như trong hợp chất
flavonoid (ví dụ: genistein và daidzein ở Hình 1.9).
O
O
A
B
C
O
O
A
B
C
HO
OH
OH
HO
OHDaidzein Genistein
1' 2'
3'
4'5'
6'
1' 2'
3'
4'5'
6'
2
3
5
6
7
8
2
3
5
6
7
8
Hình 1.9. Cấu trúc của hợp chất isoflavone
- Anthocyanidin
Anthocyanidin có bộ khung hóa học dựa trên ion pyrylium (Hình 1.10). Đặc
điểm nổi bật của hợp chất này là một ion mang điện tích dương. Hầu hết các hợp chất
của anthocyanidin là nhân tố chủ yếu tạo ra những màu sắc khác nhau ở thực vật và

10
hoa. Hai hợp chất anthocyanidin phổ biến là cyanidin, pelargonidin được trình bày ở
Hình 1.10.
O
A
B
C
OH
HO
OH
Pelargonidin
O
A
B
C
OH
HO
OH
Cyanidin
OH OH
OH
1'
2'
3'4'
5'
6'
1'
2'
3'4'
5'
6'
2
35
6
7
82
35
6
7
8
Hình 1.10. Cấu trúc của hợp chất anthocyanidin
1.1.2. Một số nguyên liệu giàu hợp chất polyphenol ở Việt Nam
1.1.2.1. Cây sa kê
Sa kê có tên khoa học là Artocarpus altilis (Hình 1.11) phân bố rộng rãi ở khu
vực Thái Bình Dương: Indonesia, Malaysia đến Hawaii. Cây thích hợp với khí hậu
nhiệt đới gió mùa nóng ẩm, mưa nhiều. Sa kê là một trong những loài cây lương thực
có sản lượng cao. Quả sa kê có chứa thành phần tinh bột, khoáng chất, các acid amin
thiết yếu. Sa kê được coi là cây đáp ứng cho nhu cầu thực phẩm của các dân tộc thiểu
số và làm nguyên liệu cho một số ngành, chủ yếu là ngành công nghệ thực phẩm. Sa
kê đã được người Pháp đưa vào Việt Nam từ Indonesia và được trồng tại miền Nam
Việt Nam. Cây không sống được trong vùng khí hậu miền Bắc Việt Nam.
Hình 1.11. Hình ảnh cây sa kê

11
Cây sa kê là một loại cây đặc biệt, tất cả các bộ phận của của cây không chỉ
chứa các thành phần cơ bản như protein, lipid, vitamin… mà còn chứa các thành phần
dược tính khác như flavonoid, stilbenoid, arylbenzofuron và jacalin [45, 57, 72, 126].
Những chất này rất có lợi cho quá trình tiêu hóa trong cơ thể con người. Lá, rễ và vỏ
của cây sa kê được sử dụng như những vị thuốc truyền thống để điều trị bệnh gút,
bệnh viêm gan, bệnh tăng huyết áp, bệnh rối loạn chức năng gan, bệnh tiểu đường và
đặc biệt là khả năng chống oxy hóa của chúng [57].
1.1.2.2. Măng cụt
Măng cụt (Hình 1.12) có tên khoa học là Garcinia mangostana L., thuộc họ Bứa
(Clusiaceae), là loài cây ăn quả rất phổ biến thuộc ở Đông Nam Á, Ấn Độ, Sri LanKa.
Cây măng cụt còn có tên gọi khác là cây măng, sơn trúc tử, người phương Tây gọi
trái măng cụt là “nữ hoàng của các loại trái cây”. Do thích hợp với khí hậu nóng ấm
nên ở Việt Nam cây măng cụt được trồng nhiều ở các tỉnh miền Đông Nam Bộ, đồng
bằng sông Cửu Long.
Hình 1.12. Hình ảnh về cây và vỏ quả măng cụt
Từ lâu vỏ măng cụt đã được sử dụng để làm thuốc điều trị các bệnh đau bụng,
tiêu chảy, kiết lỵ, nhiễm trùng vết thương và ung nhọt mãn tính [49]. Những hợp chất
chủ yếu có hoạt tính trong vỏ măng cụt là xanthone và dẫn xuất của xanthone. Chúng
thuộc loại hợp chất polyphenol và thường tìm thấy trong các cây thực vật bậc cao
[92, 135]. Một vài chất trong số các hợp chất xanthone có khả năng chống oxy hóa
cao [49, 89, 111], hoạt tính kháng viêm [15, 91], hoạt tính kháng khuẩn [26]. Vì vậy,
trong thời gian gần đây các hợp chất xanthone chiết xuất từ vỏ măng cụt được sử
dụng để sản xuất thực phẩm chức năng cũng như các sản phẩm khử trùng.

12
1.1.3. Cơ chế hoạt động của các hợp chất chống oxy hóa polyphenol
1.1.3.1. Cơ chế chuyển nguyên tử hydro (Hydrogen Atomic Transfer-HAT)
Trong cơ chế này, chất chống oxy hóa, được ký hiệu là ArOH, sẽ khống chế
các gốc tự do (ví dụ: gốc peroxyl ROO•) bằng cách chuyển một nguyên tử hydro của
nhóm OH trong ArOH sang gốc tự do ROO•:
Gốc phenoxyl (ArO•) tạo thành có thể được ổn định nhờ vào sự chuyển một
nguyên tử hydro ở xa hơn để tạo thành quinone hoặc phản ứng với gốc tự do khác
bao gồm cả các gốc phenoxyl khác. Với sự phát triển liên tục như vậy sẽ tạo ra một
chuỗi các phản ứng.
Cơ chế HAT phù hợp với kiểu phân cắt đồng ly của liên kết O-H trong hợp
chất polyphenol. Phản ứng này có thể xảy ra trên mỗi nhóm OH của hợp chất
polyphenol (ArOH) và nó phụ thuộc vào năng lượng phân ly liên kết (Bond
Dissociation Enthalpy − BDE) của nhóm OH và enthalpy của phản ứng (1.1).
Năng lượng phân ly liên kết (BDE) thể hiện độ bền nhiệt động của liên kết
OH trong hợp chất polyphenol. BDE thấp thì liên kết O−H dễ dàng bị cắt đứt và
nguyên tử hydro dễ dàng chuyển đến kết hợp với gốc tự do, điều này sẽ đóng vai trò
rất quan trọng trong phản ứng chống oxy hóa.
1.1.3.2. Cơ chế chuyển một electron chuyển proton (Single Electron Transfer-
Proton Transfer − SET−PT)
Cơ chế này gồm hai quá trình (phản ứng 1.2), trong quá trình thứ nhất một
electron từ polyphenol chuyển sang gốc tự do và quá trình thứ hai là chuyển một proton.
Cơ chế SET−PT được quyết định bởi khả năng chuyển electron, được đặc
trưng bởi năng lượng ion hóa (Ionization Energy − IE). Trong quá trình thứ hai của
cơ chế này thì liên kết O−H phân cắt theo kiểu dị ly, được đặc trưng bởi năng lượng
phân ly proton (Proton Dissociation Enthalpy – PDE), nó tỏa nhiệt mạnh đối với hợp
chất phenolic [47, 48]. Ngoài ra, dung môi cũng ảnh hưởng đến enthalpy phản ứng
của quá trình thứ nhất. Vì vậy, dung môi cần phải đưa vào nghiên cứu để thu được
một sự mô phỏng tốt về đặc điểm phản ứng oxy hóa của hợp chất polyphenol.
13
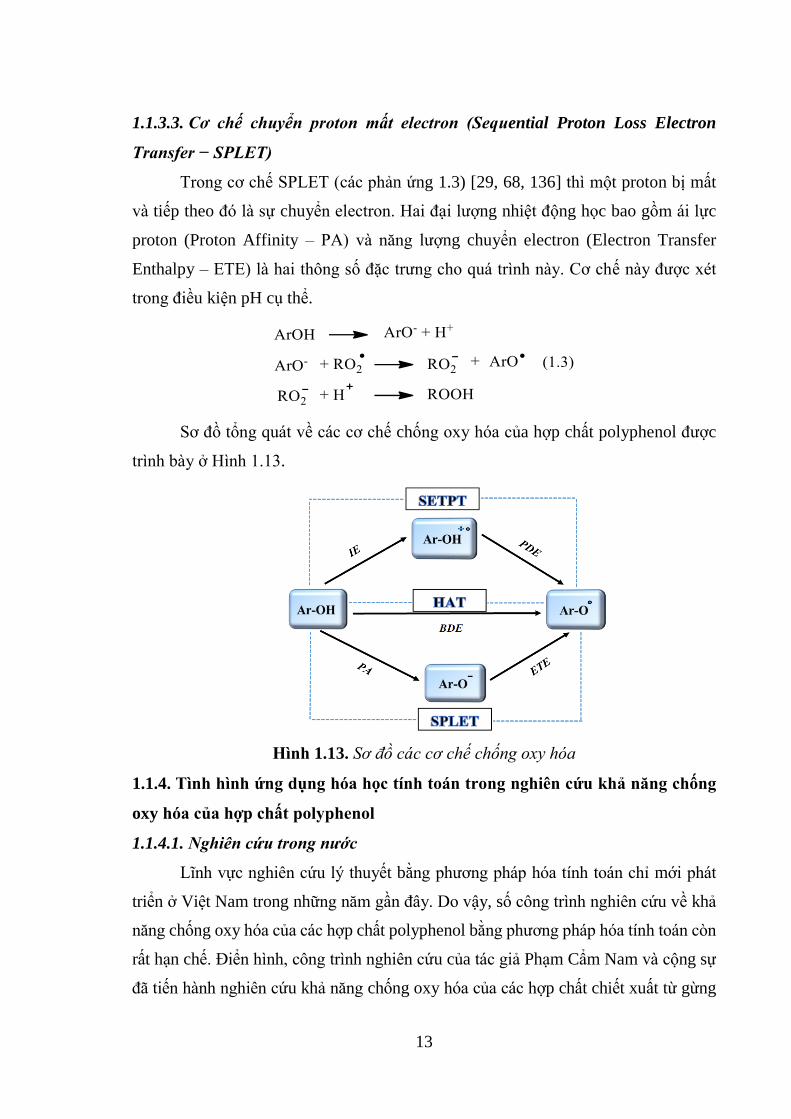
1.1.3.3. Cơ chế chuyển proton mất electron (Sequential Proton Loss Electron
Transfer − SPLET)
Trong cơ chế SPLET (các phản ứng 1.3) [29, 68, 136] thì một proton bị mất
và tiếp theo đó là sự chuyển electron. Hai đại lượng nhiệt động học bao gồm ái lực
proton (Proton Affinity – PA) và năng lượng chuyển electron (Electron Transfer
Enthalpy – ETE) là hai thông số đặc trưng cho quá trình này. Cơ chế này được xét
trong điều kiện pH cụ thể.
Sơ đồ tổng quát về các cơ chế chống oxy hóa của hợp chất polyphenol được
trình bày ở Hình 1.13.
Hình 1.13. Sơ đồ các cơ chế chống oxy hóa
1.1.4. Tình hình ứng dụng hóa học tính toán trong nghiên cứu khả năng chống
oxy hóa của hợp chất polyphenol
1.1.4.1. Nghiên cứu trong nước
Lĩnh vực nghiên cứu lý thuyết bằng phương pháp hóa tính toán chỉ mới phát
triển ở Việt Nam trong những năm gần đây. Do vậy, số công trình nghiên cứu về khả
năng chống oxy hóa của các hợp chất polyphenol bằng phương pháp hóa tính toán còn
rất hạn chế. Điển hình, công trình nghiên cứu của tác giả Phạm Cẩm Nam và cộng sự
đã tiến hành nghiên cứu khả năng chống oxy hóa của các hợp chất chiết xuất từ gừng

14
và chè xanh bằng phương pháp hóa tính toán. Trong đó, tác giả đã chọn hai hợp chất
epicatechin (EC) và epigallocatechin gallate (EGCG) để tiến hành nghiên cứu khả năng
chống oxy hóa bằng phương pháp ONIOM(ROB3LYP/6-311++G(2df,2p):AM1)
thông qua cơ chế chuyển nguyên tử hydro [85].
1.1.4.2. Nghiên cứu ngoài nước
Nenadis N. và công sự đã sử dụng phương pháp DFT/B3LYP nghiên cứu khả
năng dập tắt gốc tự do của các hợp chất 3′,4′-dihydroxy aurones có trong tự nhiên
thông qua hai đại lượng BDE(O−H) và IE [87]. Kết quả cho thấy những hợp chất
aurones là những chất chống oxy hóa hiệu quả, có thể ứng dụng trong lĩnh vực thực
phẩm và dược phẩm.
Phương pháp DFT/B3LYP cũng được nhóm tác giả Mikulski D. sử dụng để
nghiên cứu khả năng chống oxy hóa của các hợp chất polyphenol chiết xuất từ hạt
đậu phộng và sự chuyển hóa của hợp chất trans-resveratrol. Kết quả nghiên cứu đã
làm sáng tỏ về mối quan hệ giữa cấu trúc và khả năng chống oxy hóa của các hợp
chất trans-resveratrol thông qua các đại lượng BDE, IE, HOMO và mật độ spin [81].
Khả năng chống oxy hóa của hợp chất deoxybenzoins được nghiên cứu bởi
Xue Y. và cộng sự bằng phương pháp DFT thông qua ba cơ chế HAT, SET−PT và
SPLET. Tác giả đề nghị rằng cơ chế HAT ưu tiên giải thích khả năng chống oxy hóa
của hợp chất deoxybenzoins trong pha khí, còn cơ chế SPLET thì được sử dụng trong
dung môi phân cực [131].
Ngoài ra, phương pháp DFT/B3LYP được nhiều tác giả sử dụng trong nghiên
cứu khả năng chống oxy hóa của hợp chất isoflavones [59], hợp chất flavonoids [51,
62, 64, 79, 100, 103], hợp chất chalcones [52, 124, 133], các vitamin [82, 83, 86] và
anthocyanidins [63, 69] thông qua ba cơ chế HAT, SET−PT và SPLET với các đại
lượng nhiệt động đặc trưng là: BDE, IE, PA, PDE và ETE.
Qua các nghiên cứu trên cho thấy phương pháp DFT/B3LYP được sử dụng
phổ biến trong nghiên cứu khả năng chống oxy hóa của các hợp chất polyphenol. Tuy
nhiên, đối với các phân tử lớn thì đây là một trở ngại rất lớn do yêu cầu khắc khe về
cấu hình máy tính. Do đó, trong luận án này chúng tôi sử dụng phương pháp ONIOM
với sự tích hợp giữa phương pháp DFT và phương pháp PM6 để nghiên cứu khả năng
chống oxy hóa của các hợp chất polyphenol.

15
1.2. TỔNG QUAN VỀ HỢP CHẤT FULLERENE VÀ DẪN XUẤT
FULLERENE
1.2.1. Lịch sử phát hiện fullerene
Fullerene hay còn gọi là Buckyball là một phân tử chứa 60 nguyên tử carbon
(C60). Trước đó, các nhà khoa học chỉ biết đến hai dạng thuần tuý carbon, đó là kim
cương và graphit. Năm 1985, nhà hoá học Anh, Harry Kroto nhờ kính viễn vọng đã
phát hiện thấy những chuỗi nguyên tử carbon kỳ lạ ở cách xa hàng tỷ kilomet (km)
trong vũ trụ, quanh những ngôi sao lớn phát ra ánh sáng đỏ. Sau đó Kroto đã có những
cuộc tiếp xúc với Richard Smalley và Robert Curl là hai nhà khoa học Mỹ đang
nghiên cứu về những nhóm nguyên tử chỉ tồn tại ngắn ngủi. Họ đã dùng chùm laser
mạnh làm cho graphit bốc hơi trong khí hêli (mô phỏng những điều kiện tồn tại ở
ngôi sao đỏ) và phát hiện thấy nhiều phân tử carbon trước đây chưa được biết, trong
đó nhiều nhất là phân tử C60, hình dạng bên ngoài giống như quả bóng đá và các nhà
khoa học gọi đó là fullerene [55]. Thông báo về phát minh trên được đăng trong Tạp
chí Nature đã gây chấn động giới khoa học. Mọi người nhanh chóng nhận ra rằng
fullerene có thể là một phân tử đem lại nhiều ứng dụng rất hữu ích. Năm 1996, Curl,
Kroto và Smalley đã được nhận giải Nobel nhờ phát minh ra fullerene.
1.2.2. Cấu trúc và liên kết của fullerene
Fullerene là các phân tử carbon dạng lồng hình cầu, đường kính khoảng 7-15
angstrom, bao gồm một số vòng năm cạnh (ngũ giác) và sáu cạnh (lục giác). Hiện
nay, các nhà khoa học đã phát hiện ra hàng trăm hình thức kết hợp khác nhau của
những hình lục giác/ngũ giác. Để tạo một chiếc lồng kín, tất cả các phân tử fullerene
có công thức C20 + m, với m là một số nguyên. Ví dụ, cấu trúc đề xuất cho C60 là một
“Truncated icosahedron”. Một Truncated icosahedron có nguồn gốc từ một
icosahedron bằng cách thay thế mười hai đỉnh bằng các vòng năm cạnh. Đồng thời
cũng chuyển đổi hai mươi mặt hình tam giác bằng vòng sáu cạnh giống như một quả
bóng đá với 12 hình ngũ giác và 20 hình lục giác [37] (Hình 1.14).
Nhiều cấu trúc lồng rỗng có thể được xây dựng bằng hình thức kết hợp khác
nhau của những hình lục giác/ngũ giác. Điều thật thú vị là mỗi cấu trúc lồng rỗng
chứa chính xác 12 ngũ giác trong khi số lượng của hình lục giác là tùy ý. Số lượng

16
vòng ngũ giác này là cần thiết để tạo thành lồng rỗng kín [54]. Fullerene nhỏ nhất có
thể sẽ là C20, có chứa 12 ngũ giác và không có hình lục giác. Đặc điểm các liên kết
trong C60, mỗi đỉnh của Truncated icosahedron bị chiếm bởi một nguyên tử carbon.
Mỗi nguyên tử carbon tạo liên kết với ba nguyên tử carbon khác gồm một liên kết đôi
và hai liên kết đơn [112].
a b c
Hình 1.14. a) Icosahedron b) Truncated Icosahedron c) Fullerenes, C60
Có 2 loại nút giao vòng (6,6) - Hình 1.15a và (5,6) - Hình 1.15b. Trong đó liên
kết nối giữa 2 vòng 6 có độ dài trung bình là 1,391Å, còn liên kết nối giữa vòng 5 và
vòng 6 có độ dài trung bình là 1,449Å [11, 39]. Do đó, liên kết ở cầu nối vòng (6,6)
có đặc điểm giống liên kết đôi còn liên kết ở cầu nối vòng (5,6) có đặc điểm gần với
liên kết đơn. Các vòng ngũ giác không có cạnh chung với nhau (Hình 1.15).
Hình 1.15. Cấu trúc của C60: a) Liên kết ở cầu nối vòng (6,6);
b) Liên kết ở cầu nối vòng (5,6)
Các nguyên tử carbon ở trạng thái lai hóa sp2 (1 orbital 2s lai hóa với 2 orbital
2p) vì tạo ba liên kết sigma với ba nguyên tử carbon lân cận và orbital 2p còn lại tạo
liên kết pi. Góc giữa trục p và vector liên kết C-C (θp) là 101,6o [36]. Đối với nguyên

17
tử C lai hóa sp2 (dạng tam giác), các orbital lai hóa nằm trên cùng mặt phẳng và góc
θp = 0o, trong khi đó nguyên tử C lai hóa sp3 (dạng tứ diện) thì θp = 19,5o (Hình 1.16).
Vì vậy nguyên tử C trong fullerene (C60) có dạng hình học gần với cấu trúc tứ diện
hơn là cấu trúc tam giác.
Hình 1.16. Góc hình chóp (p = (π - 900) của: a) C lai hóa sp2 phẳng;
b) C lai hóa sp3 tứ diện và c) C lai hóa sp2 không phẳng
1.2.3. Tính chất vật lý
1.2.3.1. Độ bền nhiệt động và động học
Nhiệt hình thành của C60 nguyên chất được xác định bằng phương pháp đo
nhiệt lượng cả lý thuyết lẫn thực nghiệm có giá trị là 10,16 kcal.mol-1/nguyên tử C
(còn đối với graphit là 0,0 kcal.mol-1/nguyên tử C) [5]. So với kim cương (0,4
kcal.mol-1/nguyên tử C) thì C60 là một dạng thù hình của carbon có năng lượng cao.
Năng lượng tương đối cao này có nguồn gốc từ sức căng cao của C60 do các orbital
sp2 của pyramidalization (sự lồng hóa) và là nguyên nhân gây ra 80% nhiệt hình thành
[35]. Điều này làm cho C60 trở thành một trong những phân tử có sức căng lớn nhất
và bền ở điều kiện tiêu chuẩn. Ngoài ra, các fullerene-cluster ở nhiệt độ rất cao sẽ bị
phân tách thành các sản phẩm fullerene. Dưới điều kiện của chùm tia ion thì các phân
tử vòng bền giống như benzene ngay lập tức bị phân hủy [4]. Tính bền động học khác
thường này của C60 có thể quan sát trong phân tích phổ khối bằng kỹ thuật bắn phá
nguyên tử nhanh (Fast atom bombardment − FAB) của dẫn xuất fullerene, trong đó
sự phân mảnh dẫn đến sự hình thành các pic phân tử của C60 là chiếm ưu thế.

18
1.2.3.2. Độ tan
Độ tan của C60 trong dung môi hữu cơ có vai trò quan trọng trong việc tinh
chế và phản ứng hóa học. Fullerene hầu như tan rất ít trong các dung môi hữu cơ vì
C60 có khuynh hướng kết tụ cao. Ngoài ra, tương tác giữa phân tử dung môi và C60 là
rất yếu vì fullerene là phân tử không phân cực, sự phân cực khó này là do khoảng
cách năng lượng giữa HOMO và LUMO lớn. Độ tan của C60 trong dung môi phân
cực gần như bằng không. Trong dung môi ankan và benzene thì C60 có độ tan thấp,
C60 tan tốt nhất trong dung môi naphthalene (Bảng 1.1).
Bảng 1.1. Độ tan của C60 trong các dung môi phổ biến [74, 102, 106, 108]
Dung môi Độ tan (mg/mL)
Acetonitrile 0,000
Acetone 0,001
Methanol 0,000
Ethanol 0,001
n-pentane 0,005
Cyclohexane 0,036
Chloroform 0,16
Dichloromethane 0,26
Tetrachloroethylene 1,2
Nitrobenzene 3,3
Benzene 1,70
Toluen 2,80
Xylenes 5,20
Anisole 5,60
o-cresol 0,014
1,2,4-trichlorobenzene 8,5
Carbondisulfide 7,9
Tetrahydrofuran 0,000
Pyridine 0,89
1-chloronaphthalene 51
1-phenylnaphthalene 50

19
1.2.4. Đặc điểm electron và khả năng phản ứng
C60 thể hiện đặc tính điện hóa học cao điều này liên quan đến cấu hình electron.
Vì có sự lai hóa lại nên làm cho C60 có ái lực electron cao, điều này làm giảm đáng
kể mức năng lượng của orbital phân tử chưa bị chiếm thấp nhất (LUMO). Theo thuyết
orbital phân tử của Huckel thì cấu hình electron của C60 gồm 5 orbital HOMO (hu), 3
orbital LUMO (t1u) và 3 orbital LUMO+1 (t2u) với khoảng cách năng lượng là 0,757β
[10, 37, 38] (Hình 1.17).
Hình 1.17. Giản đồ orbital phân tử Huckel (HMO) của C60 và benzene
Trong phản ứng oxy hóa khử, fullerene có khả năng nhận lên đến 6 electron
[23], điều này phù hợp với sự suy biến của các orbital LUMO. Bằng chứng cho thấy
sự suy biến bậc 3 của các orbital mức LUMO của C60 đó là sự xuất hiện của các ion
âm Cn-60 (n=1-6), điều này xảy ra sau khi khử thuận nghịch một electron. Từ giản đồ
HMO cho thấy nếu sự oxy hóa C60 xảy ra bằng cách di chuyển một electron ra khỏi
orbital HOMO thì sẽ làm cho hệ thống electron π kém bền. Do đó, năng lượng ion
hóa thứ nhất của C60 xảy ra ở thế dương khá cao 1,26V. Sự khác nhau giữa thế oxy
hóa và thế khử thứ nhất của C60 ((E1/ 2ox − E1/ 2
kh = 2,32 V) được đo bằng khoảng cách
năng lượng của HOMO và LUMO trong dung dịch và nó tương quan tốt với giá trị
tính toán (1,5-2,0 eV) [134].

20
Vì C60 có bộ khung cứng nhắc trong cả trạng thái nền và trạng thái kích thích
nên năng lượng kết hợp lại là rất nhỏ [41]. Sự khử của các fullerene để kết hợp lại
với nhau có năng lượng nhỏ đối với hầu hết các phản ứng, do đó chúng dễ dàng kết
hợp với nhau tạo thành những khối phân tử lớn hơn.
1.2.5. Tính thơm
Từ tính của fullerene thể hiện khá rõ ràng là do đặc điểm không định vị của
electron π liên hợp, điều này là nguyên nhân gây ra tính thuận từ hay nghịch từ của
các vòng 5 và vòng 6. Đối với phân tử C60 trung hòa thì không thể hiện rõ tính thơm
vì nó bao gồm cả diatropic hexagon và paratropic pentagon nên tính thơm còn chưa
được xác định rõ ràng. Bühl và Hirsch đã xem hệ thống electron π như một electron
hình cầu trong pha khí để xác định bản chất tính thơm theo không gian 3 chiều của
fullerene với các hợp chất nền khác [10].
Tính thơm theo không gian 2 chiều của annulenes ở trạng thái nền singlet theo
quy tắc Huckel như sau: Nếu annulenes có 4n+2 electron π thì có tính thơm nhưng
có 4n electron π thì không có tính thơm. Đối với cấu trúc vỏ đóng, thì annulenes có
4n+2 electron π thể hiện tính nghịch từ mạnh, điều này phù hợp với quy luật (tính đối
xứng-Dnh). Nhưng đối với cấu trúc vỏ mở ở trạng thái triplet thì quy tắc này hoàn
toàn ngược lại.
Tính thơm của các icosahedral fullerene (C20, C60 và C80) và fullerene cluster
phụ thuộc vào số electron không định vị trong lớp electron hóa trị. Số electron hình
cầu lớn nhất của các cluster có thể đạt được là 2(n+1)2 electron để điền đầy vào lớp
vỏ. Quy tắc 2(n+1)2 miêu tả tương tự như quy tắc Huckel cho annulenes.
1.2.6. Tính chất hóa học
Từ khi phát hiện ra fullerene, một loạt các phản ứng hóa học với fullerene đã
được tiến hành và có thể được phân thành nhiều loại khác nhau [42, 58, 113, 119],
được mô tả ở Hình 1.18:

21
Hình 1.18. Một số phản ứng của C60
Một số phản ứng chính sau dùng để chức năng hóa fullerene tạo thành sản
phẩm exohedral fullerene như sau:
- Phản ứng cyclopropan hóa với tác nhân carbon nucleophile [7, 8].
- Phản ứng đóng vòng kiểu Diels-Alder (4+2) [53, 109].
- Phản ứng quang hóa đóng vòng (2+2) của α, β-keton chưa bão hòa [127, 128].
- Phản ứng cộng (3+2) dùng hợp chất diazo, azomethine và azides [96, 101].
- Phản ứng tạo phức với kim loại chuyển tiếp [3].
- Phản ứng cộng oxy, cộng hydro và cộng halogen.
Những nghiên cứu chính của phản ứng chức năng hóa fullerene được đề cập
trong luận án này là dựa trên methanofullerene. Trong luận án này chúng tôi quan
tâm tới phản ứng đóng vòng cyclopropan (phản ứng Bingel) của C60 với carbon
nucleophile. Đây là một trong những phản ứng phổ biến nhất để tổng hợp các dẫn
xuất fullerene cho hiệu suất cao và đa dạng. Các điều kiện ban đầu cho phản ứng
Bingel liên quan đến việc xử lý bromomalonates với NaH trong sự có mặt của C60
[7]. Phản ứng này xảy ra theo hai bước: Bước đầu tiên là sự tách proton

22
(deprotonation) của α-bromomalonate để tạo thành α-brom carbanion, sau đó tác
nhân carbanion này cộng vào C60 tạo thành phức trung gian là ion âm fullerene. Bước
thứ hai là sự tách ion âm brom do sự tương tác nucleophile nội phân tử để tạo thành
methanofullerene (Hình 1.19). Phản ứng Bingel chỉ xảy ra duy nhất ở liên kết (6,6).
Bởi vì tổng hợp các phức tiền chất bromomalonate thường rất khó khăn, do đó Hirsch
đã hiệu chỉnh phản ứng này bằng cách tổng hợp α-halomalonate từ malonate tương
ứng với sự có mặt của CBr4 và bazơ không có tính nucleophile (DBU) (vì vậy phản
ứng này còn được gọi là phản ứng Bingel - Hirsch) [12]. Trong một vài trường hợp
thì Iod có thể được sử dụng làm tác nhân halogen hóa [88]. Phản ứng này xảy ra trong
điều kiện êm dịu và có thể áp dụng cho một loạt các nhóm chức. Các
methanofullerenes tương ứng nói chung bền nhiệt do vòng cyclopropane cứng chắc.
Phản ứng tổng quát và cơ chế phản ứng Bingel được mô tả ở Hình 1.19.
Hình 1.19. Cơ chế phản ứng Bingel
1.2.7. Tình hình nghiên cứu fullerene và dẫn xuất fulerene
Gần đây, nhiều ý tưởng mới đã được thực hiện thành công trong thực nghiệm
để tạo ra các ứng dụng tiềm năng cho dẫn xuất C60 (các dẫn xuất fullerene hòa tan
trong nước) cho nhiều lĩnh vực như chất chống oxy hóa, các chất bảo vệ thần kinh,
ức chế enzyme, hoạt tính kháng khuẩn và hoạt tính kháng HIV. Các nghiên cứu cả lý
thuyết và thực nghiệm về các dẫn xuất fullerene đã chỉ ra rằng chúng thể hiện một số
loại hoạt tính sinh học, có thể ứng dụng cho mục đích y tế [18, 46]. Ví dụ, C60 và các
dẫn xuất của nó có khả năng dập tắt một số lượng lớn các gốc tự do [6, 84] chúng có
tiềm năng như một loại thuốc có khả năng hữu ích trong việc ngăn ngừa hoặc điều trị
các bệnh lý liên quan đến sự oxy hóa, cụ thể là việc điều trị bệnh thoái hóa thần kinh
[22], chất ức chế enzyme HIV-1 [30, 73, 107] hoặc trong các liệu pháp xạ trị các mô
ung thư [110].

23
CHƯƠNG 2. NỘI DUNG VÀ PHƯƠNG PHÁP NGHIÊN CỨU
2.1. NỘI DUNG NGHIÊN CỨU
- Khảo sát phương pháp tính năng lượng phân ly liên kết O−H.
- Khảo sát phương pháp tính năng lượng ion hóa.
- Nghiên cứu cấu trúc, khả năng chống oxy hóa của các hợp chất polyphenol:
+ Khả năng chống oxy hóa của các hợp chất có nguồn gốc từ cây sa kê.
+ Khả năng chống oxy hóa của các hợp chất có nguồn gốc từ vỏ măng cụt.
+ Khả năng chống oxy hóa của các hợp chất chalcone, flavone và flavanone.
- Nghiên cứu khả năng chống oxy hóa của dẫn xuất fullerene - polyphenol:
+ Chức năng hóa fullerene bằng các dẫn xuất malonate.
+ Khả năng chống oxy hóa của các dẫn xuất fullerene-polyphenol.
+ Thiết kế các hợp chất có khả năng chống oxy hóa có nguồn gốc từ cây sa
kê và măng cụt trên nền fullerene.
2.2. TỔNG QUAN VỀ PHƯƠNG PHÁP TÍNH TOÁN
2.2.1. Cơ sở phương pháp tính toán hóa lượng tử
2.3.1.1. Phương trình Schrödinger
- Dạng phụ thuộc thời gian (dạng tổng quát) [98]
Sự biến đổi trạng thái vi mô theo thời gian của hệ lượng tử được mô tả bởi
phương trình Schrödinger có dạng tổng quát:
iħ∂Ψ
∂t= ĤΨ(r,t) (2.1)
Ψ(r,t) – Hàm sóng mô tả trạng thái của hệ theo tọa độ (r) và thời gian (t).
Ĥ – Toán tử Hamilton của hệ.
- Dạng không phụ thuộc thời gian (ở trạng thái dừng) [98]
Giả sử hệ lượng tử ở vào một trường thế U không phụ thuộc thời gian, chỉ
phụ thuộc vào tọa độ Û = U(r), thì Ĥ không phụ thuộc vào thời gian. Lúc đó Ĥ chỉ
tác động lên phần phụ thuộc tọa độ của hàm Ψ(r,t). Do đó, hàm Ψ(r,t) tách làm hai
phần: Ψ(r,t) = Ψ(r)F(t)

24
Vì hệ lượng tử được xem như là một hệ ổn định theo thời gian nên hàm sóng
và năng lượng của hệ được xác định bởi phương trình Schrödinger không phụ thuộc
thời gian:
Ĥ Ψ(r) = EΨ(r) (2.2)
Trong phương trình Schrödinger không phụ thuộc thời gian của hệ lượng tử,
toán tử Hamiton bao gồm toán tử động năng T và toán tử thế năng V cho tất cả các
hạt (hạt nhân và electron)
Toán tử Ĥ dùng cho hệ bao gồm M hạt nhân và N electron được viết dưới
dạng:
Ĥ = ∑1
2
N
i=1
∇i2 − ∑
1
2MA
∇A2
M
A=1
− ∑ ∑ZA
riA
M
A=1
N
i=1
+ ∑ ∑1
rij
N
j>1
N
i=1
+ ∑ ∑ZAZB
rAB
(2.3)
M
B>𝐴
M
A=1
Ở đây A, B biểu thị cho số hạt nhân (M), còn i, j biểu thị cho số electron của hệ (N)
ZA, ZB - Điện tích của các hạt nhân A, B
rij - Khoảng cách giữa các electron thứ i và thứ j
riA - Khoảng cách giữa các electron thứ i và hạt nhân A
rAB - Khoảng cách giữa hạt nhân A và B
Do khối lượng electron nhỏ hơn hàng nghìn lần so với khối lượng hạt nhân nên
có thể coi các hạt nhân là đứng yên. Một cách gần đúng trong tính toán hóa lượng tử
người ta xem động năng của các hạt nhân bị triệt tiêu và thế năng đẩy giữa các hạt nhân
là một hằng số. Vì vậy, thực chất toán tử Ĥ ở đây là toán tử Hamilton electron – Ĥel.
Ĥel = − ∑1
2
N
i=1
∇i2 − ∑ ∑
ZA
riA
M
A=1
N
i=1
+ ∑ ∑1
rij
N
j>1
N
i=1
(2.4)
Ở đây:
∑1
2
N
i=1
∇i2 là thành phần động năng (Telectron) của các electron
∑ ∑ZA
riA
M
A=1
N
i=1
là thành phần thế năng hút electron − hạt nhân (Velectron−nhân)

25
∑ ∑1
rij
N
j>1
N
i=1
là thành phần thế năng đẩy electron − electron (Velectron−electron)
Ψ là hàm số theo tọa độ của tất cả các hạt nhân và electron, mô tả trạng thái
của toàn bộ hệ:
Ψ = Ψ(r1, r2,……., rn, R1, R2,……, Rn) với ri và Ri tương ứng là tọa độ của các
electron và hạt nhân.
Ngoại trừ những hệ một electron − một hạt nhân, phương trình Schrödinger
cho hệ nhiều electron không thể giải một cách chính xác vì không thể xác định được
toán tử thế năng đẩy giữa electron và electron.
2.3.1.2. Bộ hàm cơ cở (basis functions)
Lời giải gần đúng tốt nhất cho phương trình Schrödinger phụ thuộc không chỉ
vào việc hiệu chỉnh phương pháp tính toán, mà còn cách chọn bộ hàm cơ sở cho hệ
nghiên cứu. Bộ hàm cơ sở là biểu diễn toán học của các orbital trong hệ. Các orbital
phân tử (Molecular Orbital − MO) trong hệ được biểu diễn dưới dạng tổ hợp tuyến tính
của một tập hợp xác định gồm các hàm đơn electron, gọi là các hàm cơ sở [28, 98].
Các hàm cơ sở thường được đặt trên các nhân nguyên tử và có những nét tương tự như
các orbital nguyên tử (Atomic Orbital − AO) nhưng có tính tổng quát hơn. Các bộ hàm
cơ sở có thể là một tập hợp bất kỳ các hàm đã được định nghĩa một cách thích hợp.
Nếu bộ hàm cơ sở gồm n hàm cơ sở Ψ1, Ψ2, Ψ3… Ψn thì một MO Ψi có dạng:
Ψi = c1iΨ1 + c2iΨ2 +…. + cniΨn (2.5)
Trong đó: cμi: hệ số khai triển orbital phân tử ( = 1, 2, 3… n)
Ψμ: các hàm cơ sở chuẩn hóa
Biểu thức (2.5) được gọi là biểu thức tổ hợp tuyến tính các orbital nguyên tử
(LCAO). Tập hàm Ψμ được gọi là tập hàm cơ sở, được dùng trong tính toán hóa lượng
tử. Bộ hàm cơ sở càng lớn việc miêu tả electron trong hệ càng gần với thực tế (sự hạn
chế electron trong không gian càng giảm), mức độ gần đúng càng tốt và ngược lại. Các
hàm này được xây dựng dựa trên các hàm sóng s, p, d… đã được giải đúng trong trường
hợp nguyên tử hiđro và những hệ tương tự hiđro. Với những hệ có nhiều hơn một
electron thì áp dụng thêm các cách tính gần đúng. Có 2 loại bộ hàm cơ sở thường gặp

26
là bộ hàm kiểu Slater – STO (Slater Type Orbital) và kiểu Gaussian – GTO (Gaussian
Type Orbital). Một số bộ hàm cơ sở thường được sử dụng trong tính toán như: bộ hàm
cơ sở tối thiểu (minimal basis set); bộ hàm cơ sở hóa trị tách (split valence basis set);
bộ hàm cơ sở double zeta (double zeta basis set); bộ hàm cơ sở phân cực (polarized
basis set); bộ hàm cơ sở khuếch tán (diffusion basis set); bộ hàm cơ sở tương quan
electron của Dunning (Dunning’s correlation consistent basis set) [28].
Đối với các hợp chất hữu cơ nghiên cứu trong luận án này thì bộ hàm cơ sở 6-
31G(d), 6-311++G(2df,2p) được sử dụng.
2.2.2. Phương pháp bán thực nghiệm (semi-empirical methods)
Phương pháp này sử dụng một số tham số thực nghiệm để giản lược các phép
giải gần đúng của phương trình Schrödinger. Do vậy, các phương pháp này cần ít thời
gian tính toán hơn và có thể áp dụng cho các phân tử lớn hay rất lớn. Có nhiều phương
pháp bán thực nghiệm, chẳng hạn như AM1, PM3, PM6... được sử dụng trong bộ
phần mềm Gaussian [28]. Mỗi phương pháp sử dụng một bộ tham số khác nhau.
Đặc điểm của phương pháp bán thực nghiệm:
- Dùng cho các hệ phân tử lớn, là các hệ không thể dùng các phương pháp với
yêu cầu khắt khe về cấu hình hình máy tính và cần nhiều thời gian để tính toán.
- Dùng cho các phân tử ở trạng thái cơ bản, là các phân tử mà dựa vào đó các
phương pháp bán thực nghiệm đã được tham số hóa.
- Dùng để tính sơ bộ đối với các hệ phân tử lớn. Thí dụ, có thể tối ưu hóa bằng
phương pháp bán thực nghiệm trên một hệ phân tử lớn để có được cấu trúc dùng làm
cấu trúc bắt đầu cho phép tính năng lượng điểm đơn bằng phương pháp HF hay DFT.
- Để nhận được những thông tin định tính về một phân tử như các orbital, điện
tích, tần số dao động.
- Trong nhiều trường hợp, các phương pháp bán thực nghiệm có thể dùng rất
thành công trong việc dự đoán năng lượng ion hóa, năng lượng cấu dạng, hiệu ứng
nhóm thế… một cách định tính hay định lượng. Tuy nhiên, cần thận trọng trong các
công việc này.

27
Trong luận án này, phương pháp PM6 được sử dụng để tính năng lượng ion
hóa và ái lực electron của các hợp chất flavonoid và dẫn xuất fullerene do thời gian
tính toán hợp lý và độ chính xác chấp nhận được.
2.2.3. Phương pháp lý thuyết phiếm hàm mật độ (Density Functional Theory -
DFT methods)
Lý thuyết phiếm hàm mật độ (DFT) là lý thuyết cơ học lượng tử dựa trên mật
độ electron ρ(r) = │Ψ(r)│2 thay vì hàm sóng Ψ(r) để tính năng lượng E của hệ [118].
Trong lý thuyết này, mật độ electron chỉ phụ thuộc vào ba biến tọa độ không
gian mà không phụ thuộc vào số electron trong hệ.
Nhìn chung, kết quả tính toán theo phương pháp DFT chính xác nhất là đối
với những phép tính hiệu chỉnh gradient hoặc phép tính lai (hybrid).
Các hàm hiệu chỉnh gradient: Hàm hiệu chỉnh gradient bao gồm giá trị của cả
mật độ spin electron và gradient. Hàm trao đổi hiệu chỉnh gradient phổ biến là hàm
do Becke đề xuất vào năm 1988, hàm tương quan hiệu chỉnh gradient được sử dụng
rộng rãi là hàm LYP của Lee – Yang – Parr. Sự kết hợp của 2 dạng này tạo nên
phương pháp B-LYP. Perdew cũng đề nghị một số hàm tương quan hiệu chỉnh quan
trọng như Perdew 86 và Perdew-Wang 91 [28, 118].
Các hàm lai: Hàm lai được định nghĩa là hàm trao đổi, là sự kết hợp tuyến tính
của phép tính Hartree – Fock, phép tính cục bộ và phép tính trao đổi hiệu chỉnh
gradient. Hàm trao đổi này sau đó kết hợp với hàm cục bộ và/hoặc hàm tương quan
hiệu chỉnh gradient. Những hàm lai được biết nhiều nhất là hàm 3 tham biến của
Becke: B3LYP và B3PW91. Hàm lai của Becke được xem là có tính ưu việt so với
nhiều loại hàm truyền thống [28, 118].
Hiện nay, phương pháp hỗn hợp B3LYP là một trong những phương pháp
được sử dụng rộng rãi nhất cho các phép tính phân tử vì cho kết quả tính toán khá
chính xác trên một phạm vi rộng các hợp chất, đặc biệt là đối với các phân tử hữu cơ.
Trong luận án này, phương pháp B3LYP đã được sử dụng và thu được kết quả tốt
trong nghiên cứu cấu trúc và thuộc tính electron của các chất hữu cơ.

28
2.2.4. Phương pháp ONIOM
Một trong các nhiệm vụ của các nhà hóa học lượng tử là xây dựng và phát
triển các phương pháp tính toán sao cho đảm bảo kết quả nhận được phải có độ tin
cậy, chính xác và đúng với các quy luật tự nhiên. Cho đến nay nhiều mô hình tính
toán đã được xây dựng, phát triển và áp dụng trong các phần mềm mô phỏng trong
các hệ hóa học, hệ sinh học, các hệ vật liệu… Nổi bật trong đó là phương pháp tích
hợp đã được Keiji Morokuma phát triển. Trong phương pháp này vùng trung tâm
phản ứng tức là nơi xảy ra các quá trình hóa học (chẳng hạn sự bẻ gãy liên kết hoặc
hình thành liên kết) được xử lý với phương pháp có độ chính xác cao, trong khi đó
phần còn lại của hệ được xử lý ở mức thấp hơn. Ví dụ: loại phương pháp tích hợp
MO/MM sử dụng phương pháp orbital phân tử (MO) - phương pháp ab-initio ở mức
lý thuyết cao cho trung tâm phản ứng của hệ và sử dụng phương pháp cơ học phân tử
(MM) - phương pháp không có yêu cầu khắt khe về tính toán cho phần còn lại của hệ
phản ứng. Phương pháp QM/MM (quantum mechanics/molecular mechanics) là loại
phương pháp tích hợp được sử dụng phổ biến nhất với sự kết hợp của phương pháp
cơ học lượng tử (QM) và phương pháp cơ học phân tử (MM). Phương pháp ONIOM
là phương pháp tổng quát nhất, nó có thể kết hợp bất kỳ các phương pháp orbital phân
tử cũng như các phương pháp cơ học phân tử [121].
Phương pháp ONIOM được phát triển bởi giáo sư Keiji Morokuma và cộng
sự [19]. Phương pháp này được dùng để tính toán năng lượng, tối ưu hóa cấu trúc
hình học, dự đoán tần số dao động, đặc điểm điện tích và từ trường của các phân tử
có kích thước lớn. Phương pháp ONIOM được áp dụng cho các phân tử có kích thước
lớn trong nhiều lĩnh vực nghiên cứu khác nhau bao gồm phản ứng enzym, cơ chế
phản ứng của các hợp chất hữu cơ, quá trình quang hóa, ảnh hưởng các nhóm thế,
phản ứng của các hợp chất cơ kim, phản ứng ở bề mặt các tinh thể...
Trong phương pháp ONIOM thì hệ nghiên cứu được chia thành 2 hoặc 3 lớp
tương ứng với các mức High (cao), Medium (trung bình) và Low (thấp):
+ Lớp mức cao (High layer) tương ứng với vùng phản ứng nơi xảy ra quá trình
hình thành liên kết hoặc phá vỡ liên kết được tính toán bằng các phương pháp hóa

29
lượng tử cho kết quả có độ chính xác cao (chẳng hạn các phương pháp cơ học lượng
tử). Trong mô hình ONIOM 2 lớp, mức này được gọi là hệ mô hình (Model System).
+ Lớp mức thấp (Low layer) bao gồm toàn bộ phần còn lại của hệ phản ứng
trong mô hình ONIOM 2 lớp. Sự tính toán trong vùng này tương ứng với sự ảnh
hưởng của môi trường lên trung tâm phản ứng của hệ. Lớp mức thấp thường được áp
dụng các phương pháp cơ học phân tử, phương pháp bán thực nghiệm hoặc phương
pháp ab-initio như Hartree-Fock với các bộ hàm thích hợp.
+ Trong mô hình ONIOM 3 lớp, thì lớp trung bình (Medium layer) được xác
định và nó được xử lý bằng phương pháp có độ chính xác trung bình nằm giữa phương
pháp ở mức cao và phương pháp ở mức thấp. Lớp này cho phép mô tả sự ảnh hưởng
điện tích của môi trường phân tử lên lớp cao. Sự kết hợp lớp trung bình và lớp thấp
được gọi là hệ mô hình trung gian (Intermediate Model System). Toàn bộ phân tử
được gọi là hệ thực (Real System).
Bằng cách chia như vậy, phương pháp tính sẽ nhanh hơn nhưng kết quả và độ
chính xác của phương pháp tương đương với các phương pháp lý thuyết cao hơn đòi
hỏi cần có hệ thống máy tính với cấu hình mạnh cũng như thời gian tính toán lâu hơn.
Toàn bộ quá trình được mô tả trong Hình 2.1.
Nguyên lý cơ bản của phương pháp này là chia cấu trúc của một chất thành hai
hay ba lớp. Môt cach tông quat, trong phương phap ONIOM 2 lơp, ONIOM2 (Hình
2.1a), năng lương mưc cao cua hê thưc tê, Ehigh,real, co thê đươc tinh xâp xi băng:
Ehigh,real ≈ EONIOM2(high:low) = Ehigh,model + Elow,real − Elow,model
Sơ đô phương phap ONIOM không chi han chê ơ hai lơp ma con co thê ngoai
suy thanh ba hay nhiêu lơp. Quan sat trên Hinh 2.1b, toan bô hê "thưc" (“real”) co
thê đươc chia lam ba hê, "mô hinh" (“model”), "trung gian" (“intermediate”), "thưc"
(“real”), va sư dung ba mưc ly thuyêt tương đương, "thâp" (“low”), "trung binh"
(“medium”), "cao" (“high”). Vơi sơ đô ba mưc, ONIOM3, năng lương cua hê thưc ơ
mưc cao co thê đươc tinh xâp xi băng:
EONIOM3(high:medium:low)
= Ehigh,model + Emedium,intermediate − Emedium,model + Elow,real
− Elow,intermediate
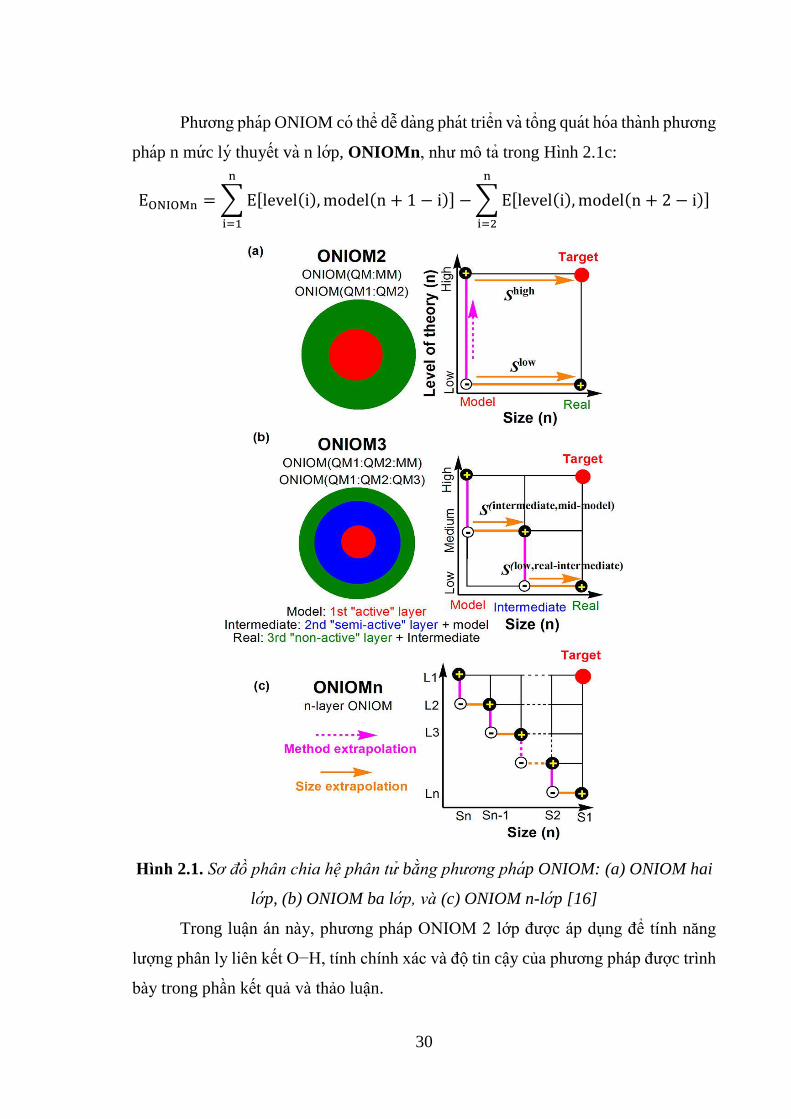
30
Phương phap ONIOM co thê dê dang phat triên va tông quat hoa thanh phương
phap n mưc ly thuyêt va n lơp, ONIOMn, như mô ta trong Hinh 2.1c:
EONIOMn = ∑ E[level(i), model(n + 1 − i)] − ∑ E[level(i), model(n + 2 − i)]
n
i=2
n
i=1
Hình 2.1. Sơ đô phân chia hê phân tư băng phương phap ONIOM: (a) ONIOM hai
lơp, (b) ONIOM ba lơp, và (c) ONIOM n-lơp [16]
Trong luận án này, phương pháp ONIOM 2 lớp được áp dụng để tính năng
lượng phân ly liên kết O−H, tính chính xác và độ tin cậy của phương pháp được trình
bày trong phần kết quả và thảo luận.

31
2.3. CÁC PHẦN MỀM TÍNH TOÁN
2.3.1. Phần mềm Gaussian 09W
Gaussian là một trong những phần mềm hiệu quả nhất để tính toán hóa lượng
tử hiện nay. Phần mềm này có hầu hết các phương pháp tính từ bán thực nghiệm đến
ab-initio và lý thuyết phiếm hàm mật độ [32]. Phần mềm này luôn được cập nhật,
nghiên cứu và phát triển bởi nhiều nhà khoa học trên thế giới, đặc biệt trong đó có
Pople J. A. là người đã đoạt giải Nobel hóa học vào năm 1998 với những đóng góp
to lớn về việc nghiên cứu phương pháp tính toán trong hóa học lượng tử.
Trên cơ sở những định luật cơ bản của cơ học lượng tử, Gaussian tính toán
năng lượng, cấu trúc phân tử và tần số dao động của hệ thống phân tử, cùng với rất
nhiều những tính chất khác của phân tử. Phần mềm Gaussian có thể được dùng để
nghiên cứu các phân tử và các phản ứng dưới các điều kiện khác nhau, bao gồm cả
những hình thái bền và những hợp chất rất khó hoặc không thể quan sát được bằng
thực nghiệm như là cấu trúc chuyển tiếp và những trạng thái trung gian có thời gian
tồn tại rất ngắn.
Phần mềm Gaussian ra đời đầu tiên vào năm 1970 với phiên bản Gaussian 70.
Sau đó, liên tục được cập nhật và hoàn thiện với các phiên bản Gaussian 76, Gaussian
80, Gaussian 82, Gaussian 86, Gaussian 88, Gaussian 90, Gaussian 92, Gaussian 94,
Gaussian 03W và Gaussian 09W. Phần mềm Gaussian 09W được cập nhật và được
bổ sung thêm nhiều tính năng mới so với phiên bản trước, như cải thiện và mở rộng
tính năng của phương pháp solvat hóa theo mô hình phân cực liên tục PCM, cho phép
xác định được nhiều thuộc tính phân tử trong dung môi, khảo sát ảnh hưởng của nhiệt
độ, áp suất, phổ hồng ngoại, phổ cộng hưởng từ hạt nhân, tăng cường khả năng dò
tìm trạng thái chuyển tiếp… Các bộ hàm cơ sở cũng rất phong phú cho phép người
sử dụng dễ dàng chọn phương án tính toán phù hợp với vấn đề của mình [28].
2.3.2. Phần mềm Gaussview 5
Gaussview 5 là phần mềm hỗ trợ cho phần mềm Gaussian 09 trong việc xây
dựng cấu trúc ban đầu. Ngoài ra, Gaussview còn được dùng để hiển thị các kết quả
tính toán của Gaussian dưới dạng đồ họa (như các cấu trúc phân tử ban đầu, các cấu
trúc phân tử đã được tối ưu hóa, kiểu dao động, phổ, hệ số orbital phân tử, mật độ
electron, điện tích ...) [31]. Nhờ đó, việc kiểm tra các kết quả tính toán trở nên đơn
giản và dễ dàng hơn.

32
2.4. HÓA HỌC TÍNH TOÁN ỨNG DỤNG CHO NGHIÊN CỨU CÁC HỢP
CHẤT POLYPHENOL VÀ DẪN XUẤT FULLERENE
2.4.1. Tối ưu hóa cấu trúc (geometry optimization)
Cấu trúc ban đầu được xây dựng bằng phần mềm Gaussview. Viêc tôi ưu hoa
câu truc phân tử va tinh tân sô dao đông đươc thưc hiên băng phương phap ban thưc
nghiêm PM6 với từ hai từ khóa “Opt” và “Freq”. Kết quả tính sẽ được kiểm tra lại
bằng phần mềm GaussView thông qua việc phân tích cấu trúc hình học và các tần số
dao động. Các cấu dạng bền (có năng lượng cực tiểu) có đặc điểm là không có tần số
âm.
Một quá trình tối ưu hoàn thành khi nó đạt những chuẩn hội tụ xác định. Bốn
chuẩn hội tụ được sử dụng mặc định trong phần mềm Gaussian [28]:
1. Lực phải bằng không (zero). Giá trị lớn nhất của lực phải nằm dưới giá trị
giới hạn 0,00045.
2. Căn bậc 2 của trị trung bình của bình phương của lực phải là không (zero)
(nhỏ hơn dung sai 0,0003).
3. Độ lệch tính toán cho bước kế tiếp phải nhỏ hơn 0,0018.
4. Căn bậc 2 trị trung bình của bình phương của độ lệch đối với bước kế tiếp
nhỏ hơn 0,0012.
Chú ý: Sự thay đổi năng lượng giữa điểm hiện tại và điểm kế tiếp không được
sử dụng là chuẩn hội tụ vì những bước nhỏ gần cực tiểu thường mang lại sự thay đổi
nhỏ trong năng lượng.
2.4.2. Xác định trạng thái chuyển tiếp và tính hàng rào năng lượng
Để xác định trạng thái chuyển tiếp, chúng tôi xây dựng cấu trúc giả định ban
đầu của trạng thái chuyển tiếp và thực hiện bằng phương pháp bán thực nghiệm PM6
với việc sử dụng đồng thời những từ khóa “opt=(ts, calcfc, noeigen) và “freq” trong
input file. Từ khóa “ts” để tối ưu tìm trạng thái chuyển tiếp, từ khóa “calcfc” dùng để
tính hằng số lực ban đầu, từ khóa “noeigen” giúp cho việc tối ưu tìm trạng thái chuyển
tiếp tự hội tụ để đưa đến một trạng thái chuyển tiếp đáng tin cậy, từ khóa “freq” được
dùng để tính toán tần số dao động bắt đầu với cấu trúc tối ưu của trạng thái chuyển tiếp.

33
Tất nhiên, cấu trúc bắt đầu để tìm trạng thái chuyển tiếp càng thích hợp thì việc đạt đến
trạng thái chuyển tiếp càng nhanh.
Để xác nhận đường phản ứng thực sự đi qua trạng thái chuyển tiếp và nối liền
hai cấu trúc có năng lượng cực tiểu là chất phản ứng và sản phẩm, chúng tôi đã tiến
hành tính tọa độ phản ứng nội (Intrinsic Reaction Coordinate - IRC) xuất phát từ điểm
yên ngựa (điểm ứng với trạng thái chuyển tiếp) cũng được tính ở cùng mức lý thuyết.
Trong một số trường hợp cần tăng số điểm trên IRC và tăng bước nhảy cho
IRC nhằm tiếp cận sát cực tiểu hơn. Từ khóa “maxpoints” nhằm xác định số điểm và
“stepsize” là bước nhảy trên đường phản ứng. Ví dụ, tính IRC với maxpoints = 10 và
stepsize = 10 có nghĩa là số điểm tính ở mỗi bên từ trạng thái chuyển tiếp hướng về
phía chất phản ứng và sản phẩm là 10 điểm, tổng số điểm trên đường IRC sẽ là 21
điểm (1 điểm ứng với trạng thái chuyển tiếp). Khoảng cách giữa hai điểm là 10 đơn
vị của 0,01 amu-1/2 bohr tức là 0,1 amu-1/2 bohr [28].
Khi khẳng định trạng thái chuyển tiếp tìm được là trạng thái chuyển tiếp mong
muốn thì tiến hành tính hàng rào năng lượng của phản ứng. Hàng rào năng lượng và
năng lượng của một phản ứng có thể được tính toán dựa vào năng lượng của các chất
phản ứng, sản phẩm và trạng thái chuyển tiếp (biểu thức 2.6, 2.7 và 2.8 và Hình 2.2).
∆Er = Echuyển tiếp - Echất phản ứng (2.6)
∆Hphản ứng = Hchất sản phẩm - Hchất phản ứng (2.7)
∆Gphản ứng = Gchất sản phẩm - Gchất phản ứng (2.8)
Hình 2.2. Giản đồ năng lượng
Chất phản ứng
Chất sản phẩm
Trạng thái chuyển tiếp
∆Er
∆Hphản ứng

34
Các thông số năng lượng tương tác được hiệu chỉnh năng lượng dao động điểm
không (Zero Point Energy − ZPE). Biến thiên enthalpy và biến thiên năng lượng tự
do Gibbs của các phản ứng được tính toán dựa trên sự khác biệt giữa tổng năng lượng
của các sản phẩm và tổng năng lượng các chất phản ứng tại nhiệt độ 298K.
2.4.3. Năng lượng điểm đơn (single point energy)
Tính toán năng lượng điểm đơn là dự đoán năng lượng và những thuộc tính
liên quan với một phân tử với cấu trúc hình học xác định. Sở dĩ ta sử dụng thuật ngữ
điểm đơn vì tính toán này được thực hiện tại một điểm duy nhất, cố định trên bề mặt
thế năng (với các thông số hình học cấu trúc đã tối ưu). Năng lượng điểm đơn là tổng
năng lượng electron và năng lượng đẩy hạt nhân của phân tử tại một cấu hình hạt
nhân cụ thể. Tuy nhiên, những dự đoán năng lượng chính xác và đầy đủ đòi hỏi phải
hiệu chỉnh năng lượng dao động điểm không−ZPE hoặc năng lượng dao động nhiệt
đối với enthalpy−TCE (Thermal Correction to Enthalpy) hoặc năng lượng dao động
nhiệt đối với năng lượng tự do Gibbs−TCGFE (Thermal Correction to Gibbs Free
Energy). Độ chính xác của kết quả phụ thuộc nhiều vào cấu trúc hợp lý của phân tử.
Việc tính năng lượng điểm đơn được thực hiện cho nhiều mục đích khác nhau, cơ
bản gồm những mục đích như sau:
- Để đạt được thông tin cơ bản về phân tử
- Để kiểm tra tính phù hợp của một phân tử sử dụng cho điểm bắt đầu của quá
trình tối ưu hóa.
- Để tính toán những giá trị gần đúng chính xác nhất của năng lượng và những
thuộc tính khác đối với một hình học được tối ưu hóa tại mức lý thuyết thấp hơn.
- Khi cần so sánh độ bền của các cấu dạng khác nhau hoặc phân tử khác nhau.
Trong luận án này, từ cấu trúc tối ưu ở mức lý thuyết PM6, các giá trị năng
lượng điểm đơn cho các phản ứng cụ thể được tính ở các mức lý thuyết như sau:
+ Năng lượng phân ly liên kết (BDE) được tiến hành ở mức lý thuyết
ONIOM(ROB3LYP/6-311++G(2df,2p):PM6). Giản đồ năng lượng liên quan đến
phản ứng giữa polyphenol và gốc tự do peroxyl, trạng thái trung gian, trạng thái
chuyển tiếp và sản phẩm được tính ở cùng mức lý thuyết trong pha khí.
+ Năng lượng ion hóa (IE) của các hợp chất nghiên cứu được tiến hành ở mức

35
lý thuyết PM6.
+ Bề mặt thế năng của phản ứng giữa fullerene và các dẫn xuất malonate, các
trạng thái trung gian, trạng thái chuyển tiếp và sản phẩm được tính ở mức lý thuyết
B3LYP/6-31G(d) trong pha khí.
Ngoài ra, chung tôi còn sư dung mô hình IEFPCM đê khao sat sư anh hương
cua dung môi đên cac đai lương enthalpy [13, 14]. Tân sô dao đông ơ mưc lý thuyết
PM6 đươc nhân thêm hê sô hiêu chinh la 1,078 vao năng lương dao đông điêm không
(ZPE) [1].
2.4.4. Các đại lượng nhiệt động xác định khả năng chống oxy hóa của hợp chất
polyphenol
Dựa trên 3 cơ chế chính được dùng để đánh giá khả năng chống oxy hóa: Cơ
chế chuyển nguyên tử hydro (HAT), cơ chế chuyển electron-chuyển proton (SET-
PT) và cơ chế mất proton chuyển electron (SPLET). Các đại lượng tương ứng: Năng
lượng đứt liên kết (BDE), năng lượng ion hóa (IE), ái lực proton (PA), năng lượng
phân ly proton (PDE) và năng lượng chuyển electron (ETE) sẽ được tính toán trong
cả pha khí và dung môi như sau:
- Năng lượng phân ly liên kết (BDE) - cơ chế HAT
BDE(O−H) = H(ArO) + H(H) - H(ArOH)
- Năng lượng ion hóa (IE) và năng lượng phân ly proton (PDE) - cơ chế SET-PT
IE = H(ArOH+) + H(e-) - H(ArOH)
PDE = H(ArO) + H(H+) - H(ArOH+)
- Ái lực proton (PA) và năng lượng chuyển electron (ETE) - cơ chế SPLET
PA = H(ArO−) + H(H+) - H(ArOH)
ETE = H(ArOH) + H(e-) - H(ArO−)
Trong đó:
H(ArOH) là enthalpy của hợp chất phenolic
H(ArO) là enthalpy của gốc tự do hợp chất phenolic
H(ArOH+) là enthalpy của ion dương gốc tự do (radical cation) hợp chất
phenolic

36
H(ArO-) là enthalpy của ion âm hợp chất phenolic
H(H) là enthalpy của gốc tự do hydro
H(H+) là enthalpy của ion dương hydro
H(e-) là enthalpy của electron
Tổng enthalpy của cấu tử X, H(X), ở nhiệt độ T được tính bằng biểu thức sau:
H(X) = Eelec + ZPE + Htrans + Hrot + Hvib + RT. Trong đó: Htrans, Hrot, và Hvib lần lượt
là enthalpy của sự tịnh tiến, sự quay và sự dao động; Eelec là tổng năng lượng electron
của hệ ở 0 K và ZPE là năng lượng dao động điểm không.
Enthalpy trong dung môi của H, H+, và e- được tính bằng các biểu thức sau:
H(H)solvat = H(H)pha khí + ∆solvatH(H)
H(H+)solvat = H(H+)pha khí + ∆solvatH(H+)
H(e-)solvat = H(e-)pha khí + ∆solvatH(e-)
2.4.5. Mô hình hóa các hệ phân tử trong dung môi
Trong phần mềm Gaussian nếu không dùng từ khóa yêu cầu tính toán trong
dung môi thì tính chất của các phân tử sẽ được mô phỏng như ở trạng thái khí. Tuy
nhiên, các tính chất và hình dạng của các phân tử chất tan đang nghiên cứu có thể sẽ
thay đổi rất nhiều khi chuyển từ trạng thái khí sang trạng thái trong dung dịch. Chính
vì vậy, khi tính toán trong phần mềm Gaussian, để có kết quả tính toán chính xác hơn,
cần thực hiện các phép tính trong dung môi phù hợp với phản ứng xảy ra trong thực tế
[28, 98].
2.5.5.1. Mô hình trường phản ứng Onsager
Mô hình trường phản ứng tự hợp (Self Consistent Reaction Field − SCRF) đơn
giản nhất là mô hình trường phản ứng Onsager. Trong phương pháp này, chất tan
chiếm một hốc hình cầu cố định có bán kính a0 trong dung môi (Hình 2.3). Lưỡng
cực điện trong phân tử sẽ bị cảm ứng bởi một lưỡng cực trong môi trường và điện
trường của lưỡng cực này sẽ tương tác trở lại lưỡng cực của phân tử để cuối cùng dẫn
đến trạng thái ổn định.

37
Hình 2.3. Mô hình Onsager (SCRF = Dipole)
Chú ý rằng, những hệ có moment lưỡng cực bằng không sẽ không thể hiện các
hiệu ứng dung môi đối với mô hình Onsager SCRF và do đó tính toán theo mô hình
Onsager (SCRF = Dipole) sẽ cho các kết quả đều như nhau đối với pha khí. Đây là
một giới hạn của phương pháp Onsager.
2.5.5.2. Mô hình IEFPCM
Mô hình phân cực liên tục sử dụng phương trình tích phân (Integral Equation
Formalism Polarizable Continuum Model − IEFPCM) hay còn gọi là mô hình phân
cực liên tục (PCM) là sự kết hợp của một chuỗi các hình cầu đại diện cho các nguyên
tử gắn chặt với nhau (Hình 2.4). Hiệu ứng của độ phân cực dung môi được số hóa:
nó được tính bằng phép lấy tích phân số chứ không theo cách tính gần đúng dưới
dạng giải tích như trong mô hình Onsager, do đó đã giải quyết được hạn chế của mô
hình Onsager.
Mô hình IEFPCM thường cho kết quả tính toán tốt và có khả năng mô tả bất
kì chất tan nào, do đó đây là một mô hình được sử dụng rộng rãi.
Hình 2.4. Mô hình IEFPCM (SCRF = IEFPCM) Mô hình IPCM
Mô hình đẳng mật độ phân cực liên tục (Isodensity Polarized Continuum
Model - IPCM) định nghĩa hốc như là một bề mặt đồng mật độ của phân tử (Hình

38
2.5). Sự đồng mật độ được xác định bởi một quá trình lặp mà trong đó vòng lặp SCF
được thực hiện và hội tụ bằng việc sử dụng hốc đồng mật độ hiện thời. Kế đó, hàm
sóng tổng hợp được dùng để tính toán bề mặt đồng mật độ đã được cập nhật và vòng
lặp SCF được lặp đi lặp lại cho đến khi hình dạng hốc không còn thay đổi nữa. Một
bề mặt đồng mật độ là mô hình rất tự nhiên và trực quan cho hốc vì nó tương ứng với
mô hình phản ứng của phân tử (hơn là một mô hình đơn giản, được định trước chẳng
hạn như một hình cầu hay một tập hợp các hình cầu phủ lên nhau).
Hình 2.5. Mô hình IPCM (SCRF = IPCM)
2.5.5.4. Mô hình SCI-PCM
Mô hình đẳng mật độ tự hợp phân cực liên tục (Self Consistent Isodensity
Polarized Continuum Model − SCIPCM) tương tự với mô hình IPCM nhưng khác
nhau trong cách thực hiện (Hình 2.6). Nó bao gồm hiệu ứng solvat hóa trong dung
dịch của bài toán SCF. Các hiệu ứng solvat hóa được đặt trong tính toán lặp SCF thay
vì sử dụng bước tính toán phụ sau đó. Mô hình SCI-PCM giải thích một cách đầy đủ
sự kết hợp giữa mô hình hốc và mật độ electron và bao gồm việc kết hợp các số hạng
mà mô hình IPCM bỏ qua.
Hình 2.6. Mô hình SCIPCM (SCRF = SCIPCM)

39
2.5.5.5. Mô hình dung môi được sử dụng trong luận án
Luận án đã chọn mô hình phân cực liên tục sử dụng phương trình tích phân
của loại trường phản ứng tự hợp để thực hiện các tính toán trên hệ phản ứng khảo sát
với từ khóa SCRF = (IEFPCM, solvent = water) áp dụng cho dung môi nước (H2O)
và từ khóa SCRF = (IEFPCM, solvent = methanol) áp dụng cho dung môi methanol
(CH3OH).

40
CHƯƠNG 3. KẾT QUẢ VÀ THẢO LUẬN
3.1. KHẢO SÁT PHƯƠNG PHÁP TÍNH NĂNG LƯỢNG PHÂN LY LIÊN KẾT
O−H
3.1.1. Cách chọn mô hình ONIOM 2 lớp
Các phương pháp sử dụng lý thuyết phiếm hàm mật độ (DFT) đã được nhiều
tác giả sử dụng để xác định đại lượng năng lượng liên kết của các hợp chất phenol,
thiophenol, aniline [70, 97]. Tuy nhiên, các phương pháp đã nêu gặp nhiều khó khăn
trong tính toán cho các phân tử có kích thước lớn (với số lượng lớn các nguyên tử)
do hạn chế về khả năng của máy tính, vì vậy cần phải có các phương pháp phù hợp
hơn. Để giải quyết vấn đề này, phương pháp ONIOM của giáo sư Keiji Morokuma
được đề nghị áp dụng [16].
Nhiều công trình nghiên cứu đã dựa trên phương pháp ONIOM để xác định
các tính chất quan trọng của các chất trong đó có năng lượng phân ly liên kết [33, 67,
123]. Guo và cộng sự đã sử dụng ONIOM-G3B3 để xác định chính xác năng lượng
phân ly liên kết OH của một số chất chống oxy hóa phổ biến như ubiquinol,
flavonoid, olive, curcuminoid cũng như các hợp chất phenolic [65]. Một số lớn các
công trình sử dụng phương pháp ONIOM để tính toán cho thấy khả năng ứng dụng
của phương pháp này cho các hệ có kích thước lớn trong sinh học và hợp chất thiên
nhiên.
Trong phương pháp ONIOM thì các giá trị enthalpy ở phương pháp mức cao
sẽ được tính từ năng lượng điểm đơn dựa trên cấu trúc đã tối ưu bằng phương pháp
PM6. Trong Hình 3.1, chúng tôi mô tả 7 cách chọn các lớp trong mô hình ONIOM.
Trong đó, mỗi phân tử được chia thành 2 lớp: các nguyên tử ở vị trí phân ly liên kết
được chọn làm lớp cao, các nguyên tử phần còn lại được chọn làm lớp thấp. Tương
ứng với lớp cao thì được tính bằng phương pháp (RO)B3LYP/6-311++G(2df,2p),
còn lớp thấp thì được tính bằng phương pháp PM6.
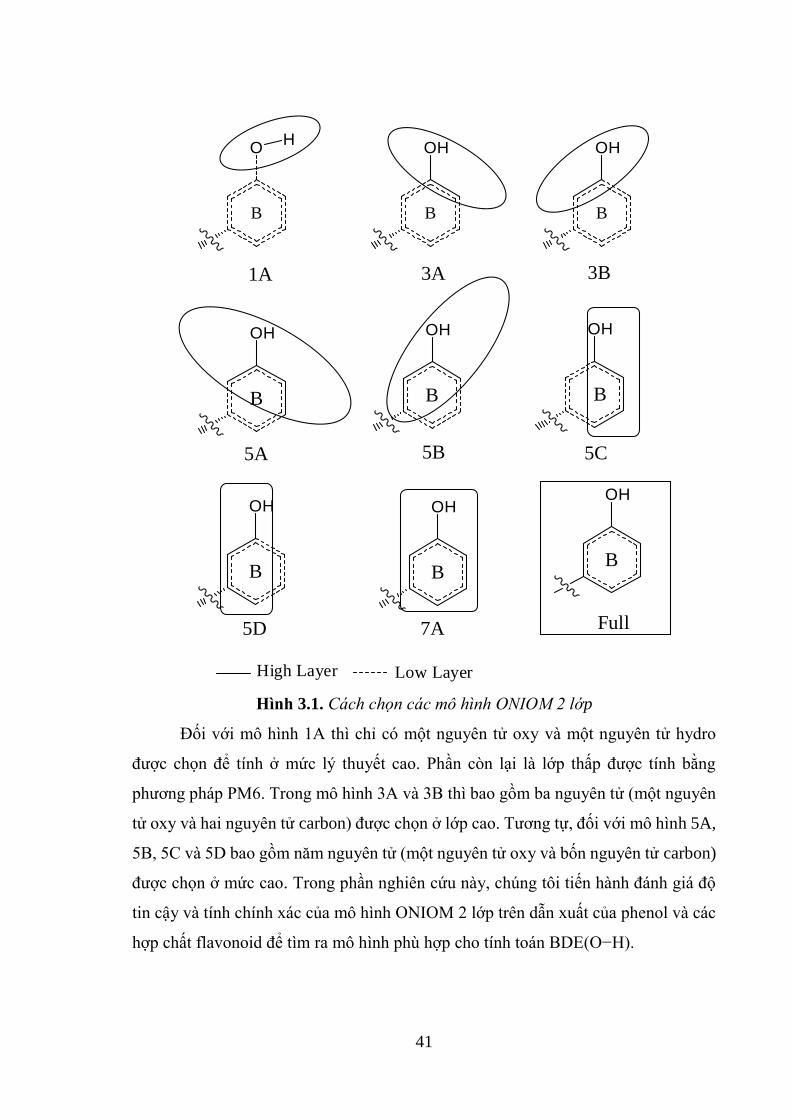
41
B
OH
1A
B
OH
3A
B
OH
3B
B
OH
5A
B
OH
5B
B
OH
5C
B
OH
5D
B
OH
Full
B
OH
7A
High Layer Low Layer
Hình 3.1. Cách chọn các mô hình ONIOM 2 lớp
Đối với mô hình 1A thì chỉ có một nguyên tử oxy và một nguyên tử hydro
được chọn để tính ở mức lý thuyết cao. Phần còn lại là lớp thấp được tính bằng
phương pháp PM6. Trong mô hình 3A và 3B thì bao gồm ba nguyên tử (một nguyên
tử oxy và hai nguyên tử carbon) được chọn ở lớp cao. Tương tự, đối với mô hình 5A,
5B, 5C và 5D bao gồm năm nguyên tử (một nguyên tử oxy và bốn nguyên tử carbon)
được chọn ở mức cao. Trong phần nghiên cứu này, chúng tôi tiến hành đánh giá độ
tin cậy và tính chính xác của mô hình ONIOM 2 lớp trên dẫn xuất của phenol và các
hợp chất flavonoid để tìm ra mô hình phù hợp cho tính toán BDE(O−H).

42
3.1.2. Áp dụng cho các dẫn xuất của phenol
Chúng tôi chọn một số dẫn xuất của phenol (Y-C6H4OH với Y = H, F, CH3,
NH2, NO2, và OH) là tiêu chuẩn so sánh giá trị BDE(O−H) tính toán với các mô hình
được mô tả ở Hình 3.1 với giá trị BDE(O−H) thực nghiệm. Chúng tôi khảo sát các
nhóm thế của phenol ở các vị trí ortho, meta và para (trừ vị trí o-NO2 và o-NH2). Kết
quả tính toán BDE(O−H) với các nhóm thế của phenol được trình bày ở Hình 3.2.
Kết quả tính toán ở Hình 3.2 cho thấy độ lệch lớn nhất giữa giá trị BDE(O-H)
tính toán sử dụng mô hình 1A và giá trị BDE(O-H) thực nghiệm là 1,3 kcal/mol đối
với trường hợp của o-CH3C6H4OH. Đối với các nhóm thế còn lại của hợp chất phenol,
độ lệch giữa giá trị tính toán lý thuyết và thực nghiệm rất nhỏ, nằm trong khoảng từ
0,5 kcal/mol đến 1,0 kcal/mol. Tuy nhiên, khi chúng tôi áp dụng mô hình 3A và 3B
thì sự khác nhau giữa giá trị tính toán lý thuyết và giá trị thực nghiệm tăng lên một
các đáng kể, độ lệch trung bình là 5,2 kcal/mol. Với mô hình 5A, 5B, 5C và 5D thì
kết quả cho sai số lớn nhất, độ lệch trung bình nằm trong khoảng 5,6 kcal/mol đến
11,2 kcal/mol.
Các kết quả tính toán trong Hình 3.2 chỉ ra rằng các giá trị BDE(O−H) sử dụng
mô hình 1A có độ chính xác cao và gần với giá trị thực nghiệm. Để đánh giá mức độ
hợp lý của cách chọn các lớp cao và lớp thấp trong mô hình 1A, chúng tôi tiến hành
tính giá trị ∆S là sai số về năng lượng tính bằng phương pháp ONIOM so với năng
lượng của toàn phân tử tính ở mức lý thuyết cao [122]. Biểu thức của ∆S được tính
như sau:
∆S = BDE(ONIOM) - BDE(high,real)
= [BDE(low,real) - BDE(low,model)] - [BDE(high,real) - BDE(high,model)]
= S(low) - S(high)
Trong đó: S(level) = BDE(level,real) - BDE(level,model) là “ảnh hưởng của
các nhóm thế” lên năng lượng phân ly liên kết được tính ở các mức lý thuyết khác
nhau. Nếu ảnh hưởng của nhóm thế được tính ở mức thấp gọi là S(low), tương tự như
vậy khi tính ở mức cao gọi là S(high). Sai số ∆S của ONIOM thì bằng zero.
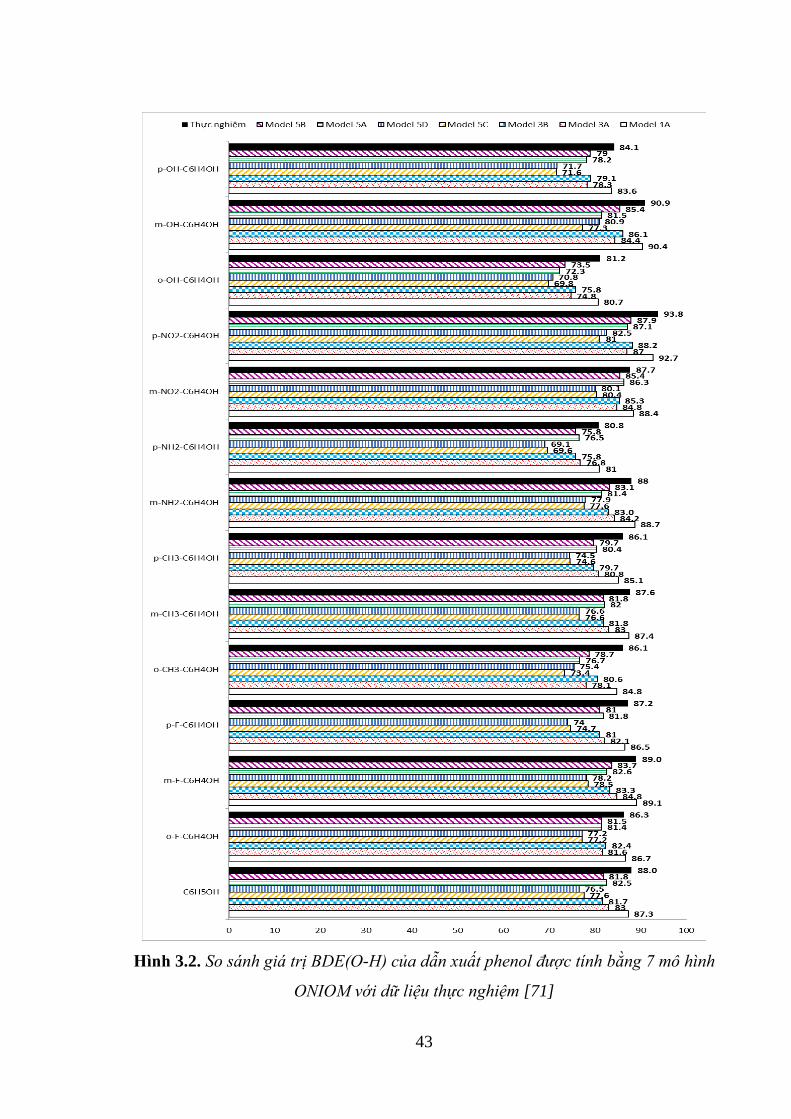
43
Hình 3.2. So sánh giá trị BDE(O-H) của dẫn xuất phenol được tính bằng 7 mô hình
ONIOM với dữ liệu thực nghiệm [71]
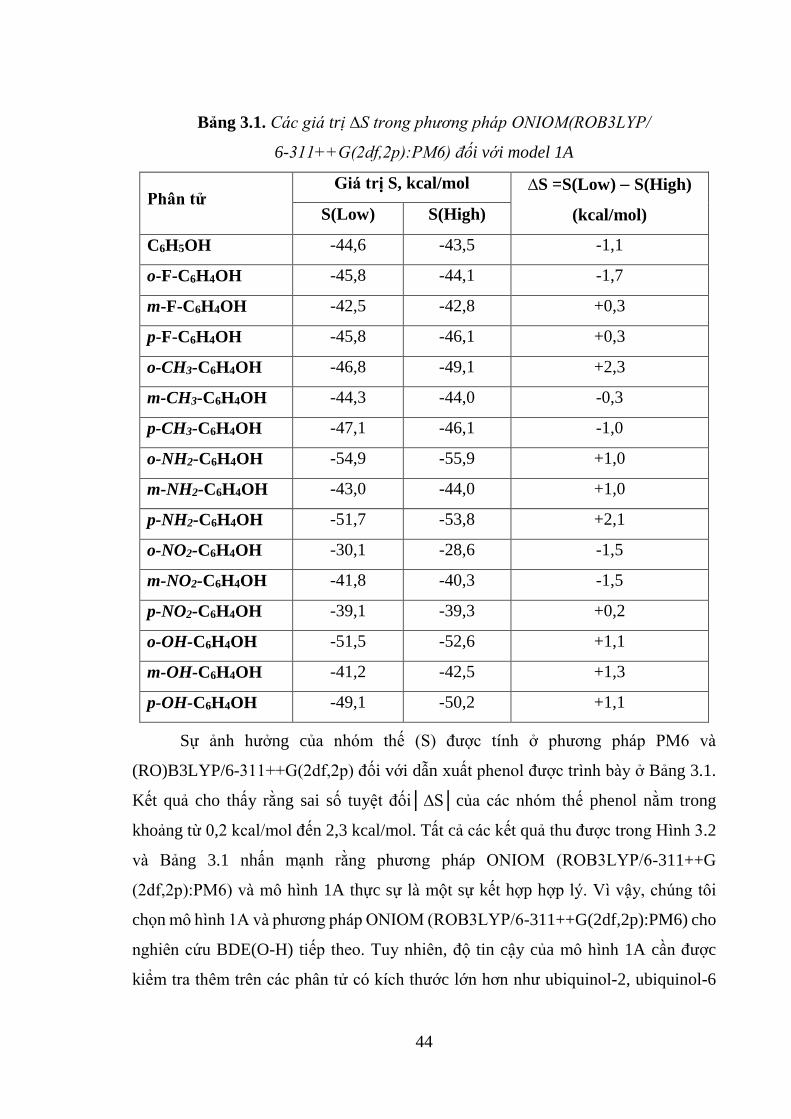
44
Bảng 3.1. Các giá trị ∆S trong phương pháp ONIOM(ROB3LYP/
6-311++G(2df,2p):PM6) đối với model 1A
Phân tử Giá trị S, kcal/mol ∆S =S(Low) S(High)
(kcal/mol) S(Low) S(High)
C6H5OH -44,6 -43,5 -1,1
o-F-C6H4OH -45,8 -44,1 -1,7
m-F-C6H4OH -42,5 -42,8 +0,3
p-F-C6H4OH -45,8 -46,1 +0,3
o-CH3-C6H4OH -46,8 -49,1 +2,3
m-CH3-C6H4OH -44,3 -44,0 -0,3
p-CH3-C6H4OH -47,1 -46,1 -1,0
o-NH2-C6H4OH -54,9 -55,9 +1,0
m-NH2-C6H4OH -43,0 -44,0 +1,0
p-NH2-C6H4OH -51,7 -53,8 +2,1
o-NO2-C6H4OH -30,1 -28,6 -1,5
m-NO2-C6H4OH -41,8 -40,3 -1,5
p-NO2-C6H4OH -39,1 -39,3 +0,2
o-OH-C6H4OH -51,5 -52,6 +1,1
m-OH-C6H4OH -41,2 -42,5 +1,3
p-OH-C6H4OH -49,1 -50,2 +1,1
Sự ảnh hưởng của nhóm thế (S) được tính ở phương pháp PM6 và
(RO)B3LYP/6-311++G(2df,2p) đối với dẫn xuất phenol được trình bày ở Bảng 3.1.
Kết quả cho thấy rằng sai số tuyệt đối│∆S│của các nhóm thế phenol nằm trong
khoảng từ 0,2 kcal/mol đến 2,3 kcal/mol. Tất cả các kết quả thu được trong Hình 3.2
và Bảng 3.1 nhấn mạnh rằng phương pháp ONIOM (ROB3LYP/6-311++G
(2df,2p):PM6) và mô hình 1A thực sự là một sự kết hợp hợp lý. Vì vậy, chúng tôi
chọn mô hình 1A và phương pháp ONIOM (ROB3LYP/6-311++G(2df,2p):PM6) cho
nghiên cứu BDE(O-H) tiếp theo. Tuy nhiên, độ tin cậy của mô hình 1A cần được
kiểm tra thêm trên các phân tử có kích thước lớn hơn như ubiquinol-2, ubiquinol-6

45
và ubiquinol-10 (trong đó ubiquinol-10 là một chất oxy hóa mạnh). Hình 3.3 mô tả
cách cấu trúc của các ubiquinol với mô hình 1A, trong đó lớp cao có định dạng là các
quả cầu liên kết còn lớp thấp có định dạng hình que. Kết quả tính BDE(O−H) của các
ubiquinol được trình bày trong Bảng 3.2 cho thấy rằng giá trị BDE(O−H) tính toán
lý thuyết có sự tương đồng rất tốt so với giá trị thực nghiệm với độ lệch chỉ ±1
kcal/mol.
Bảng 3.2. Giá trị BDE(O−H) của các ubiquinol (kcal/mol)
Phân tử BDE(O–H) Giá trị thực nghiệma BDE(O–H)b
Ubiquinol-2 81,9 82,3 -0,4
Ubiquinol-6 81,9 82,3 -0,4
Ubiquinol-10 77,8 78,5 -0,7
aTham khảo từ tài liệu [71].
bBDE(O–H) = BDE(O–H)ONIOM 1A – BDE(O–H)thực nghiệm.
Hình 3.3. Cấu trúc và mô hình ONIOM 2 lớp của ubiquinol-n (n = 2, 6, 10)
3.1.3. Áp dụng cho các hợp chất flavonoid
Trong Hình 3.4, một loạt các flavonoid bao gồm chalcone, flavone và
flavanone được dùng để tính BDE(O-H) với các mô hình ONIOM khác nhau. Đây là
những hợp chất phenolic thu hút nhiều sự quan tâm của các nhà khoa học bởi vì chúng
có khả năng chống oxy hóa mạnh [16-19].
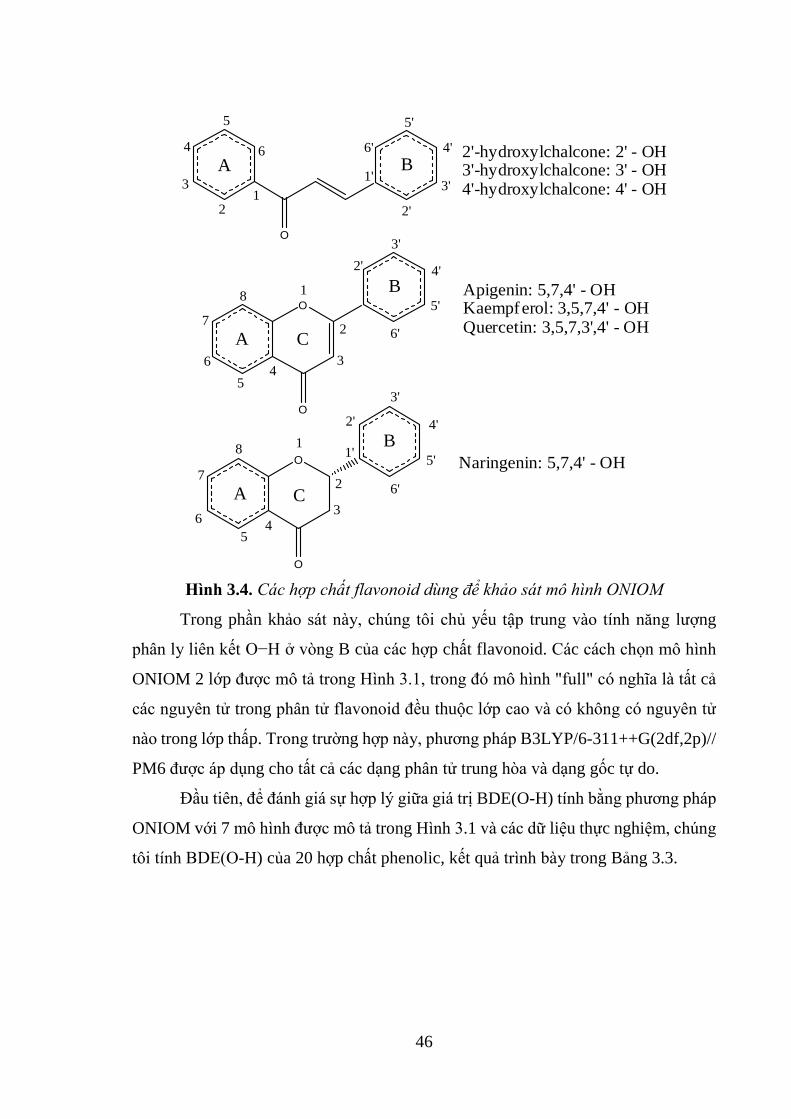
46
O
1'
2'
3'
4'
5'
6'
A B
12
3
4
5
6 2'-hydroxylchalcone: 2' - OH3'-hydroxylchalcone: 3' - OH4'-hydroxylchalcone: 4' - OH
O
A C
B
2'
3'
4'
5'
6'2
34
5
6
7
8
O
Apigenin: 5,7,4' - OHKaempferol: 3,5,7,4' - OHQuercetin: 3,5,7,3',4' - OH
O
A C
B
2'
3'
4'
5'
6'2
3
45
6
7
8
O
Naringenin: 5,7,4' - OH
11'
1
Hình 3.4. Các hợp chất flavonoid dùng để khảo sát mô hình ONIOM
Trong phần khảo sát này, chúng tôi chủ yếu tập trung vào tính năng lượng
phân ly liên kết O−H ở vòng B của các hợp chất flavonoid. Các cách chọn mô hình
ONIOM 2 lớp được mô tả trong Hình 3.1, trong đó mô hình "full" có nghĩa là tất cả
các nguyên tử trong phân tử flavonoid đều thuộc lớp cao và có không có nguyên tử
nào trong lớp thấp. Trong trường hợp này, phương pháp B3LYP/6-311++G(2df,2p)//
PM6 được áp dụng cho tất cả các dạng phân tử trung hòa và dạng gốc tự do.
Đầu tiên, để đánh giá sự hợp lý giữa giá trị BDE(O-H) tính bằng phương pháp
ONIOM với 7 mô hình được mô tả trong Hình 3.1 và các dữ liệu thực nghiệm, chúng
tôi tính BDE(O-H) của 20 hợp chất phenolic, kết quả trình bày trong Bảng 3.3.
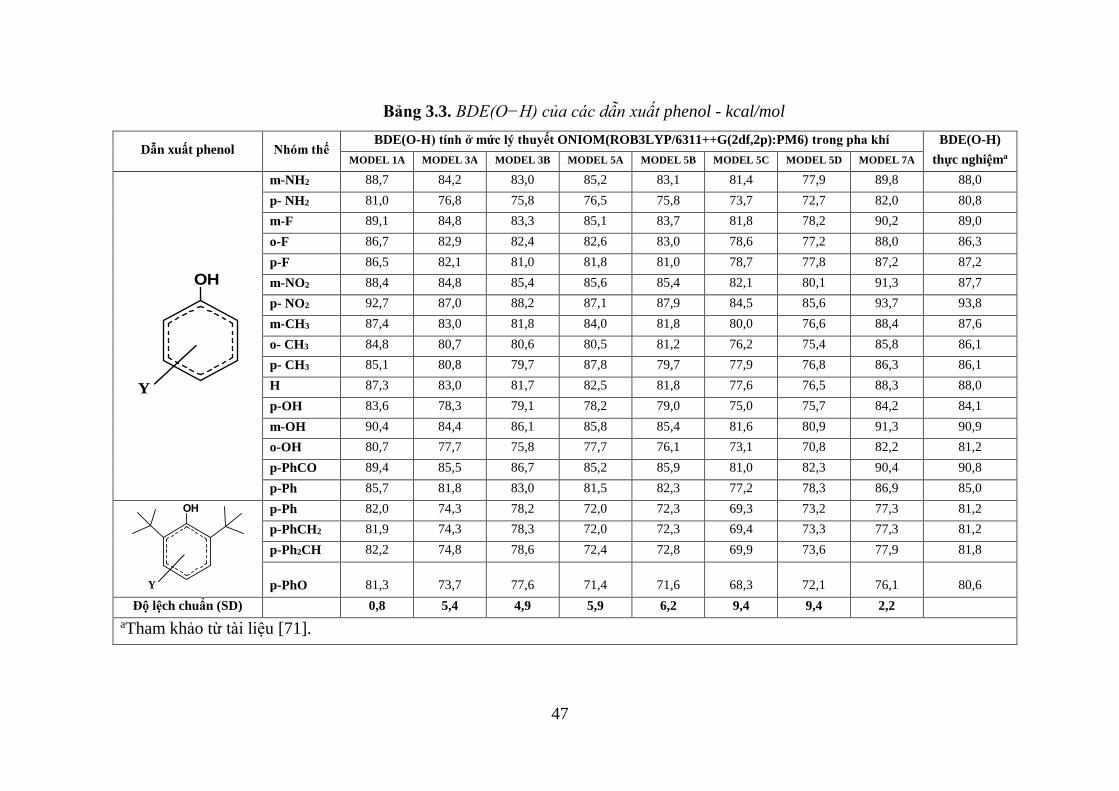
47
Bảng 3.3. BDE(O−H) của các dẫn xuất phenol - kcal/mol
Dẫn xuất phenol Nhóm thế BDE(O-H) tính ở mức lý thuyết ONIOM(ROB3LYP/6311++G(2df,2p):PM6) trong pha khí BDE(O-H)
thực nghiệma MODEL 1A MODEL 3A MODEL 3B MODEL 5A MODEL 5B MODEL 5C MODEL 5D MODEL 7A
OH
Y
m-NH2 88,7 84,2 83,0 85,2 83,1 81,4 77,9 89,8 88,0
p- NH2 81,0 76,8 75,8 76,5 75,8 73,7 72,7 82,0 80,8
m-F 89,1 84,8 83,3 85,1 83,7 81,8 78,2 90,2 89,0
o-F 86,7 82,9 82,4 82,6 83,0 78,6 77,2 88,0 86,3
p-F 86,5 82,1 81,0 81,8 81,0 78,7 77,8 87,2 87,2
m-NO2 88,4 84,8 85,4 85,6 85,4 82,1 80,1 91,3 87,7
p- NO2 92,7 87,0 88,2 87,1 87,9 84,5 85,6 93,7 93,8
m-CH3 87,4 83,0 81,8 84,0 81,8 80,0 76,6 88,4 87,6
o- CH3 84,8 80,7 80,6 80,5 81,2 76,2 75,4 85,8 86,1
p- CH3 85,1 80,8 79,7 87,8 79,7 77,9 76,8 86,3 86,1
H 87,3 83,0 81,7 82,5 81,8 77,6 76,5 88,3 88,0
p-OH 83,6 78,3 79,1 78,2 79,0 75,0 75,7 84,2 84,1
m-OH 90,4 84,4 86,1 85,8 85,4 81,6 80,9 91,3 90,9
o-OH 80,7 77,7 75,8 77,7 76,1 73,1 70,8 82,2 81,2
p-PhCO 89,4 85,5 86,7 85,2 85,9 81,0 82,3 90,4 90,8
p-Ph 85,7 81,8 83,0 81,5 82,3 77,2 78,3 86,9 85,0
OH
Y
p-Ph 82,0 74,3 78,2 72,0 72,3 69,3 73,2 77,3 81,2
p-PhCH2 81,9 74,3 78,3 72,0 72,3 69,4 73,3 77,3 81,2
p-Ph2CH 82,2 74,8 78,6 72,4 72,8 69,9 73,6 77,9 81,8
p-PhO 81,3 73,7 77,6 71,4 71,6 68,3 72,1 76,1 80,6
Độ lệch chuẩn (SD) 0,8 5,4 4,9 5,9 6,2 9,4 9,4 2,2
aTham khảo từ tài liệu [71].
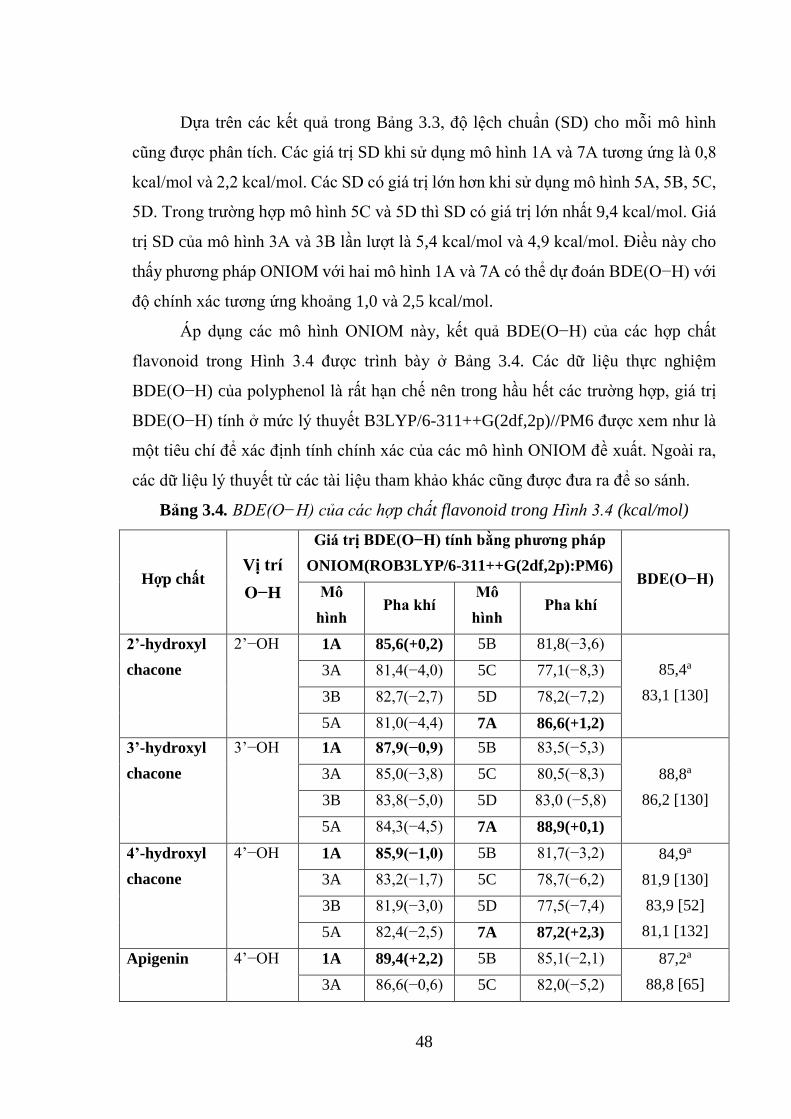
48
Dựa trên các kết quả trong Bảng 3.3, độ lệch chuẩn (SD) cho mỗi mô hình
cũng được phân tích. Các giá trị SD khi sử dụng mô hình 1A và 7A tương ứng là 0,8
kcal/mol và 2,2 kcal/mol. Các SD có giá trị lớn hơn khi sử dụng mô hình 5A, 5B, 5C,
5D. Trong trường hợp mô hình 5C và 5D thì SD có giá trị lớn nhất 9,4 kcal/mol. Giá
trị SD của mô hình 3A và 3B lần lượt là 5,4 kcal/mol và 4,9 kcal/mol. Điều này cho
thấy phương pháp ONIOM với hai mô hình 1A và 7A có thể dự đoán BDE(O−H) với
độ chính xác tương ứng khoảng 1,0 và 2,5 kcal/mol.
Áp dụng các mô hình ONIOM này, kết quả BDE(O−H) của các hợp chất
flavonoid trong Hình 3.4 được trình bày ở Bảng 3.4. Các dữ liệu thực nghiệm
BDE(O−H) của polyphenol là rất hạn chế nên trong hầu hết các trường hợp, giá trị
BDE(O−H) tính ở mức lý thuyết B3LYP/6-311++G(2df,2p)//PM6 được xem như là
một tiêu chí để xác định tính chính xác của các mô hình ONIOM đề xuất. Ngoài ra,
các dữ liệu lý thuyết từ các tài liệu tham khảo khác cũng được đưa ra để so sánh.
Bảng 3.4. BDE(O−H) của các hợp chất flavonoid trong Hình 3.4 (kcal/mol)
Hợp chất Vị trí
O−H
Giá trị BDE(O−H) tính bằng phương pháp
ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) BDE(O−H)
Mô
hình Pha khí
Mô
hình Pha khí
2’-hydroxyl
chacone
2’−OH 1A 85,6(+0,2) 5B 81,8(−3,6)
85,4a
83,1 [130]
3A 81,4(−4,0) 5C 77,1(−8,3)
3B 82,7(−2,7) 5D 78,2(−7,2)
5A 81,0(−4,4) 7A 86,6(+1,2)
3’-hydroxyl
chacone
3’−OH 1A 87,9(−0,9) 5B 83,5(−5,3)
88,8a
86,2 [130]
3A 85,0(−3,8) 5C 80,5(−8,3)
3B 83,8(−5,0) 5D 83,0 (−5,8)
5A 84,3(−4,5) 7A 88,9(+0,1)
4’-hydroxyl
chacone
4’−OH 1A 85,9(−1,0) 5B 81,7(−3,2) 84,9a
81,9 [130]
83,9 [52]
81,1 [132]
3A 83,2(−1,7) 5C 78,7(−6,2)
3B 81,9(−3,0) 5D 77,5(−7,4)
5A 82,4(−2,5) 7A 87,2(+2,3)
Apigenin 4’−OH 1A 89,4(+2,2) 5B 85,1(−2,1) 87,2a
88,8 [65] 3A 86,6(−0,6) 5C 82,0(−5,2)
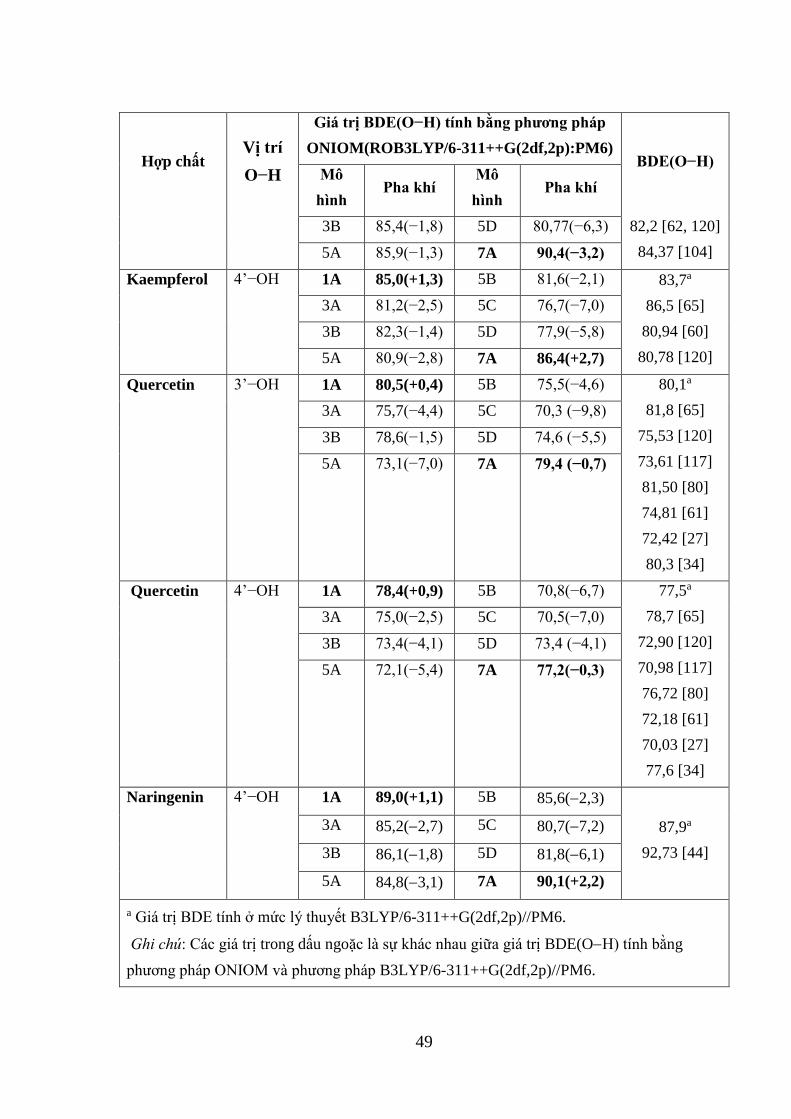
49
Hợp chất Vị trí
O−H
Giá trị BDE(O−H) tính bằng phương pháp
ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) BDE(O−H)
Mô
hình Pha khí
Mô
hình Pha khí
3B 85,4(−1,8) 5D 80,77(−6,3) 82,2 [62, 120]
84,37 [104] 5A 85,9(−1,3) 7A 90,4(−3,2)
Kaempferol 4’−OH 1A 85,0(+1,3) 5B 81,6(−2,1) 83,7a
86,5 [65]
80,94 [60]
80,78 [120]
3A 81,2(−2,5) 5C 76,7(−7,0)
3B 82,3(−1,4) 5D 77,9(−5,8)
5A 80,9(−2,8) 7A 86,4(+2,7)
Quercetin 3’−OH 1A 80,5(+0,4) 5B 75,5(−4,6) 80,1a
81,8 [65]
75,53 [120]
73,61 [117]
81,50 [80]
74,81 [61]
72,42 [27]
80,3 [34]
3A 75,7(−4,4) 5C 70,3 (−9,8)
3B 78,6(−1,5) 5D 74,6 (−5,5)
5A 73,1(−7,0) 7A 79,4 (−0,7)
Quercetin 4’−OH 1A 78,4(+0,9) 5B 70,8(−6,7) 77,5a
78,7 [65]
72,90 [120]
70,98 [117]
76,72 [80]
72,18 [61]
70,03 [27]
77,6 [34]
3A 75,0(−2,5) 5C 70,5(−7,0)
3B 73,4(−4,1) 5D 73,4 (−4,1)
5A 72,1(−5,4) 7A 77,2(−0,3)
Naringenin 4’−OH 1A 89,0(+1,1) 5B 85,6(2,3)
87,9a
92,73 [44]
3A 85,2(2,7) 5C 80,7(7,2)
3B 86,1(1,8) 5D 81,8(6,1)
5A 84,8(3,1) 7A 90,1(+2,2)
a Giá trị BDE tính ở mức lý thuyết B3LYP/6-311++G(2df,2p)//PM6.
Ghi chú: Các giá trị trong dấu ngoặc là sự khác nhau giữa giá trị BDE(OH) tính bằng
phương pháp ONIOM và phương pháp B3LYP/6-311++G(2df,2p)//PM6.

50
Các kết quả thu được trong Bảng 3.4 cho thấy sự khác biệt giữa hai giá trị
BDE(O−H) tính bằng phương pháp ONIOM và phương pháp B3LYP/6-311++
G(2df,2p)//PM6, các giá trị này thay đổi tùy theo các mô hình ONIOM. Tuy nhiên,
mỗi mô hình ONIOM thì có một xu hướng tương tự nhau. Nói chung, các giá trị
BDE(O−H) khi sử dụng các mô hình 3A, 3B, 5A, 5B, 5C và 5D thường thấp so với
giá trị BDE(O−H) tính ở mức lý thuyết B3LYP/6-311++G(2df,2p)//PM6. Độ lệch
lớn nhất khi sử dụng mô hình 5C là 9,1 kcal/mol. Giá trị BDE sử dụng mô hình 1A
và 7A cao hơn so với dữ liệu từ tài liệu tham khảo [23, 26-29] nhưng phù hợp với dữ
liệu của Guo và cộng sự tính bằng phương pháp ONIOM-B3G3. Độ lệch tuyệt đối
của giá trị BDE(O−H) khi sử dụng mô hình 1A so với dữ liệu tính ở mức lý thuyết
B3LYP/6-311++G(2df,2p)//PM6 là khoảng 1,0 kcal/mol (số liệu trong ngoặc đơn của
Bảng 3.4). Theo nghiên cứu của Wright, những hợp chất polyphenol có BDE(O−H)
nhỏ hơn giá trị BDE(O−H) thực nghiệm của phenol (88,30,7 kcal/mol) [71]) khoảng
10 kcal/mol được chứng minh là có khả năng chống oxy hóa theo cơ chế HAT [129].
3.1.4. Nhận xét
Qua kết quả tính toán ở trên, chúng tôi có thể kết luận rằng sử dụng phương
pháp ONIOM với mô hình 1A là sự lựa chọn tốt nhất cho tính BDE(O−H) của các
dẫn xuất phenol và các hợp chất flavonoid có độ chính xác cao, thời gian tính toán
nhanh và tiết kiệm được tài nguyên máy tính. Do đó, chúng tôi sẽ sử dụng phương
pháp này để tính BDE(O−H) của các hợp chất polyphenol có nguồn gốc từ cây sa kê,
vỏ măng cụt và hợp chất flavonoid trong các phần nghiên cứu tiếp theo.
3.2. KHẢO SÁT PHƯƠNG PHÁP TÍNH NĂNG LƯỢNG ION HÓA
3.2.1. Giới thiệu
Khi thảo luận về cơ chế chuyển electron chuyển proton thì giá trị IE là một
thông số quan trọng cần được xem xét khi nghiên cứu khả năng chống oxy hóa của
các hợp chất. Trước hết, chúng tôi kiểm tra độ chính xác của phương pháp PM6 để
sử dụng tính các giá trị IE. Giá trị IE của các dẫn xuất phenol được tính bằng phương
pháp PM6 và so sánh với giá trị thực nghiệm. Trong đó IE adiabatic (đoạn nhiệt) là
giá trị mà ở đó ion dương tạo ra được tối ưu hình học cân bằng mới, còn IE vertical
(thẳng đứng) là giá trị mà ở đó ion dương tạo ra nhưng vẫn có hình học của phân tử
trung hòa.

51
3.2.2. Đánh giá tính chính xác của phương pháp tính IE ở mức lý thuyết PM6
Các giá trị IE tính toán trong Bảng 3.5 chỉ ra rằng phương pháp PM6 có thể
dự đoán tốt giá trị IE adiabatic với độ lệch trung bình 0,091 eV so với giá trị thực
nghiệm (trừ trường hợp của BHT và 2,4,6-trimethyl phenol thì không có sẵn giá trị
IE adiabatic thực nghiệm).
Bảng 3.5. Giá trị năng lượng ion hóa (eV) tính bằng phương pháp PM6 của các
dẫn xuất phenol và giá trị thực nghiệm
Hợp chất IE
IEb PM6 Thực nghiệma
Phenol 8,38(8,61) 8,490,02(8,70) 0,11(0,09)
1,4-Benzenediol 8,00(8,28) 7,940,01(8,44) -0,06(0,16)
1,3-Benzenediol 8,29(8,56) 8,20 (8,63) -0,09(0,07)
1,2-Benzenediol 8,10(8,44) 8,15(8,56) 0,05(0,12)
BHT 7,39(7,74) N/A(7,80) N/A(0,06)
2-Methyl phenol 8,17(8,46) 8,14(8,460,06) -0,03(0,00)
2-Nitro phenol 9,02(9,43) 9,1(9,29) 0,08(-0,14)
2,4-Dimethyl phenol 7,84(8,17) 8,0(8,18) 0,16(0,01)
2,4,6-Trimethyl phenol 7,64(7,98) N/A(8,0) N/A(0,02)
2,6-Diclo phenol 8,60(8,84) 8,650,02(N/A) 0,06(N/A)
2,6-Dimethyl phenol 7,95(8,25) 8,050.02(8,26) 0,1(0,01)
3 Methyl phenol 8,26(8,57) 8,290,02(N/A) 0,03(N/A)
3,4-Dimethyl phenol 7,93(8,24) 8,09(N/A) 0,16(N/A)
4-Nitro phenol 9,26(9,57) 9,1(9,38) -0,16(-0,19)
Ghi chú: Giá trị trong dấu ngoặc là giá trị IE vertical.
a Giá trị thực nghiệm được tham khảo từ NIST Chemistry web book, số 69,
http://webbook.nist.gov/chemistry/.
b IE = IEthực nghiệm – IEPM6.
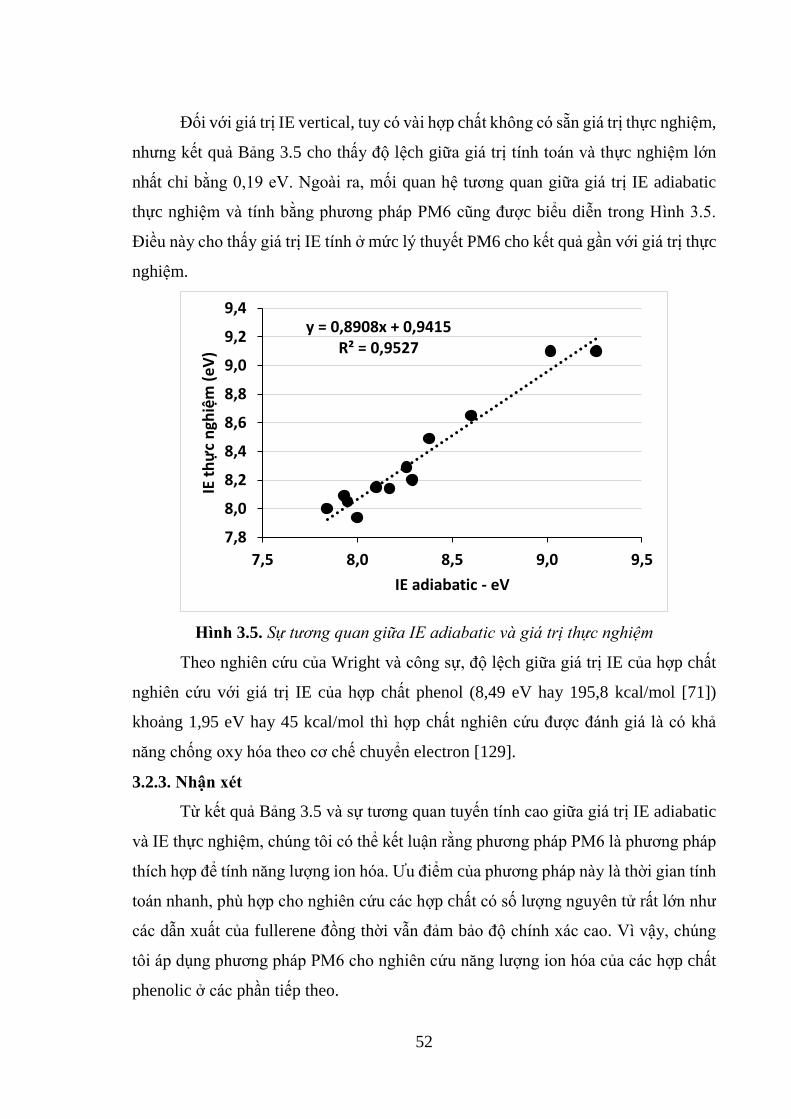
52
Đối với giá trị IE vertical, tuy có vài hợp chất không có sẵn giá trị thực nghiệm,
nhưng kết quả Bảng 3.5 cho thấy độ lệch giữa giá trị tính toán và thực nghiệm lớn
nhất chỉ bằng 0,19 eV. Ngoài ra, mối quan hệ tương quan giữa giá trị IE adiabatic
thực nghiệm và tính bằng phương pháp PM6 cũng được biểu diễn trong Hình 3.5.
Điều này cho thấy giá trị IE tính ở mức lý thuyết PM6 cho kết quả gần với giá trị thực
nghiệm.
Hình 3.5. Sự tương quan giữa IE adiabatic và giá trị thực nghiệm
Theo nghiên cứu của Wright và công sự, độ lệch giữa giá trị IE của hợp chất
nghiên cứu với giá trị IE của hợp chất phenol (8,49 eV hay 195,8 kcal/mol [71])
khoảng 1,95 eV hay 45 kcal/mol thì hợp chất nghiên cứu được đánh giá là có khả
năng chống oxy hóa theo cơ chế chuyển electron [129].
3.2.3. Nhận xét
Từ kết quả Bảng 3.5 và sự tương quan tuyến tính cao giữa giá trị IE adiabatic
và IE thực nghiệm, chúng tôi có thể kết luận rằng phương pháp PM6 là phương pháp
thích hợp để tính năng lượng ion hóa. Ưu điểm của phương pháp này là thời gian tính
toán nhanh, phù hợp cho nghiên cứu các hợp chất có số lượng nguyên tử rất lớn như
các dẫn xuất của fullerene đồng thời vẫn đảm bảo độ chính xác cao. Vì vậy, chúng
tôi áp dụng phương pháp PM6 cho nghiên cứu năng lượng ion hóa của các hợp chất
phenolic ở các phần tiếp theo.
y = 0,8908x + 0,9415R² = 0,9527
7,8
8,0
8,2
8,4
8,6
8,8
9,0
9,2
9,4
7,5 8,0 8,5 9,0 9,5
IE t
hự
c n
ghiệ
m (
eV
)
IE adiabatic - eV

53
3.3. KHẢ NĂNG CHỐNG OXY HÓA CỦA CÁC HỢP CHẤT POLYPHENOL
CÓ NGUỒN GỐC TỪ CÂY SA KÊ (ARTOCARPUS ALTILIS)
3.3.1. Lựa chọn một số hợp chất polyphenol có chứa trong cây sa kê
Nhiều hợp chất polyphenol đã được chiết xuất từ cây sa kê. Tuy nhiên, trong
phần nghiên cứu này, chúng tôi chỉ chọn 9 hợp chất geranyl flavonoid mới được phân
lập bởi nhóm nghiên cứu của Yu Wang [126] và 3 hợp chất geranyl aurone mới được
phát hiện bởi tác giả Nguyễn Thị Thanh Mai và cộng sự [72] để nghiên cứu khả năng
chống oxy hóa, bởi vì chưa có nghiên cứu lý thuyết nào phân tích khả năng chống
oxy hóa của các hợp chất này cũng như xác định hợp chất có hiệu quả dập tắt gốc tự
do cao nhất. Kết quả nghiên cứu này sẽ làm tiền đề và định hướng cho các nghiên
cứu thực nghiệm tiếp theo về thử nghiệm hoạt tính chống oxy hóa của các hợp chất
có nguồn gốc từ cây sa kê.
Cấu trúc của 12 hợp chất nghiên cứu được trình bày ở Hình 3.6. Các hợp chất
từ S1 đến S12 có danh pháp như sau: 1-(2,4-dihydroxyphenyl)-3-[8-hydroxy-2-
methyl-2-(4-methyl-3-pentenyl)-2H-1-benzopyran-5-yl]-1-propanone (S1), 1-(2,4-
dihydroxyphenyl)-3-4-hydroxy-6,6,9-trimethyl-6a,7,8,10a-tetrahydro-6H-dibenzo
[b,d] pyran-5-yl-1-propanone (S2), 2-geranyl-2',3,4,4'-tetrahydroxy dihydrochalcone
(S3), 1-(2,4-dihydroxyphenyl)-3-[3,4-dihydro-3,8-dihydroxy -2-methyl-2-(4-
methyl-3-pentenyl)-2H-1-benzopyran-5-yl]-1-propanone (S4), 1-(2,4-
dihydroxyphenyl)-3-[8-hydroxy-2-methyl-2-(3,4-epoxy-4-methyl-1-pentenyl)-2H-
1-benzopyran-5-yl]-1-propanone (S5), 2'-geranyl-3',4',7-trihydroxyflavanone (S6),
cycloaltilisin 6 (S7), 1-(2,4-dihydroxyphenyl)-3-[8-hydroxy-2-methyl-2-(4-hydroxy-
4-methyl-2-pentenyl)-2H-1-benzopyran-5-yl]-1-propanone (S8) và 2-[6-hydroxy-
3,7-dimethylocta-2(E), 7-dienyl]-2',3,4,4'-tetrahydroxydihydrochalcone (S9),
altilisin H (S10), altilisin I (S11), altilisin J (S12) [72, 126].
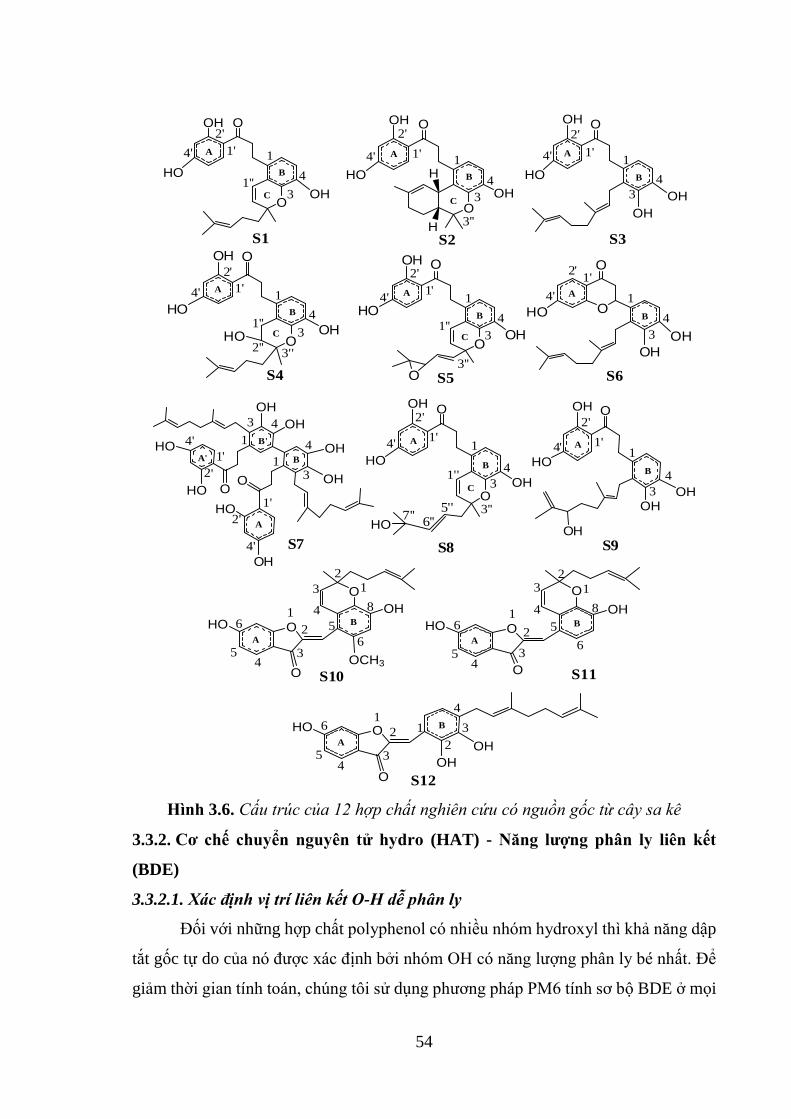
54
OH
HO
O
OH
OH
OH
HO
O
OHOH
OH
HO
O
OH
OH
O
OH
OH
OH
OHO
HO
OH
OHO
HO
O
OOH
HO
OH
A
B
C
1'
2'
4' 1
3
41''
S1
A 1'
2'
4'
B
1
3
4
S3
O
OOH
HO
OH
A
B
C
1'
2'
4' 1
3
4
3''
S2H
H
O
OOH
HO
OH
A
B
C
1'
2'
4' 1
3
41''
3''
S4
HO2'' O
OOH
HO
OH
A
B
C
1'
2'
4' 1
3
41''
3''
S5O
A
B
1'2'
4' 1
3
4
S6
O
OOH
HO
OH
A
B
C
1'
2'
4' 1
3
41''
3''
S8
A
B
1'
2'
4' 1
3
4
HO
5''6''
7''A
BA'
B'
S7 S9
41'
1'
2'
2'
4'
4'
1
1
3
3
4
O
O
HO
OOCH3
OH
O
O
HO
O
OH
OHO
OOH
OH
A
B
A
B
1
23
4
5
6
81
2
34
5
6
12
3
4
5
6
81
2
34
5
6
1
2
41
2
34
5
6
A
B 3
S10 S11
S12
Hình 3.6. Cấu trúc của 12 hợp chất nghiên cứu có nguồn gốc từ cây sa kê
3.3.2. Cơ chế chuyển nguyên tử hydro (HAT) - Năng lượng phân ly liên kết
(BDE)
3.3.2.1. Xác định vị trí liên kết O-H dễ phân ly
Đối với những hợp chất polyphenol có nhiều nhóm hydroxyl thì khả năng dập
tắt gốc tự do của nó được xác định bởi nhóm OH có năng lượng phân ly bé nhất. Để
giảm thời gian tính toán, chúng tôi sử dụng phương pháp PM6 tính sơ bộ BDE ở mọi
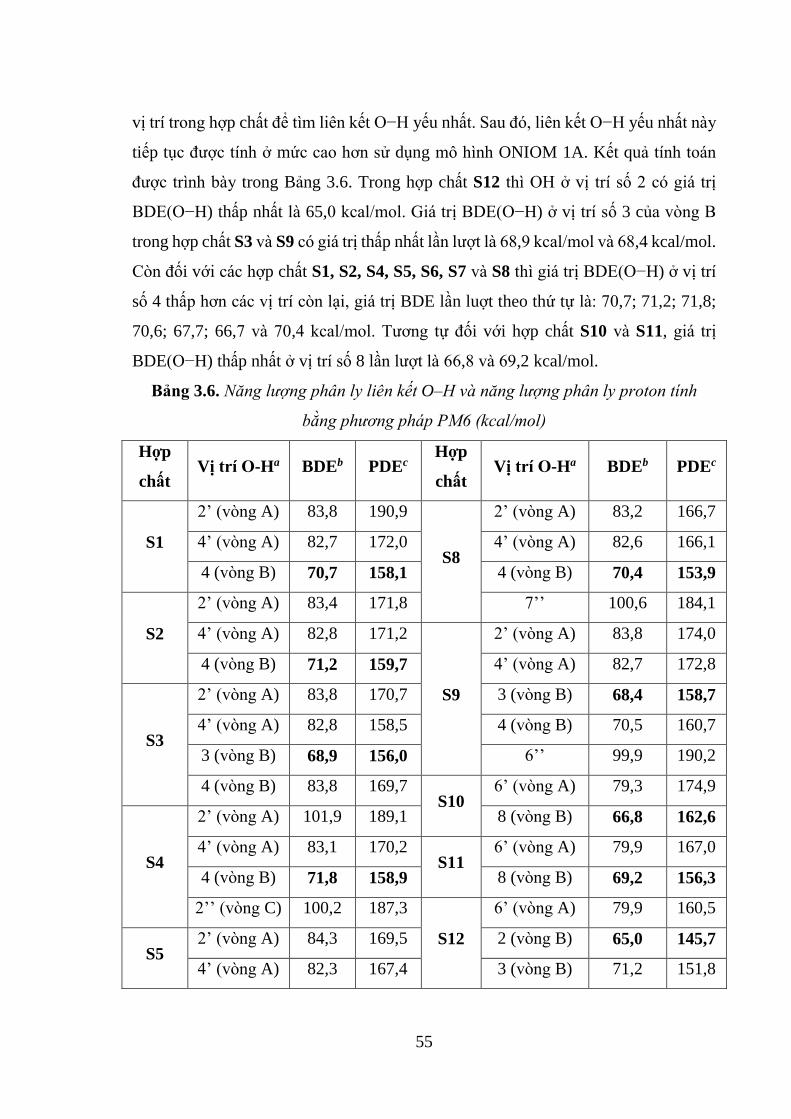
55
vị trí trong hợp chất để tìm liên kết O−H yếu nhất. Sau đó, liên kết O−H yếu nhất này
tiếp tục được tính ở mức cao hơn sử dụng mô hình ONIOM 1A. Kết quả tính toán
được trình bày trong Bảng 3.6. Trong hợp chất S12 thì OH ở vị trí số 2 có giá trị
BDE(O−H) thấp nhất là 65,0 kcal/mol. Giá trị BDE(O−H) ở vị trí số 3 của vòng B
trong hợp chất S3 và S9 có giá trị thấp nhất lần lượt là 68,9 kcal/mol và 68,4 kcal/mol.
Còn đối với các hợp chất S1, S2, S4, S5, S6, S7 và S8 thì giá trị BDE(O−H) ở vị trí
số 4 thấp hơn các vị trí còn lại, giá trị BDE lần luợt theo thứ tự là: 70,7; 71,2; 71,8;
70,6; 67,7; 66,7 và 70,4 kcal/mol. Tương tự đối với hợp chất S10 và S11, giá trị
BDE(O−H) thấp nhất ở vị trí số 8 lần lượt là 66,8 và 69,2 kcal/mol.
Bảng 3.6. Năng lượng phân ly liên kết O–H và năng lượng phân ly proton tính
bằng phương pháp PM6 (kcal/mol)
Hợp
chất Vị trí O-Ha BDEb PDEc
Hợp
chất Vị trí O-Ha BDEb PDEc
S1
2’ (vòng A) 83,8 190,9
S8
2’ (vòng A) 83,2 166,7
4’ (vòng A) 82,7 172,0 4’ (vòng A) 82,6 166,1
4 (vòng B) 70,7 158,1 4 (vòng B) 70,4 153,9
S2
2’ (vòng A) 83,4 171,8 7’’ 100,6 184,1
4’ (vòng A) 82,8 171,2
S9
2’ (vòng A) 83,8 174,0
4 (vòng B) 71,2 159,7 4’ (vòng A) 82,7 172,8
S3
2’ (vòng A) 83,8 170,7 3 (vòng B) 68,4 158,7
4’ (vòng A) 82,8 158,5 4 (vòng B) 70,5 160,7
3 (vòng B) 68,9 156,0 6’’ 99,9 190,2
4 (vòng B) 83,8 169,7 S10
6’ (vòng A) 79,3 174,9
S4
2’ (vòng A) 101,9 189,1 8 (vòng B) 66,8 162,6
4’ (vòng A) 83,1 170,2 S11
6’ (vòng A) 79,9 167,0
4 (vòng B) 71,8 158,9 8 (vòng B) 69,2 156,3
2’’ (vòng C) 100,2 187,3
S12
6’ (vòng A) 79,9 160,5
S5 2’ (vòng A) 84,3 169,5 2 (vòng B) 65,0 145,7
4’ (vòng A) 82,3 167,4 3 (vòng B) 71,2 151,8

56
Hợp
chất Vị trí O-Ha BDEb PDEc
Hợp
chất Vị trí O-Ha BDEb PDEc
4 (vòng B) 70,6 155,9
a Cách đánh số được mô tả ở Hình 3.6.
b BDE(O–H) cho hợp chất polyphenol.
c PDE cho các ion dương gốc tự do của
hợp chất polyphenol.
S6
4’ (vòng A) 80,2 156,7
3 (vòng B) 71,9 148,4
4 (vòng B) 67,7 144,2
S7
2’ (vòng A) 85,1 171,8
4’ (vòng A) 81,6 168,4
3 (vòng B) 70,9 157,8
4 (vòng B) 66,7 153,6
2’(vòng A’) 81,2 167,9
4’ (vòng A’) 81,9 168,7
3 (vòng B’) 68,9 155,8
4 (vòng B’) 71,8 158,7
3.3.2.2. BDE(O-H) của 12 hợp chất polyphenol có nguồn gốc từ cây sa kê và ảnh
hưởng của dung môi
Khả năng cho một nguyên tử hydro và hình thành gốc tự do của các hợp chất
polyphenol được đặc trưng bằng giá trị BDE(O−H). Giá trị này đặc trưng cho khả
năng phân ly liên kết O−H (tách nguyên tử hydro) và đây là đại lượng mô tả sự ổn
định của liên kết hydroxyl. Vì vậy, phân tử nào có giá trị BDE thấp nhất thì có khả
năng tách H• dễ dàng nhất. Bảng 3.7 trình bày kết quả BDE tính trong pha khí, dung
môi methanol và nước sử dụng phương pháp ONIOM với mô hình 1A. Dựa vào kết
quả BDE(O−H) trong Bảng 3.7 cho thấy khả năng cho nguyên tử hydro của các hợp
chất polyphenol theo thứ tự sau: S12 > S9 ≈ S7 > S10 ≈ S6 > S3 > S11 > S1 > S8 >
S5 > S2 > S4. Hơn nữa, trong số các nhóm hydroxyl polyphenol ở các vị trí khác
nhau thì nhóm hydroxyl ở vị trí số 2 trong hợp chất S12 có giá trị BDE thấp nhất với
giá trị lần lượt là 77,3; 79,0 và 78,3 kcal/mol tương ứng với pha khí, dung môi
methanol và nước.

57
Bảng 3.7 cho thấy rằng các giá trị BDE của mỗi nhóm O−H của tất cả các hợp
chất polyphenol nhỏ hơn giá trị BDE(O−H) của phenol khi được tính cùng mức lý
thuyết. Điều này chứng tỏ hầu hết các hợp chất polyphenol có khả năng cho nguyên
tử hydro mạnh hơn so với phenol. Kết quả ở Bảng 3.7 cũng cho thấy BDE(O−H) ở
vị trí số 2 của hợp chất S12 (77,3 kcal/mol trong pha khí) tương tự với giá trị
BDE(O−H) của ubiquinol-10 (78,5 kcal/mol).
Bảng 3.7. BDE(O–H) của 12 hợp chất polyphenol có nguồn gốc từ cây sa kê tính
bằng phương pháp ONIOM(ROB3LYP/6-311++G(2df,2p):PM6)
Hợp chất Vị trí O−H BDE(O–H) (kcal/mol)
Pha khí Methanol Nước
S1 4−OH (vòng B) 83,2 83,5 82,8
S2 4−OH (vòng B) 84,5 83,2 83,1
S3 3−OH (vòng B) 80,8 82,4 82,2
S4 4−OH (vòng B) 85,1 83,7 83,1
S5 4−OH (vòng B) 83,9 83,8 83,1
S6 4−OH (vòng B) 80,3 83,9 83,1
S7 4−OH (vòng B) 79,3 82,9 82,1
S8 4−OH (vòng B) 83,7 82,8 81,4
S9 3−OH (vòng B) 79,3 81,3 80,5
S10 8−OH (vòng B) 80,3 82,7 81,6
S11 8−OH (vòng B) 82,5 83,9 83,2
S12 2−OH (vòng B) 77,3 79,0 78,3
3.3.3. Cơ chế chuyển electron chuyển proton (SET-PT)
3.3.3.1. Năng lượng ion hóa (IE)
Giá trị IE vertical và IE adiabatic của 12 hợp chất polyphenol có nguồn gốc từ
cây sa kê được trình bày ở Bảng 3.8. Trong đó, hợp chất S10 có giá trị IE nhỏ nhất,
điều này chứng tỏ hợp chất S10 dễ cho electron hơn các hợp chất khác. Vì vậy, hợp
chất S10 được xem là chất chống oxy hóa tiềm năng theo cơ chế chuyển electron.
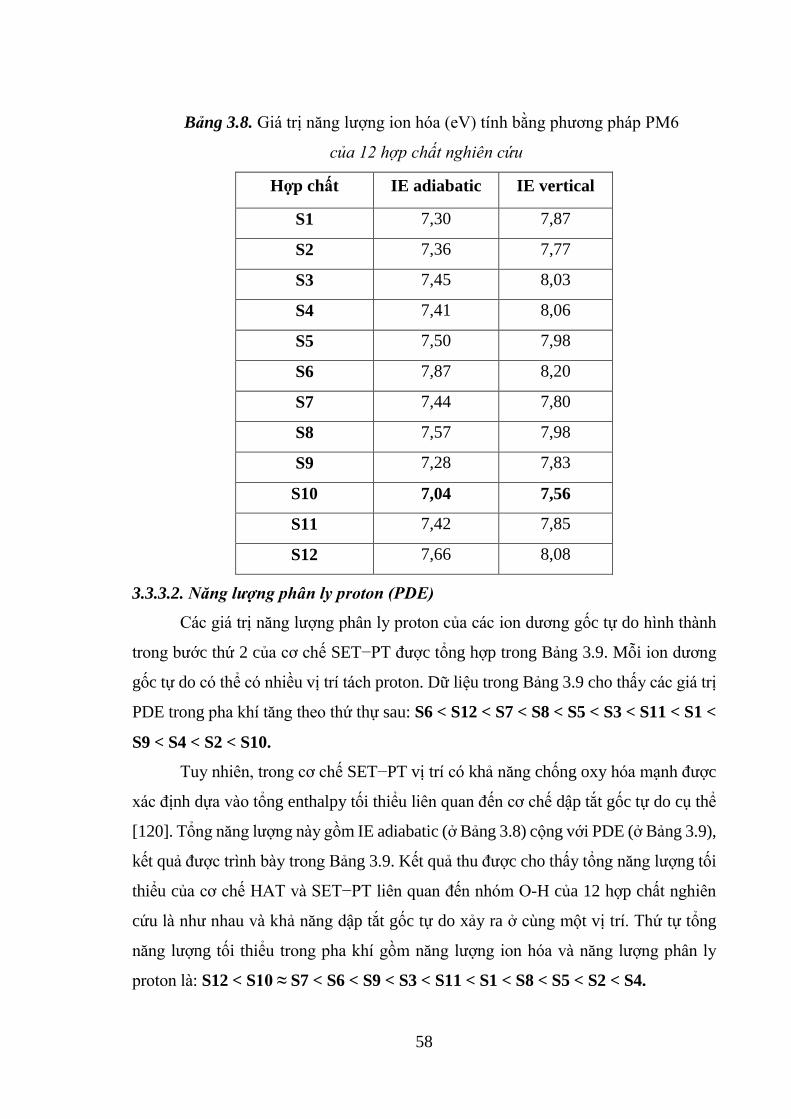
58
Bảng 3.8. Giá trị năng lượng ion hóa (eV) tính bằng phương pháp PM6
của 12 hợp chất nghiên cứu
Hợp chất IE adiabatic IE vertical
S1 7,30 7,87
S2 7,36 7,77
S3 7,45 8,03
S4 7,41 8,06
S5 7,50 7,98
S6 7,87 8,20
S7 7,44 7,80
S8 7,57 7,98
S9 7,28 7,83
S10 7,04 7,56
S11 7,42 7,85
S12 7,66 8,08
3.3.3.2. Năng lượng phân ly proton (PDE)
Các giá trị năng lượng phân ly proton của các ion dương gốc tự do hình thành
trong bước thứ 2 của cơ chế SET−PT được tổng hợp trong Bảng 3.9. Mỗi ion dương
gốc tự do có thể có nhiều vị trí tách proton. Dữ liệu trong Bảng 3.9 cho thấy các giá trị
PDE trong pha khí tăng theo thứ thự sau: S6 < S12 < S7 < S8 < S5 < S3 < S11 < S1 <
S9 < S4 < S2 < S10.
Tuy nhiên, trong cơ chế SET−PT vị trí có khả năng chống oxy hóa mạnh được
xác định dựa vào tổng enthalpy tối thiểu liên quan đến cơ chế dập tắt gốc tự do cụ thể
[120]. Tổng năng lượng này gồm IE adiabatic (ở Bảng 3.8) cộng với PDE (ở Bảng 3.9),
kết quả được trình bày trong Bảng 3.9. Kết quả thu được cho thấy tổng năng lượng tối
thiểu của cơ chế HAT và SET−PT liên quan đến nhóm O-H của 12 hợp chất nghiên
cứu là như nhau và khả năng dập tắt gốc tự do xảy ra ở cùng một vị trí. Thứ tự tổng
năng lượng tối thiểu trong pha khí gồm năng lượng ion hóa và năng lượng phân ly
proton là: S12 < S10 ≈ S7 < S6 < S9 < S3 < S11 < S1 < S8 < S5 < S2 < S4.
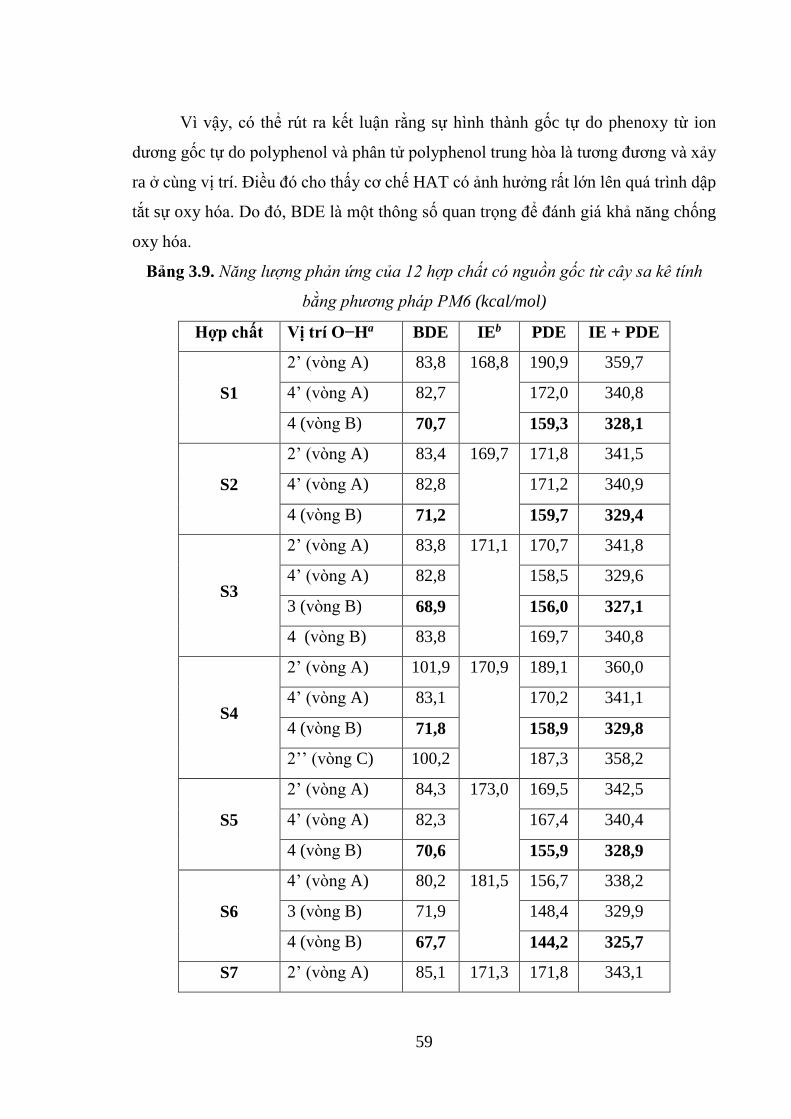
59
Vì vậy, có thể rút ra kết luận rằng sự hình thành gốc tự do phenoxy từ ion
dương gốc tự do polyphenol và phân tử polyphenol trung hòa là tương đương và xảy
ra ở cùng vị trí. Điều đó cho thấy cơ chế HAT có ảnh hưởng rất lớn lên quá trình dập
tắt sự oxy hóa. Do đó, BDE là một thông số quan trọng để đánh giá khả năng chống
oxy hóa.
Bảng 3.9. Năng lượng phản ứng của 12 hợp chất có nguồn gốc từ cây sa kê tính
bằng phương pháp PM6 (kcal/mol)
Hợp chất Vị trí O−Ha BDE IEb PDE IE + PDE
S1
2’ (vòng A) 83,8 168,8 190,9 359,7
4’ (vòng A) 82,7 172,0 340,8
4 (vòng B) 70,7 159,3 328,1
S2
2’ (vòng A) 83,4 169,7 171,8 341,5
4’ (vòng A) 82,8 171,2 340,9
4 (vòng B) 71,2 159,7 329,4
S3
2’ (vòng A) 83,8 171,1 170,7 341,8
4’ (vòng A) 82,8 158,5 329,6
3 (vòng B) 68,9 156,0 327,1
4 (vòng B) 83,8 169,7 340,8
S4
2’ (vòng A) 101,9 170,9 189,1 360,0
4’ (vòng A) 83,1 170,2 341,1
4 (vòng B) 71,8 158,9 329,8
2’’ (vòng C) 100,2 187,3 358,2
S5
2’ (vòng A) 84,3 173,0 169,5 342,5
4’ (vòng A) 82,3 167,4 340,4
4 (vòng B) 70,6 155,9 328,9
S6
4’ (vòng A) 80,2 181,5 156,7 338,2
3 (vòng B) 71,9 148,4 329,9
4 (vòng B) 67,7 144,2 325,7
S7 2’ (vòng A) 85,1 171,3 171,8 343,1
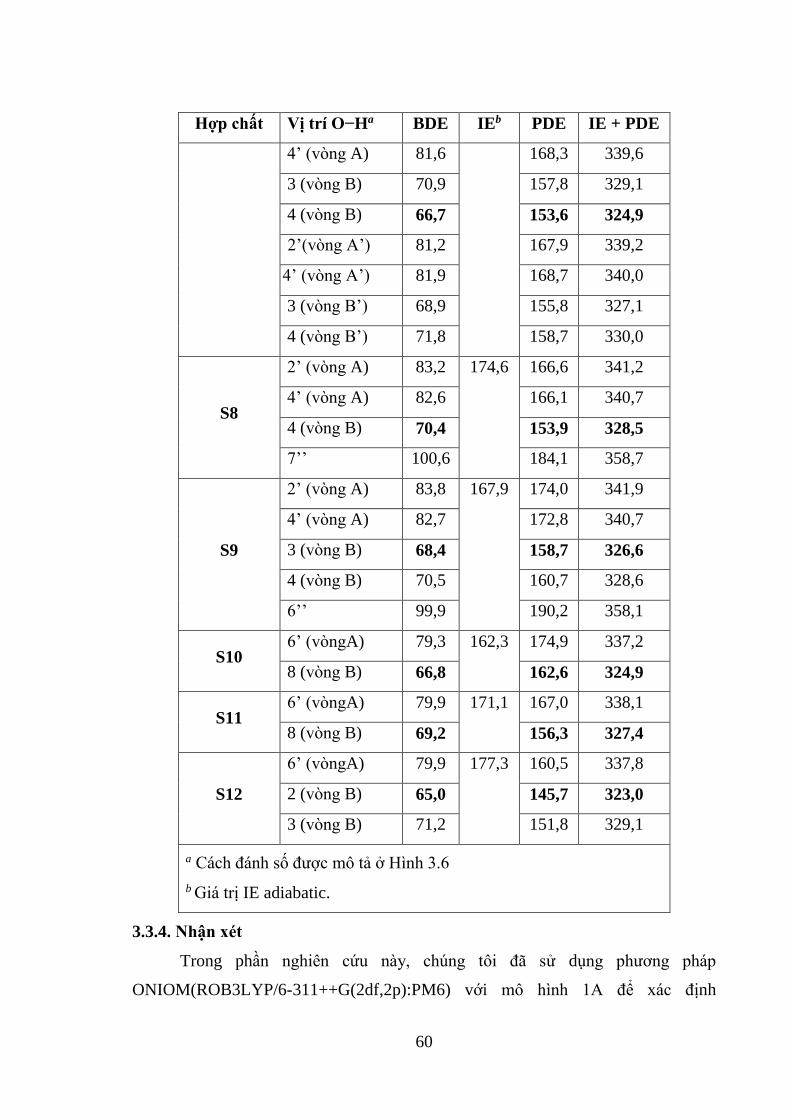
60
Hợp chất Vị trí O−Ha BDE IEb PDE IE + PDE
4’ (vòng A) 81,6 168,3 339,6
3 (vòng B) 70,9 157,8 329,1
4 (vòng B) 66,7 153,6 324,9
2’(vòng A’) 81,2 167,9 339,2
4’ (vòng A’) 81,9 168,7 340,0
3 (vòng B’) 68,9 155,8 327,1
4 (vòng B’) 71,8 158,7 330,0
S8
2’ (vòng A) 83,2 174,6 166,6 341,2
4’ (vòng A) 82,6 166,1 340,7
4 (vòng B) 70,4 153,9 328,5
7’’ 100,6 184,1 358,7
S9
2’ (vòng A) 83,8 167,9 174,0 341,9
4’ (vòng A) 82,7 172,8 340,7
3 (vòng B) 68,4 158,7 326,6
4 (vòng B) 70,5 160,7 328,6
6’’ 99,9 190,2 358,1
S10 6’ (vòngA) 79,3 162,3 174,9 337,2
8 (vòng B) 66,8 162,6 324,9
S11 6’ (vòngA) 79,9 171,1 167,0 338,1
8 (vòng B) 69,2 156,3 327,4
S12
6’ (vòngA) 79,9 177,3 160,5 337,8
2 (vòng B) 65,0 145,7 323,0
3 (vòng B) 71,2 151,8 329,1
a Cách đánh số được mô tả ở Hình 3.6
b Giá trị IE adiabatic.
3.3.4. Nhận xét
Trong phần nghiên cứu này, chúng tôi đã sử dụng phương pháp
ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) với mô hình 1A để xác định

61
BDE(O−H) của các hợp chất polyphenol có nguồn gốc từ cây sa kê. Trong đó,
BDE(O−H) của các hợp chất S3, S6, S7, S9, S10 và S12 lần lượt là: 80,8; 80,3; 79,3;
79,3; 80,3 và 77,3 kcal/mol, chúng được xem như là những chất chống oxy hóa tiềm
năng. Năng lượng ion hóa adiabatic của các hợp chất polyphenol được xác định bằng
phương pháp PM6. Sự hình thành gốc tự do phenoxy từ ion dương gốc tự do phenolic
và phân tử trung hòa tương ứng ưu tiên xảy ra ở cùng một vị trí. Dựa trên kết quả thu
được có thể kết luận rằng giá trị BDE được xem như là một thông số hóa lý chính
trong việc xác định khả năng dập tắt gốc tự do của các hợp chất polyphenol.
3.4. KHẢ NĂNG CHỐNG OXY HÓA CỦA CÁC HỢP CHẤT POLYPHENOL
CÓ NGUỒN GỐC TỪ VỎ MĂNG CỤT (GARCINIA MANGOSTANA)
3.4.1. Các hợp chất polyphenol có chứa trong vỏ măng cụt
Kinghorn và cộng sự [49] đã sử dụng phương pháp sắc ký để phân lập các hợp
chất xanthone chiết xuất tử vỏ măng cụt (Hình 3.7). Ngoài ra, khả năng chống oxy
hóa của tất cả các hợp chất cũng được xác định dựa vào sự chuyển hóa của
peroxynitrat. Mặc dù, các kết quả thực nghiệm về khả năng chống oxy hóa của các
xanthone đã được báo cáo nhưng chưa có nghiên cứu lý thuyết nào phân tích khả
năng dập tắt gốc tự do của các hợp chất này cũng như xác định cơ chế của phản ứng.
Vì vậy, trong nghiên cứu này, chúng tôi sẽ xác định khả năng chống oxy hóa của các
hợp chất có trong vỏ măng cụt trên Hình 3.7 bằng cách phân tích năng lượng chuyển
electron, năng lượng chuyển hydro và xác định chất có hiệu quả dập tắt gốc tự do cao
nhất, đồng thời làm rõ mối quan hệ giữa nghiên cứu lý thuyết và thực nghiệm về xác
định khả năng chống oxy hóa của hợp chất polyohenol.
Các hợp chất từ M1 đến M14 chỉ ra trong Hình 3.7 có danh pháp như sau: 8-
hydroxycudraxanthone G (M1), mangostingone [7-methoxy-2-(3-methyl-2-butenyl)-8-
(3-methyl-2-oxo-3-butenyl)-1,3,6-trihydroxyxanthone] (M2), cudraxanthone G (M3),
8-deoxygartanin (M4), garcimangosone B (M5), garcinone D (M6), garcinone E (M7),
gartanin (M8), 1-isomangostin (M9), α-mangostin (M10), γ-mangostin (M11),
mangostinone (M12), smeathxanthone A (M13), and tovophyllin A (M14) [49].
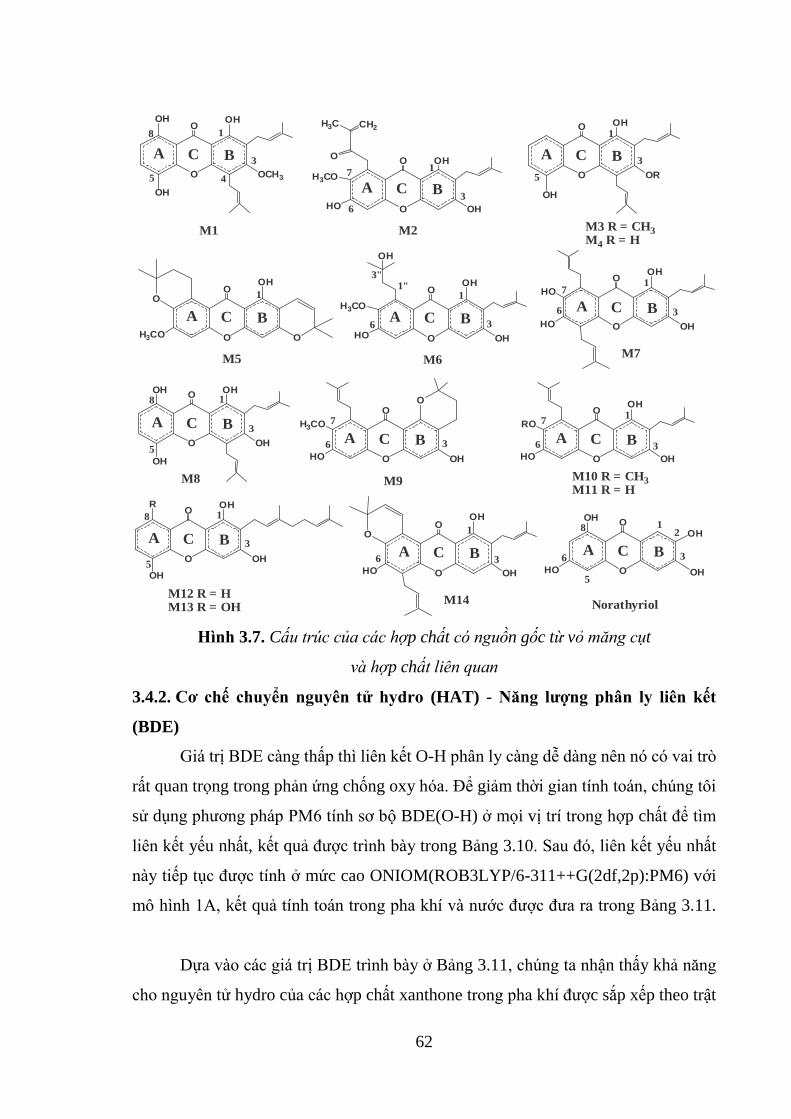
62
O OCH3
OHO
OH
OH
O OH
OHO
O OR
OHO
OH
O
H3C CH2
H3CO
HO
M3 R = CH3M4 R = H
O
OHO
H3CO O
O
O OH
OHO
H3CO
HO
OH
O OH
OHO
HO
HO
O OH
OHO
OH O OH
O
H3CO
HO O OH
OHO
RO
HO
OHO
M10 R = CH3M11 R = H
O OH
OHO
OH
R
O OH
OHO
HO
O
M12 R = HM13 R = OH
A C B
A C B
A C B
A C B A C BA C B
A C BA C B
A C B
A C BA C B
8 1
3
451
3
6
7
1
3
5
1 1
36
3"1" 7
6
1
3
1
36
7
36
3
18
5
1
3
5
8
7
6
1
3O OH
OHO
OH
Norathyriol
A C B 3
8
5HO
12
6
M1 M2
M7M6M5
M8 M9
M14
Hình 3.7. Cấu trúc của các hợp chất có nguồn gốc từ vỏ măng cụt
và hợp chất liên quan
3.4.2. Cơ chế chuyển nguyên tử hydro (HAT) - Năng lượng phân ly liên kết
(BDE)
Giá trị BDE càng thấp thì liên kết O-H phân ly càng dễ dàng nên nó có vai trò
rất quan trọng trong phản ứng chống oxy hóa. Để giảm thời gian tính toán, chúng tôi
sử dụng phương pháp PM6 tính sơ bộ BDE(O-H) ở mọi vị trí trong hợp chất để tìm
liên kết yếu nhất, kết quả được trình bày trong Bảng 3.10. Sau đó, liên kết yếu nhất
này tiếp tục được tính ở mức cao ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) với
mô hình 1A, kết quả tính toán trong pha khí và nước được đưa ra trong Bảng 3.11.
Dựa vào các giá trị BDE trình bày ở Bảng 3.11, chúng ta nhận thấy khả năng
cho nguyên tử hydro của các hợp chất xanthone trong pha khí được sắp xếp theo trật

63
tự sau: M11 > M10 > M7 > M1 > M9 ≈ Norathyriol > M8 > M13 > M6 >M14 >
M2 > M3 > M4 > M12 > M5. Trong tất cả các hợp chất nghiên cứu thì BDE thấp
nhất được tìm thấy ở nhóm OH của vòng A (trừ hợp chất M5 và Norathyriol).
Kinghorn và cộng sự đã nghiên cứu thực nghiệm về khả năng dập tắt gốc tự do của
13 hợp chất (M1 và M3 − M14) dựa vào giá trị IC50 của ONOO−, kết quả được trình
bày trong Bảng 3.11 [49]. Năm trong số 13 hợp chất xanthone là M1, M8, M10, M11
và M13 được chứng minh là chất có tiềm năng chống oxy hóa trong cả 2 phương
pháp thử nghiệm. Đáng chú ý nhất là hợp chất M10 và M11, chúng có giá trị
BDE(O−H) ở vị trí số 6 thấp hơn so với các vị trí khác, với giá trị lần lượt là 82,3 và
82,8 kcal/mol. Điều này chỉ ra rằng sự chuyển nguyên tử hydro từ 6−OH của hợp
chất M10 và M11 cho gốc tự do là dễ dàng hơn so với nhóm OH của các hợp chất
khác. Vì vậy, nhóm OH ở vị trí số 6 của hợp chất M10 và M11 đóng vai trò quan
trọng trong cơ chế chống oxy hóa HAT và điều này cũng được khẳng định bởi nghiên
cứu thực nghiệm trước đó [49].
Quan sát các kết quả trong Bảng 3.11 cho thấy sự ảnh hưởng của dung môi
nước lên giá trị BDE là rất ít. Độ lệch lớn nhất giữa giá trị BDE trong pha khí và dung
môi nước là 5,9 kcal/mol. Trật tự của giá trị BDE trong dung môi nước khác so với
trong pha khí: M1 < M8 < M13 < M10 < M9 < M11 < M14 < Norathyriol < M3 <
M6 < M4 < M7 < M2 < M12 < M5.
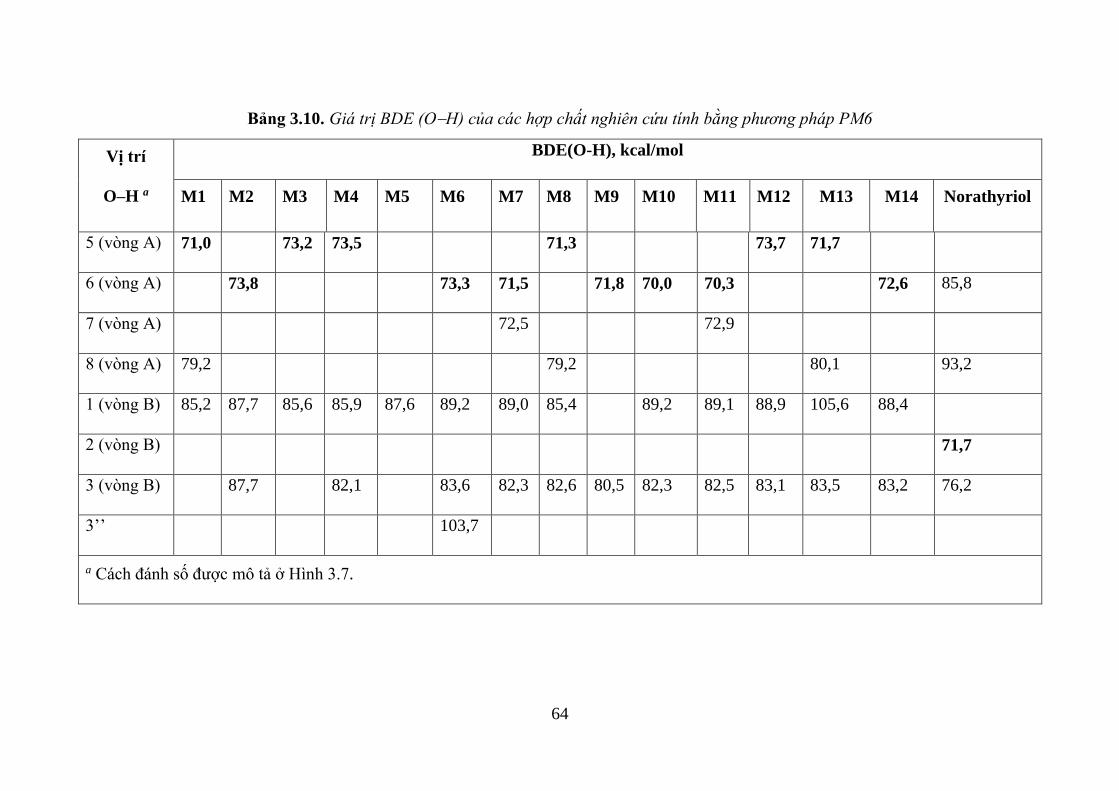
64
Bảng 3.10. Giá trị BDE (OH) của các hợp chất nghiên cứu tính bằng phương pháp PM6
Vị trí
O–H a
BDE(O-H), kcal/mol
M1 M2 M3 M4 M5 M6 M7 M8 M9 M10 M11 M12 M13 M14 Norathyriol
5 (vòng A) 71,0 73,2 73,5 71,3 73,7 71,7
6 (vòng A) 73,8 73,3 71,5 71,8 70,0 70,3 72,6 85,8
7 (vòng A) 72,5 72,9
8 (vòng A) 79,2 79,2 80,1 93,2
1 (vòng B) 85,2 87,7 85,6 85,9 87,6 89,2 89,0 85,4 89,2 89,1 88,9 105,6 88,4
2 (vòng B) 71,7
3 (vòng B) 87,7 82,1 83,6 82,3 82,6 80,5 82,3 82,5 83,1 83,5 83,2 76,2
3’’ 103,7
a Cách đánh số được mô tả ở Hình 3.7.
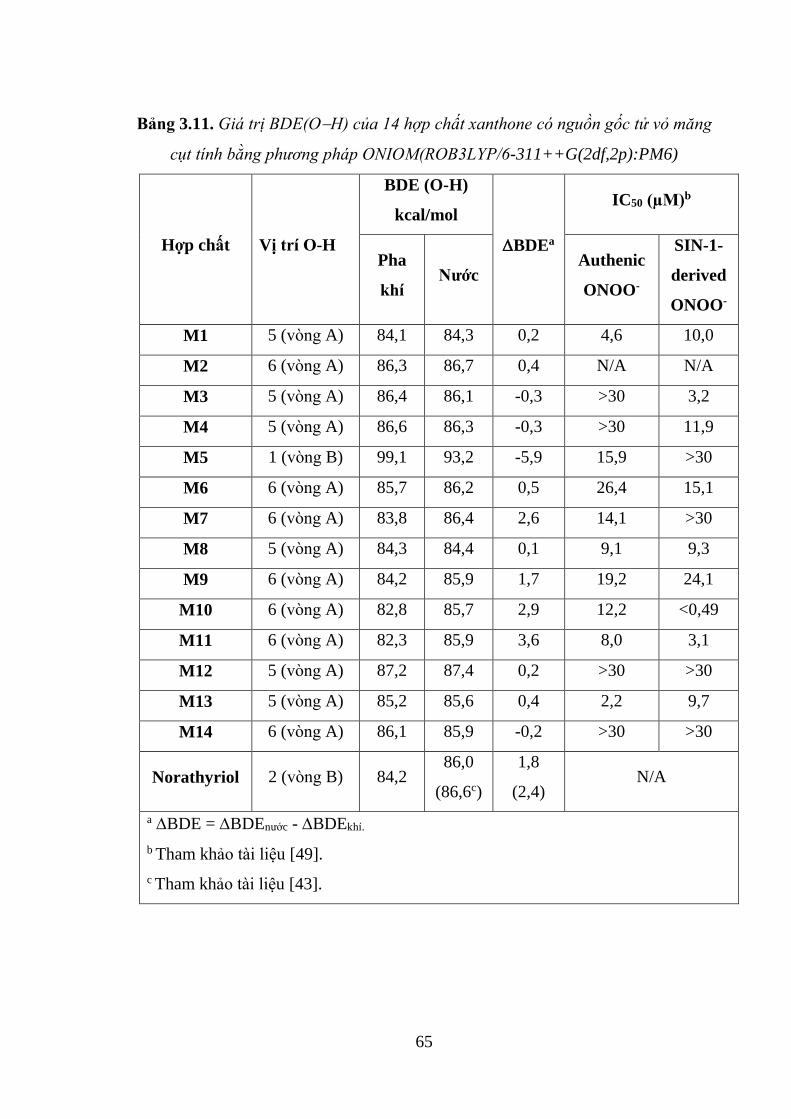
65
Bảng 3.11. Giá trị BDE(OH) của 14 hợp chất xanthone có nguồn gốc tử vỏ măng
cụt tính bằng phương pháp ONIOM(ROB3LYP/6-311++G(2df,2p):PM6)
Hợp chất Vị trí O-H
BDE (O-H)
kcal/mol
BDEa
IC50 (µM)b
Pha
khí Nước
Authenic
ONOO-
SIN-1-
derived
ONOO-
M1 5 (vòng A) 84,1 84,3 0,2 4,6 10,0
M2 6 (vòng A) 86,3 86,7 0,4 N/A N/A
M3 5 (vòng A) 86,4 86,1 -0,3 >30 3,2
M4 5 (vòng A) 86,6 86,3 -0,3 >30 11,9
M5 1 (vòng B) 99,1 93,2 -5,9 15,9 >30
M6 6 (vòng A) 85,7 86,2 0,5 26,4 15,1
M7 6 (vòng A) 83,8 86,4 2,6 14,1 >30
M8 5 (vòng A) 84,3 84,4 0,1 9,1 9,3
M9 6 (vòng A) 84,2 85,9 1,7 19,2 24,1
M10 6 (vòng A) 82,8 85,7 2,9 12,2 <0,49
M11 6 (vòng A) 82,3 85,9 3,6 8,0 3,1
M12 5 (vòng A) 87,2 87,4 0,2 >30 >30
M13 5 (vòng A) 85,2 85,6 0,4 2,2 9,7
M14 6 (vòng A) 86,1 85,9 -0,2 >30 >30
Norathyriol 2 (vòng B) 84,2 86,0
(86,6c)
1,8
(2,4) N/A
a BDE = BDEnước - BDEkhí.
b Tham khảo tài liệu [49].
c Tham khảo tài liệu [43].
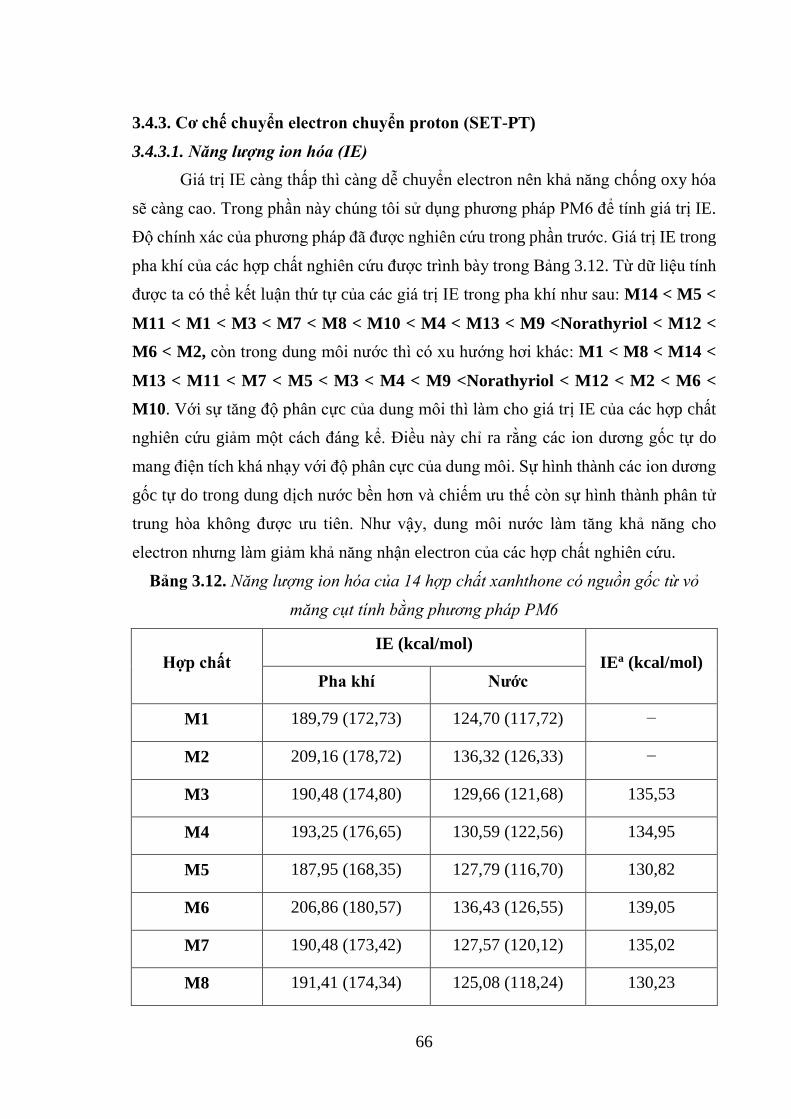
66
3.4.3. Cơ chế chuyển electron chuyển proton (SET-PT)
3.4.3.1. Năng lượng ion hóa (IE)
Giá trị IE càng thấp thì càng dễ chuyển electron nên khả năng chống oxy hóa
sẽ càng cao. Trong phần này chúng tôi sử dụng phương pháp PM6 để tính giá trị IE.
Độ chính xác của phương pháp đã được nghiên cứu trong phần trước. Giá trị IE trong
pha khí của các hợp chất nghiên cứu được trình bày trong Bảng 3.12. Từ dữ liệu tính
được ta có thể kết luận thứ tự của các giá trị IE trong pha khí như sau: M14 < M5 <
M11 < M1 < M3 < M7 < M8 < M10 < M4 < M13 < M9 <Norathyriol < M12 <
M6 < M2, còn trong dung môi nước thì có xu hướng hơi khác: M1 < M8 < M14 <
M13 < M11 < M7 < M5 < M3 < M4 < M9 <Norathyriol < M12 < M2 < M6 <
M10. Với sự tăng độ phân cực của dung môi thì làm cho giá trị IE của các hợp chất
nghiên cứu giảm một cách đáng kể. Điều này chỉ ra rằng các ion dương gốc tự do
mang điện tích khá nhạy với độ phân cực của dung môi. Sự hình thành các ion dương
gốc tự do trong dung dịch nước bền hơn và chiếm ưu thế còn sự hình thành phân tử
trung hòa không được ưu tiên. Như vậy, dung môi nước làm tăng khả năng cho
electron nhưng làm giảm khả năng nhận electron của các hợp chất nghiên cứu.
Bảng 3.12. Năng lượng ion hóa của 14 hợp chất xanhthone có nguồn gốc từ vỏ
măng cụt tính bằng phương pháp PM6
Hợp chất IE (kcal/mol)
IEa (kcal/mol) Pha khí Nước
M1 189,79 (172,73) 124,70 (117,72) −
M2 209,16 (178,72) 136,32 (126,33) −
M3 190,48 (174,80) 129,66 (121,68) 135,53
M4 193,25 (176,65) 130,59 (122,56) 134,95
M5 187,95 (168,35) 127,79 (116,70) 130,82
M6 206,86 (180,57) 136,43 (126,55) 139,05
M7 190,48 (173,42) 127,57 (120,12) 135,02
M8 191,41 (174,34) 125,08 (118,24) 130,23
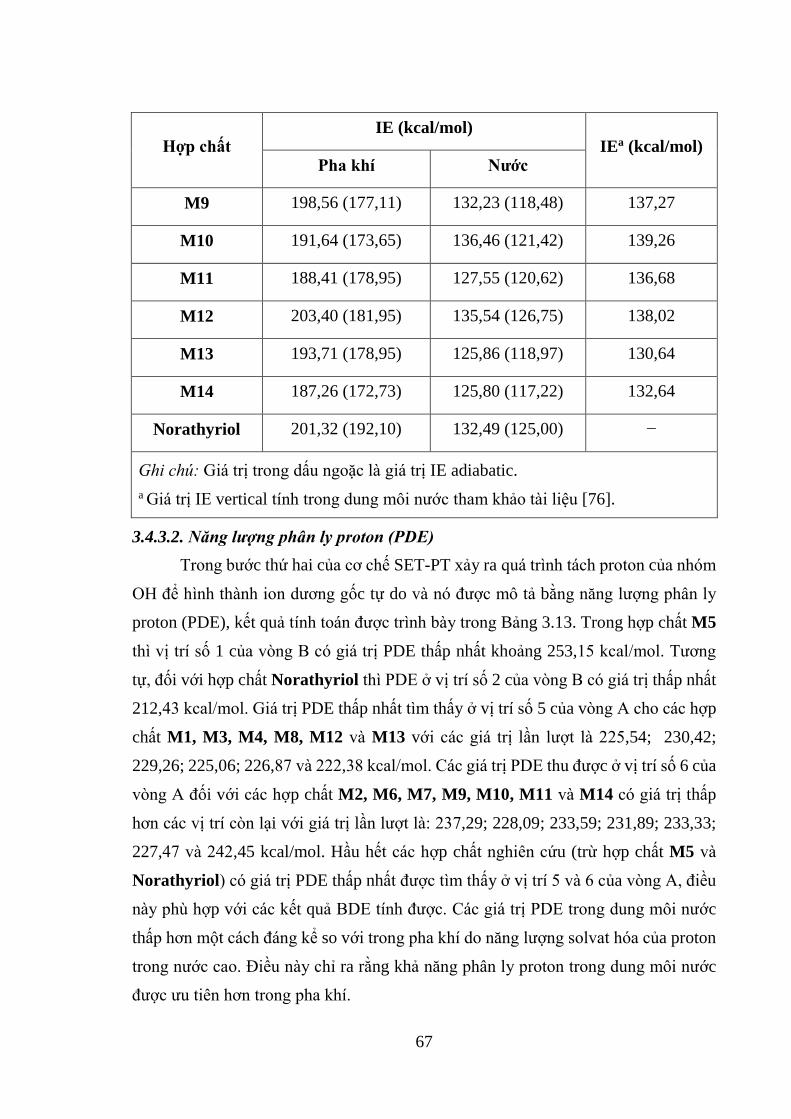
67
Hợp chất IE (kcal/mol)
IEa (kcal/mol) Pha khí Nước
M9 198,56 (177,11) 132,23 (118,48) 137,27
M10 191,64 (173,65) 136,46 (121,42) 139,26
M11 188,41 (178,95) 127,55 (120,62) 136,68
M12 203,40 (181,95) 135,54 (126,75) 138,02
M13 193,71 (178,95) 125,86 (118,97) 130,64
M14 187,26 (172,73) 125,80 (117,22) 132,64
Norathyriol 201,32 (192,10) 132,49 (125,00) −
Ghi chú: Giá trị trong dấu ngoặc là giá trị IE adiabatic.
a Giá trị IE vertical tính trong dung môi nước tham khảo tài liệu [76].
3.4.3.2. Năng lượng phân ly proton (PDE)
Trong bước thứ hai của cơ chế SET-PT xảy ra quá trình tách proton của nhóm
OH để hình thành ion dương gốc tự do và nó được mô tả bằng năng lượng phân ly
proton (PDE), kết quả tính toán được trình bày trong Bảng 3.13. Trong hợp chất M5
thì vị trí số 1 của vòng B có giá trị PDE thấp nhất khoảng 253,15 kcal/mol. Tương
tự, đối với hợp chất Norathyriol thì PDE ở vị trí số 2 của vòng B có giá trị thấp nhất
212,43 kcal/mol. Giá trị PDE thấp nhất tìm thấy ở vị trí số 5 của vòng A cho các hợp
chất M1, M3, M4, M8, M12 và M13 với các giá trị lần lượt là 225,54; 230,42;
229,26; 225,06; 226,87 và 222,38 kcal/mol. Các giá trị PDE thu được ở vị trí số 6 của
vòng A đối với các hợp chất M2, M6, M7, M9, M10, M11 và M14 có giá trị thấp
hơn các vị trí còn lại với giá trị lần lượt là: 237,29; 228,09; 233,59; 231,89; 233,33;
227,47 và 242,45 kcal/mol. Hầu hết các hợp chất nghiên cứu (trừ hợp chất M5 và
Norathyriol) có giá trị PDE thấp nhất được tìm thấy ở vị trí 5 và 6 của vòng A, điều
này phù hợp với các kết quả BDE tính được. Các giá trị PDE trong dung môi nước
thấp hơn một cách đáng kể so với trong pha khí do năng lượng solvat hóa của proton
trong nước cao. Điều này chỉ ra rằng khả năng phân ly proton trong dung môi nước
được ưu tiên hơn trong pha khí.
68
3.4.4. Cơ chế chuyển proton chuyển electron (SPLET)
3.4.4.1. Ái lực proton (PA)
Trong bước đầu tiên của cơ chế SPLET, nhóm hydroxyl của hợp chất
polyphenol bị phân ly để hình thành ion âm phenolat và một proton. Quá trình mất
một proton là bước quan trọng trong cơ chế này, nó được mô tả bằng các giá trị PA.
Các giá trị PA của các hợp chất nghiên cứu được trình bày ở Bảng 3.13. Trong mỗi
phân tử, giá trị PA thấp nhất được in đậm.
Nhiều công trình nghiên cứu đã sử dụng mô hình IEFPCM để nghiên cứu sự
ảnh hưởng của dung môi lên enthalpy [43, 99, 120, 131]. Vì vậy, chúng tôi sử dụng
mô hình này để nghiên cứu sự ảnh hưởng của dung môi lên các đại lượng BDE, IE,
PA, ETE và PDE của hợp chất polyphenol. Kết quả cho thấy dung môi nước ảnh
hưởng rất lớn đến giá trị PA và PDE.
Quá trình mất proton là một bước quan trọng trong cơ chế SPLET, nó được
đặc trưng bằng đại lượng PA. Vì vậy những hợp chất nghiên cứu có giá trị PA càng
thấp thì sẽ có khả năng chống oxy hóa càng cao. Dữ liệu trong Bảng 3.13 cho thấy
dung môi nước ảnh hưởng đáng kể đến các giá trị PA vì năng lượng solvat hóa của
proton trong nước cao. Sự khác nhau giữa năng lượng PA trong pha khí và dung môi
nước nằm trong khoảng từ -295,75 đến -378,23 kcal/mol. Điều này có nghĩa là quá
trình tách proton xảy ra trong dung môi nước sẽ được ưu tiên hơn. Trong pha khí thì
các giá trị PA cao hơn so với các giá trị BDE và IE. Tuy nhiên, trong dung môi nước
thì giá trị PA thấp hơn rất nhiều so với các giá trị BDE và IE tương ứng. Như vậy,
các kết quả thu được cho thấy trong dung môi nước thì cơ chế SPLET được ưu tiên
hơn theo quan điểm nhiệt động.
Bảng 3.13. Các giá trị PA, ETE và PDE của 14 hợp chất xanhthone có nguồn gốc từ
vỏ măng cụt tính ở mức lý thuyết B3LYP/6-31+G(d,p)//PM6 trong pha khí và nước
Hợp chất
PA (kcal/mol)
PAa
ETE (kcal/mol)
ETEb
PDE (kcal/mol)
PDEc
Pha khí Nước Pha khí Nước Pha khí Nước
M1
5 (vòng A) 336,75 33,47 -303,28 56,49 69,74 13,25 225,54 -6,25 -231,79
8 (vòng A) 347,64 40,67 -306,97 58,45 73,12 14,67 238,39 4,33 -234,06
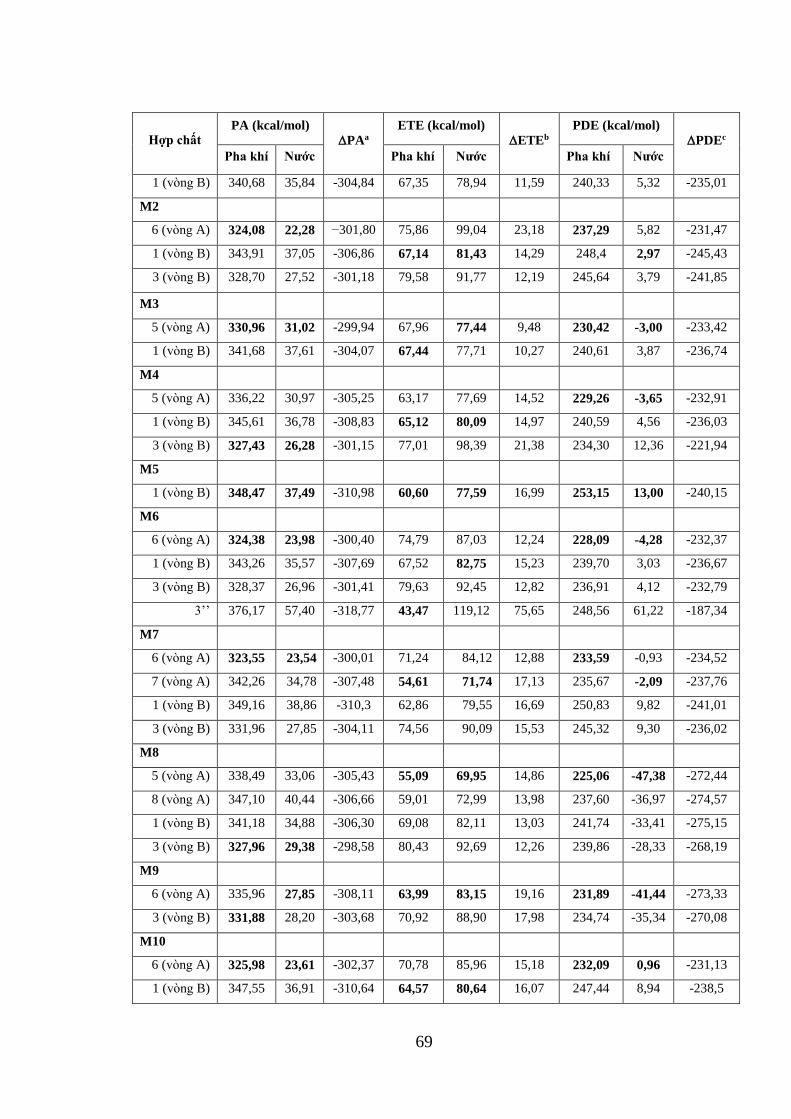
69
Hợp chất PA (kcal/mol)
PAa
ETE (kcal/mol)
ETEb
PDE (kcal/mol)
PDEc
Pha khí Nước Pha khí Nước Pha khí Nước
1 (vòng B) 340,68 35,84 -304,84 67,35 78,94 11,59 240,33 5,32 -235,01
M2
6 (vòng A) 324,08 22,28 −301,80 75,86 99,04 23,18 237,29 5,82 -231,47
1 (vòng B) 343,91 37,05 -306,86 67,14 81,43 14,29 248,4 2,97 -245,43
3 (vòng B) 328,70 27,52 -301,18 79,58 91,77 12,19 245,64 3,79 -241,85
M3
5 (vòng A) 330,96 31,02 -299,94 67,96 77,44 9,48 230,42 -3,00 -233,42
1 (vòng B) 341,68 37,61 -304,07 67,44 77,71 10,27 240,61 3,87 -236,74
M4
5 (vòng A) 336,22 30,97 -305,25 63,17 77,69 14,52 229,26 -3,65 -232,91
1 (vòng B) 345,61 36,78 -308,83 65,12 80,09 14,97 240,59 4,56 -236,03
3 (vòng B) 327,43 26,28 -301,15 77,01 98,39 21,38 234,30 12,36 -221,94
M5
1 (vòng B) 348,47 37,49 -310,98 60,60 77,59 16,99 253,15 13,00 -240,15
M6
6 (vòng A) 324,38 23,98 -300,40 74,79 87,03 12,24 228,09 -4,28 -232,37
1 (vòng B) 343,26 35,57 -307,69 67,52 82,75 15,23 239,70 3,03 -236,67
3 (vòng B) 328,37 26,96 -301,41 79,63 92,45 12,82 236,91 4,12 -232,79
3’’ 376,17 57,40 -318,77 43,47 119,12 75,65 248,56 61,22 -187,34
M7
6 (vòng A) 323,55 23,54 -300,01 71,24 84,12 12,88 233,59 -0,93 -234,52
7 (vòng A) 342,26 34,78 -307,48 54,61 71,74 17,13 235,67 -2,09 -237,76
1 (vòng B) 349,16 38,86 -310,3 62,86 79,55 16,69 250,83 9,82 -241,01
3 (vòng B) 331,96 27,85 -304,11 74,56 90,09 15,53 245,32 9,30 -236,02
M8
5 (vòng A) 338,49 33,06 -305,43 55,09 69,95 14,86 225,06 -47,38 -272,44
8 (vòng A) 347,10 40,44 -306,66 59,01 72,99 13,98 237,60 -36,97 -274,57
1 (vòng B) 341,18 34,88 -306,30 69,08 82,11 13,03 241,74 -33,41 -275,15
3 (vòng B) 327,96 29,38 -298,58 80,43 92,69 12,26 239,86 -28,33 -268,19
M9
6 (vòng A) 335,96 27,85 -308,11 63,99 83,15 19,16 231,89 -41,44 -273,33
3 (vòng B) 331,88 28,20 -303,68 70,92 88,90 17,98 234,74 -35,34 -270,08
M10
6 (vòng A) 325,98 23,61 -302,37 70,78 85,96 15,18 232,09 0,96 -231,13
1 (vòng B) 347,55 36,91 -310,64 64,57 80,64 16,07 247,44 8,94 -238,5
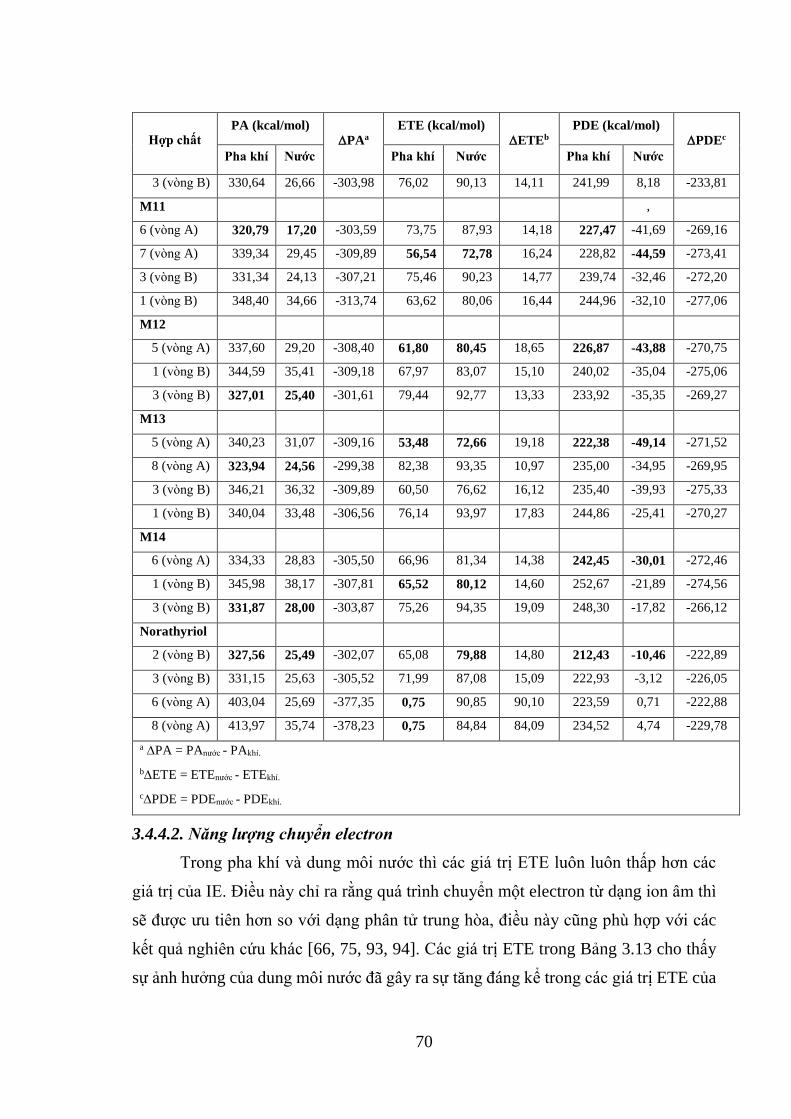
70
Hợp chất PA (kcal/mol)
PAa
ETE (kcal/mol)
ETEb
PDE (kcal/mol)
PDEc
Pha khí Nước Pha khí Nước Pha khí Nước
3 (vòng B) 330,64 26,66 -303,98 76,02 90,13 14,11 241,99 8,18 -233,81
M11 ,
6 (vòng A) 320,79 17,20 -303,59 73,75 87,93 14,18 227,47 -41,69 -269,16
7 (vòng A) 339,34 29,45 -309,89 56,54 72,78 16,24 228,82 -44,59 -273,41
3 (vòng B) 331,34 24,13 -307,21 75,46 90,23 14,77 239,74 -32,46 -272,20
1 (vòng B) 348,40 34,66 -313,74 63,62 80,06 16,44 244,96 -32,10 -277,06
M12
5 (vòng A) 337,60 29,20 -308,40 61,80 80,45 18,65 226,87 -43,88 -270,75
1 (vòng B) 344,59 35,41 -309,18 67,97 83,07 15,10 240,02 -35,04 -275,06
3 (vòng B) 327,01 25,40 -301,61 79,44 92,77 13,33 233,92 -35,35 -269,27
M13
5 (vòng A) 340,23 31,07 -309,16 53,48 72,66 19,18 222,38 -49,14 -271,52
8 (vòng A) 323,94 24,56 -299,38 82,38 93,35 10,97 235,00 -34,95 -269,95
3 (vòng B) 346,21 36,32 -309,89 60,50 76,62 16,12 235,40 -39,93 -275,33
1 (vòng B) 340,04 33,48 -306,56 76,14 93,97 17,83 244,86 -25,41 -270,27
M14
6 (vòng A) 334,33 28,83 -305,50 66,96 81,34 14,38 242,45 -30,01 -272,46
1 (vòng B) 345,98 38,17 -307,81 65,52 80,12 14,60 252,67 -21,89 -274,56
3 (vòng B) 331,87 28,00 -303,87 75,26 94,35 19,09 248,30 -17,82 -266,12
Norathyriol
2 (vòng B) 327,56 25,49 -302,07 65,08 79,88 14,80 212,43 -10,46 -222,89
3 (vòng B) 331,15 25,63 -305,52 71,99 87,08 15,09 222,93 -3,12 -226,05
6 (vòng A) 403,04 25,69 -377,35 0,75 90,85 90,10 223,59 0,71 -222,88
8 (vòng A) 413,97 35,74 -378,23 0,75 84,84 84,09 234,52 4,74 -229,78
a PA = PAnước - PAkhí.
bETE = ETEnước - ETEkhí.
cPDE = PDEnước - PDEkhí.
3.4.4.2. Năng lượng chuyển electron
Trong pha khí và dung môi nước thì các giá trị ETE luôn luôn thấp hơn các
giá trị của IE. Điều này chỉ ra rằng quá trình chuyển một electron từ dạng ion âm thì
sẽ được ưu tiên hơn so với dạng phân tử trung hòa, điều này cũng phù hợp với các
kết quả nghiên cứu khác [66, 75, 93, 94]. Các giá trị ETE trong Bảng 3.13 cho thấy
sự ảnh hưởng của dung môi nước đã gây ra sự tăng đáng kể trong các giá trị ETE của

71
các ion âm phenolat, điều đó có nghĩa là dung môi nước không thuận lợi cho quá
trình chuyển electron. Độ lệch trung bình giữa các giá trị ETE trong pha khí và dung
môi nước xấp xỉ 19,07 kcal/mol.
3.4.5. Nhận xét
Khả năng chống oxy hóa của 14 hợp chất xanhthone có nguồn gốc từ vỏ măng
cụt được nghiên cứu trong cả pha khí và dung môi nước thông qua ba cơ chế HAT,
SET−PT và SPLET. Chúng tôi đã tính toán các thông số BDE, IE, PA, ETE và PDE
tương ứng với từng cơ chế dập tắt gốc tự do. Dựa trên kết quả thu được trong pha khí
và dung môi nước có thể rút ra một vài kết luận sau:
- Trong số 3 cơ chế, xét theo quan điểm nhiệt động học thì cơ chế HAT được
ưu tiên trong pha khí còn cơ chế SPLET ưu tiên xảy ra trong dung môi nước.
- Sự khác nhau đáng kể giữa các giá trị enthalpy trong pha khí và dung môi
nước đặc trưng cho những phản ứng liên quan đến phần tử mang điện tích.
- Trong số các hợp chất xanhthone nghiên cứu thì hợp chất M10 và M11 có
khả năng chống oxy hóa cao nhất, kết quả này cũng phù hợp với các nghiên cứu trước
[49, 75].
3.5. KHẢ NĂNG DẬP TẮT GỐC TỰ DO CỦA MỘT SỐ HỢP CHẤT
POLYPHENOL
3.5.1. Giới thiệu
Trong tự nhiên, chúng ta có thể tìm thấy nhiều hợp chất polyphenol được chiết
xuất từ lá, thân hoặc các bộ phận khác của cây (chẳng hạn như lá sa kê, vỏ măng
cụt...) được xem như những chất chống oxy hóa tiềm năng [9, 45, 49]. Ưu điểm nổi
bật của các hợp chất này là có khả năng chống oxy hóa cao, không độc hại với cơ thể
con người và an toàn với môi trường [95].
Trong phần này, chúng tôi tiến hành nghiên cứu một cách hệ thống để làm
sáng tỏ hơn đặc tính chống oxy hóa của một số hợp chất polyphenol thường được sử
dụng (Hình 3.8). Phản ứng của gốc tự do peroxyl (ROO•) với các chất chống oxy hóa
polyphenol (ArOH) cũng được làm sáng tỏ thông qua cơ chế chuyển nguyên tử hydro
(HAT). Cấu trúc phân tử, năng lượng và trạng thái chuyển tiếp của mỗi phản ứng
được phân tích cụ thể. Ngoài ra, mối quan hệ giữa hàng rào năng lượng và khả năng
chống oxy hóa cũng được thảo luận trong phần nghiên cứu này.
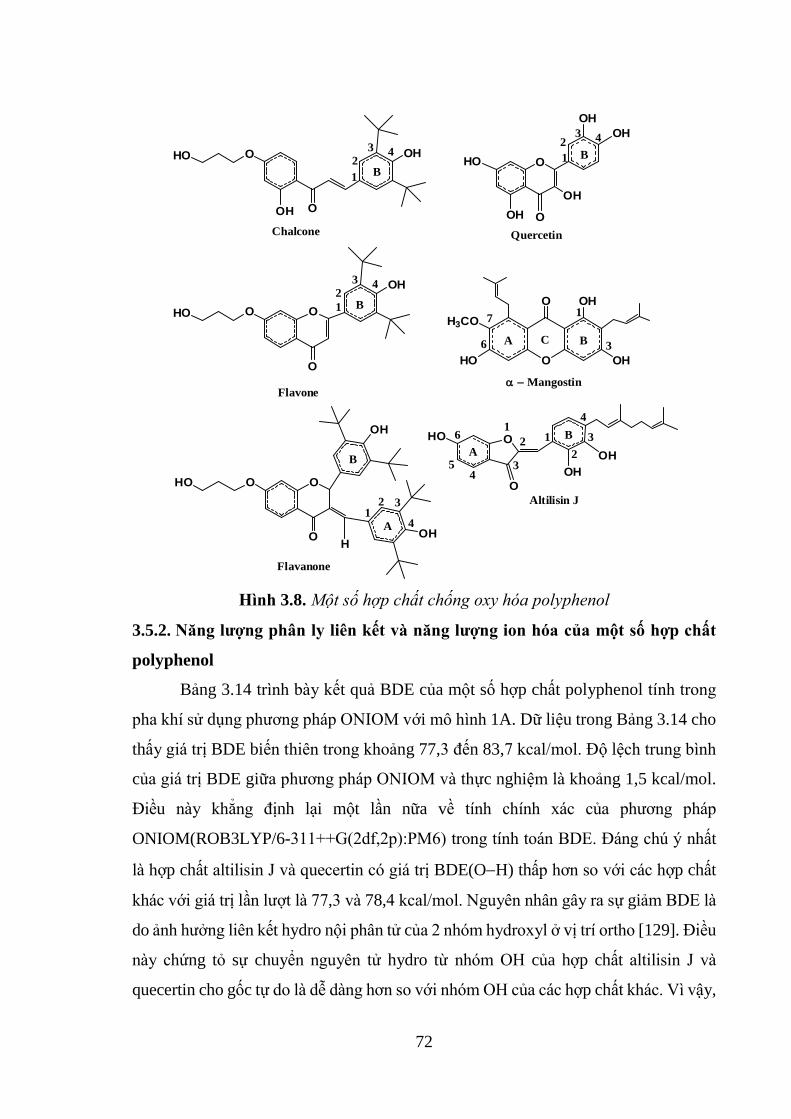
72
OH
OH
OOH
OH
O
OH
O
O O
O
O O
H
HO
HO
HO
B
B
B
A
O OH
OHO
H3CO
HO
A C B
1
36
7
OHO
O
OH
OH
1
2
41
2
34
5
6
A
B 3
Altilisin J
Flavanone
Flavone
Chalcone
Mangostin
O
OOH
HO
OH
OH
Quercetin
OH
B
3 43 4
3 4
3
41
1
1
1
2
2
2
2
Hình 3.8. Một số hợp chất chống oxy hóa polyphenol
3.5.2. Năng lượng phân ly liên kết và năng lượng ion hóa của một số hợp chất
polyphenol
Bảng 3.14 trình bày kết quả BDE của một số hợp chất polyphenol tính trong
pha khí sử dụng phương pháp ONIOM với mô hình 1A. Dữ liệu trong Bảng 3.14 cho
thấy giá trị BDE biến thiên trong khoảng 77,3 đến 83,7 kcal/mol. Độ lệch trung bình
của giá trị BDE giữa phương pháp ONIOM và thực nghiệm là khoảng 1,5 kcal/mol.
Điều này khẳng định lại một lần nữa về tính chính xác của phương pháp
ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) trong tính toán BDE. Đáng chú ý nhất
là hợp chất altilisin J và quecertin có giá trị BDE(OH) thấp hơn so với các hợp chất
khác với giá trị lần lượt là 77,3 và 78,4 kcal/mol. Nguyên nhân gây ra sự giảm BDE là
do ảnh hưởng liên kết hydro nội phân tử của 2 nhóm hydroxyl ở vị trí ortho [129]. Điều
này chứng tỏ sự chuyển nguyên tử hydro từ nhóm OH của hợp chất altilisin J và
quecertin cho gốc tự do là dễ dàng hơn so với nhóm OH của các hợp chất khác. Vì vậy,
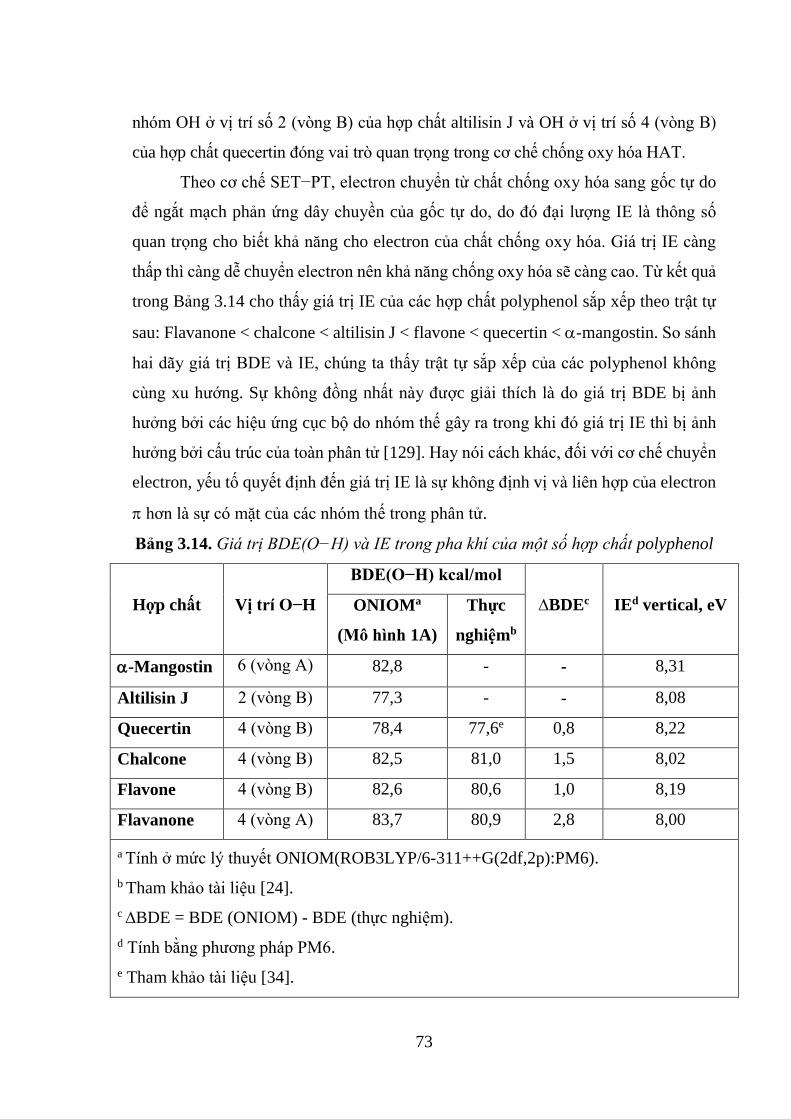
73
nhóm OH ở vị trí số 2 (vòng B) của hợp chất altilisin J và OH ở vị trí số 4 (vòng B)
của hợp chất quecertin đóng vai trò quan trọng trong cơ chế chống oxy hóa HAT.
Theo cơ chế SET−PT, electron chuyển từ chất chống oxy hóa sang gốc tự do
để ngắt mạch phản ứng dây chuyền của gốc tự do, do đó đại lượng IE là thông số
quan trọng cho biết khả năng cho electron của chất chống oxy hóa. Giá trị IE càng
thấp thì càng dễ chuyển electron nên khả năng chống oxy hóa sẽ càng cao. Từ kết quả
trong Bảng 3.14 cho thấy giá trị IE của các hợp chất polyphenol sắp xếp theo trật tự
sau: Flavanone < chalcone < altilisin J < flavone < quecertin < -mangostin. So sánh
hai dãy giá trị BDE và IE, chúng ta thấy trật tự sắp xếp của các polyphenol không
cùng xu hướng. Sự không đồng nhất này được giải thích là do giá trị BDE bị ảnh
hưởng bởi các hiệu ứng cục bộ do nhóm thế gây ra trong khi đó giá trị IE thì bị ảnh
hưởng bởi cấu trúc của toàn phân tử [129]. Hay nói cách khác, đối với cơ chế chuyển
electron, yếu tố quyết định đến giá trị IE là sự không định vị và liên hợp của electron
hơn là sự có mặt của các nhóm thế trong phân tử.
Bảng 3.14. Giá trị BDE(O−H) và IE trong pha khí của một số hợp chất polyphenol
Hợp chất Vị trí O−H
BDE(O−H) kcal/mol
∆BDEc IEd vertical, eV ONIOMa
(Mô hình 1A)
Thực
nghiệmb
-Mangostin 6 (vòng A) 82,8 - - 8,31
Altilisin J 2 (vòng B) 77,3 - - 8,08
Quecertin 4 (vòng B) 78,4 77,6e 0,8 8,22
Chalcone 4 (vòng B) 82,5 81,0 1,5 8,02
Flavone 4 (vòng B) 82,6 80,6 1,0 8,19
Flavanone 4 (vòng A) 83,7 80,9 2,8 8,00
a Tính ở mức lý thuyết ONIOM(ROB3LYP/6-311++G(2df,2p):PM6).
b Tham khảo tài liệu [24].
c ∆BDE = BDE (ONIOM) - BDE (thực nghiệm).
d Tính bằng phương pháp PM6.
e Tham khảo tài liệu [34].

74
3.5.3. Phản ứng dập tắt gốc tự do peroxyl (CH3OO•) của một số hợp chất
polyphenol
Cơ chế phản ứng giữa gốc tự do CH3OO• và một số hợp chất polyphenol sẽ
được thảo luận chi tiết để có một cái nhìn rõ hơn về khả năng chống oxy hóa của hợp
chất polyphenol. Một cách tổng quát, phản ứng này bao gồm 3 bước cơ bản: đầu tiên
hình thành trạng thái trung gian 1 (Int‒1), tiếp theo phản ứng chuyển nguyên tử hydro
đi qua trạng thái chuyển tiếp (TS) và cuối cùng là hình thành sản phẩm gốc tự do
phenoxyl từ trạng thái trung gian 2 (Int‒2) mà không đi qua trạng thái chuyển tiếp
nào. Trong trạng thái trung gian Int‒1 và Int‒2, các liên kết hydro được hình thành
giữa hợp chất polyphenol với gốc tự do CH3OO• và giữa gốc tự do phenoxyl và
CH3OOH.
Các cấu tử tham gia phản ứng giữa hợp chất polyphenol với gốc tự do CH3OO•
được tối ưu bằng phương pháp PM6. Cấu trúc hình học của các trạng thái trung gian
và trạng thái chuyển tiếp của phản ứng quecertin và CH3OO• được minh họa ở Hình
3.9 như là một ví dụ tiêu biểu cho các hợp chất polyphenol.
Hình 3.9. Cấu trúc của trạng thái trung gian Int-1, Int-2 và trạng thái chuyển tiếp
(TS) của phản ứng giữa quecertin và gốc tự do CH3OO•
Từ cấu trúc tối ưu, năng lượng điểm đơn của các cấu tử tham gia phản ứng
giữa hợp chất polyphenol với gốc tự do CH3OO• được tính ở mức lý thuyết ONIOM
(B3LYP/6-311G++(2df,2p):PM6). Giản đồ năng lượng được minh họa trong Hình
3.10. Các giá trị năng lượng tính bằng hartree của các phần tử tham gia phản ứng giữa
hợp chất polyphenol và gốc tự do CH3OO• được trình bày trong Bảng 3.15.

75
Quan sát ở Hình 3.10 ta thấy khuynh hướng phản ứng của các polyphenol với
gốc tự do CH3OO• là khá giống nhau. Các trạng thái trung gian Int‒1 tạo thành bền
hơn so với chất tham gia phản ứng khoảng 3,5 − 7,9 kcal/mol. Năng lượng của các
trạng thái trung gian Int‒2 có giá trị thấp hơn năng lượng của các trạng thái Int‒1
tương ứng khoảng 8,2 − 15,1 kcal/mol. Điều này cho thấy liên kết hydro
ArOHOOCH3 trong trạng thái trung gian Int‒2 bền hơn so với liên kết hydro
ArOHOOCH3 trong trạng thái trung gian Int‒1.
Bảng 3.15. Giá trị năng lượng của các phần tử tham gia phản ứng giữa
một số hợp chất polyphenol và gốc tự do CH3OO• tính ở mức lý thuyết
ONIOM (B3LYP/6-311G++(2df,2p):PM6)
Hợp chất ZPE
(hartree)
ZPE hiệu chỉnh
(hartree) E (hartree)
Năng lượng
tương đối
(kcal/mol)a
CH3OO• 0,03767 0,04060826 -190,2691219
CH3OOH 0,04754 0,05124812 -190,9285618
-Mangostin 0,41706 0,44959068 -76,72711529
Int-1 0,45545 0,49097510 -267,0057137 -5,5
TS 0,452765 0,48808067 -266,970410 14,9
Int-2 0,456321 0,491914038 -267,0219036 -15
P 0,407544 0,439332432 -76,08338184 -9,6
Chalcone 0,504228 0,543557784 -76,70875091
Int-1 0,542737 0,585070486 -266,9886356 -6,2
TS 0,53973 0,58182894 -266,9557105 12,4
Int-2 0,542051 0,584330978 -267,0045054 -16,6
P 0,493281 0,531756918 -76,06789605 -12,4
Flavanone 0,795411 0,857453058 -76,79739956
Int-1 0,834045 0,899100510 -267,0769596 -5,9
TS 0,831277 0,896116606 -267,0446452 12,5
Int-2 0,833634 0,898657452 -267,0927824 -16,1
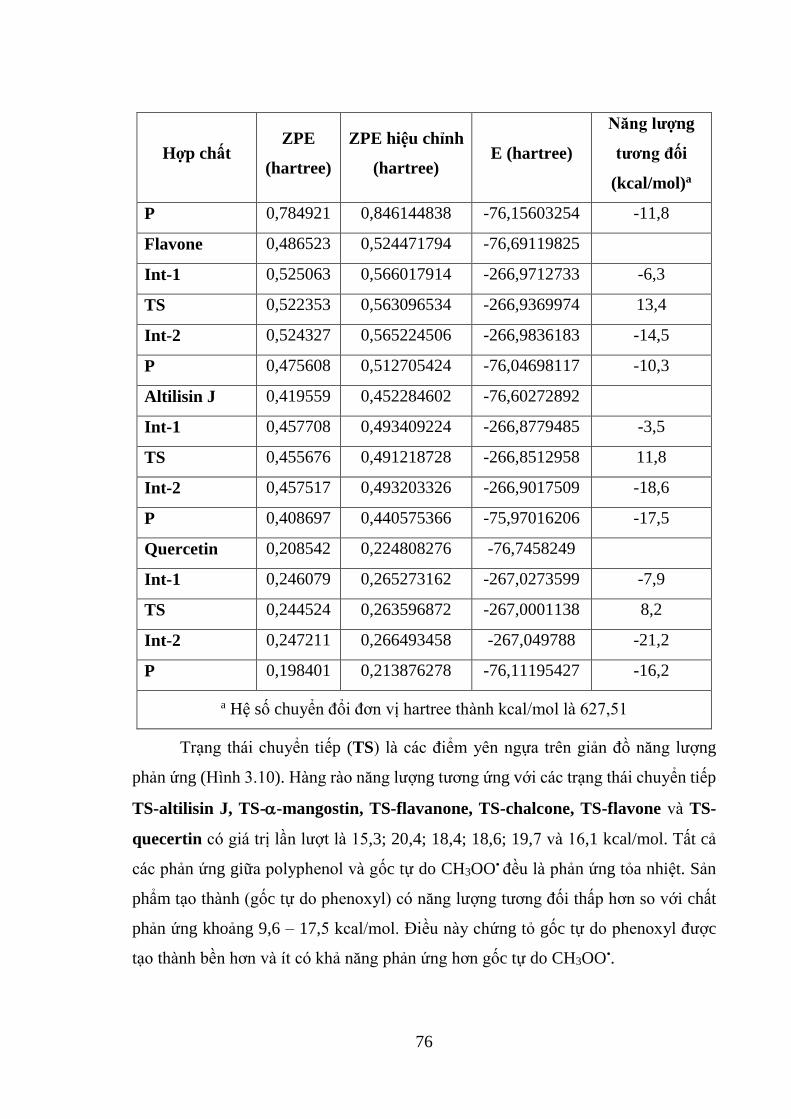
76
Hợp chất ZPE
(hartree)
ZPE hiệu chỉnh
(hartree) E (hartree)
Năng lượng
tương đối
(kcal/mol)a
P 0,784921 0,846144838 -76,15603254 -11,8
Flavone 0,486523 0,524471794 -76,69119825
Int-1 0,525063 0,566017914 -266,9712733 -6,3
TS 0,522353 0,563096534 -266,9369974 13,4
Int-2 0,524327 0,565224506 -266,9836183 -14,5
P 0,475608 0,512705424 -76,04698117 -10,3
Altilisin J 0,419559 0,452284602 -76,60272892
Int-1 0,457708 0,493409224 -266,8779485 -3,5
TS 0,455676 0,491218728 -266,8512958 11,8
Int-2 0,457517 0,493203326 -266,9017509 -18,6
P 0,408697 0,440575366 -75,97016206 -17,5
Quercetin 0,208542 0,224808276 -76,7458249
Int-1 0,246079 0,265273162 -267,0273599 -7,9
TS 0,244524 0,263596872 -267,0001138 8,2
Int-2 0,247211 0,266493458 -267,049788 -21,2
P 0,198401 0,213876278 -76,11195427 -16,2
a Hệ số chuyển đổi đơn vị hartree thành kcal/mol là 627,51
Trạng thái chuyển tiếp (TS) là các điểm yên ngựa trên giản đồ năng lượng
phản ứng (Hình 3.10). Hàng rào năng lượng tương ứng với các trạng thái chuyển tiếp
TS-altilisin J, TS--mangostin, TS-flavanone, TS-chalcone, TS-flavone và TS-
quecertin có giá trị lần lượt là 15,3; 20,4; 18,4; 18,6; 19,7 và 16,1 kcal/mol. Tất cả
các phản ứng giữa polyphenol và gốc tự do CH3OO• đều là phản ứng tỏa nhiệt. Sản
phẩm tạo thành (gốc tự do phenoxyl) có năng lượng tương đối thấp hơn so với chất
phản ứng khoảng 9,6 – 17,5 kcal/mol. Điều này chứng tỏ gốc tự do phenoxyl được
tạo thành bền hơn và ít có khả năng phản ứng hơn gốc tự do CH3OO•.
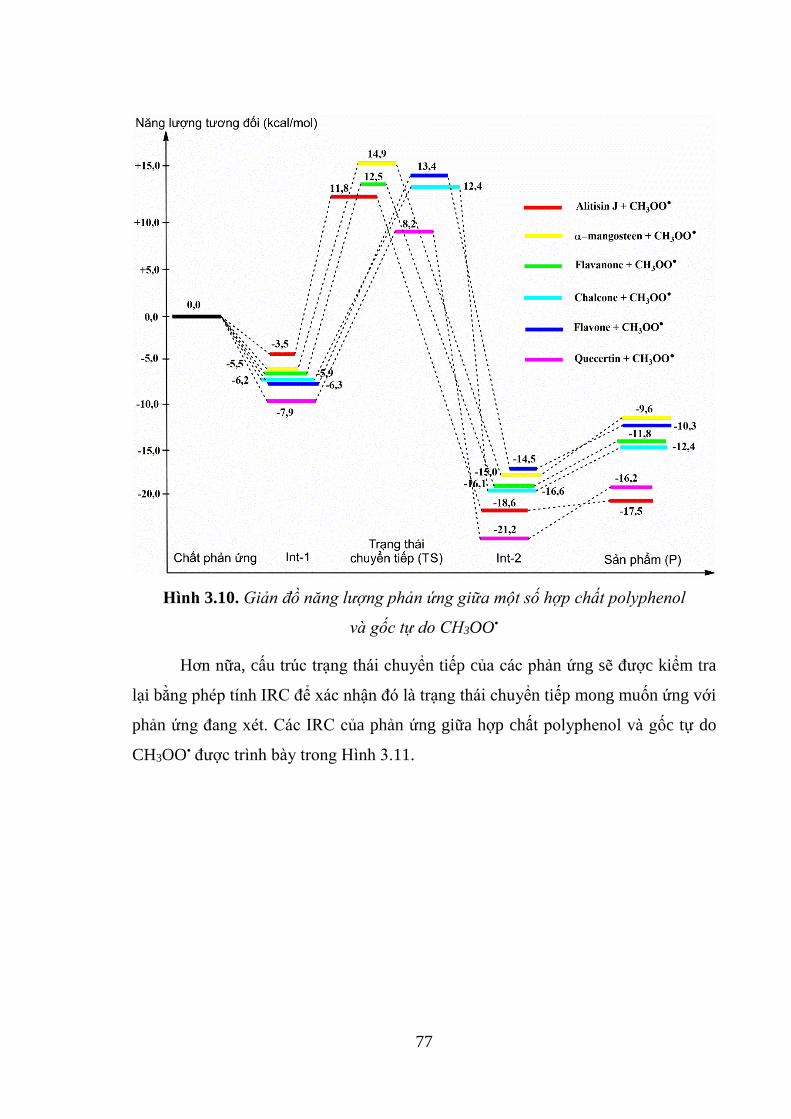
77
Hình 3.10. Giản đồ năng lượng phản ứng giữa một số hợp chất polyphenol
và gốc tự do CH3OO•
Hơn nữa, cấu trúc trạng thái chuyển tiếp của các phản ứng sẽ được kiểm tra
lại bằng phép tính IRC để xác nhận đó là trạng thái chuyển tiếp mong muốn ứng với
phản ứng đang xét. Các IRC của phản ứng giữa hợp chất polyphenol và gốc tự do
CH3OO• được trình bày trong Hình 3.11.
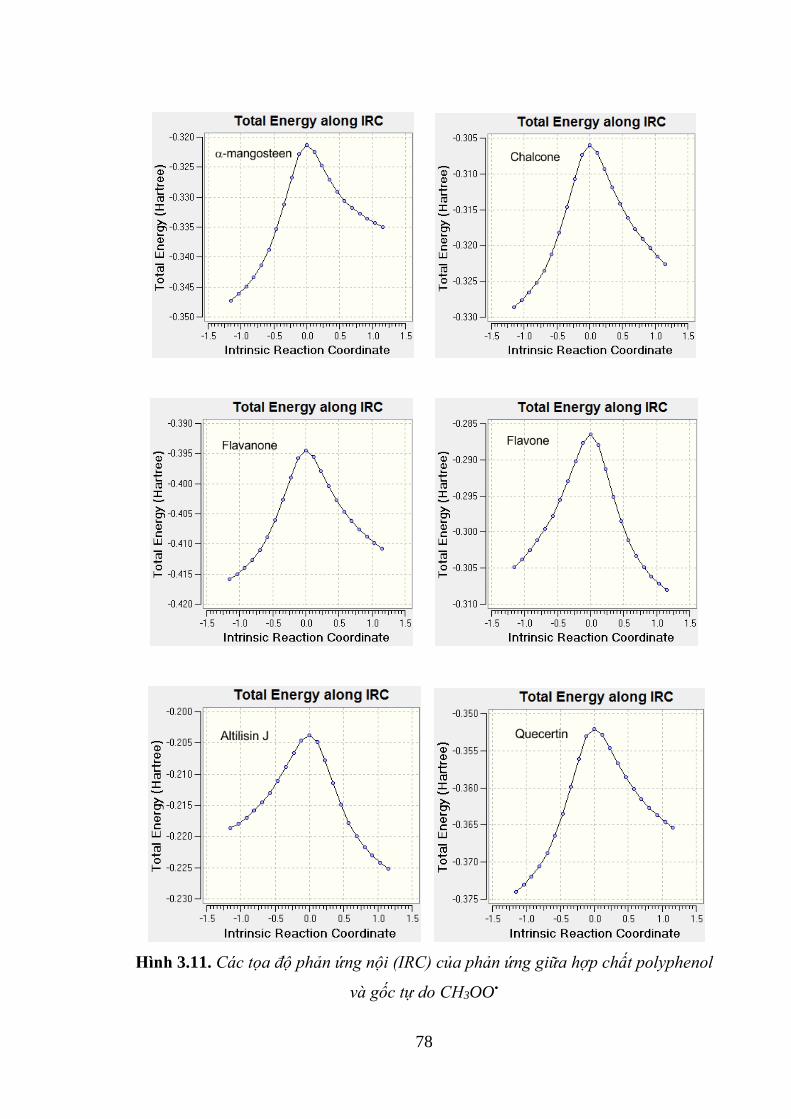
78
Hình 3.11. Các tọa độ phản ứng nội (IRC) của phản ứng giữa hợp chất polyphenol
và gốc tự do CH3OO•
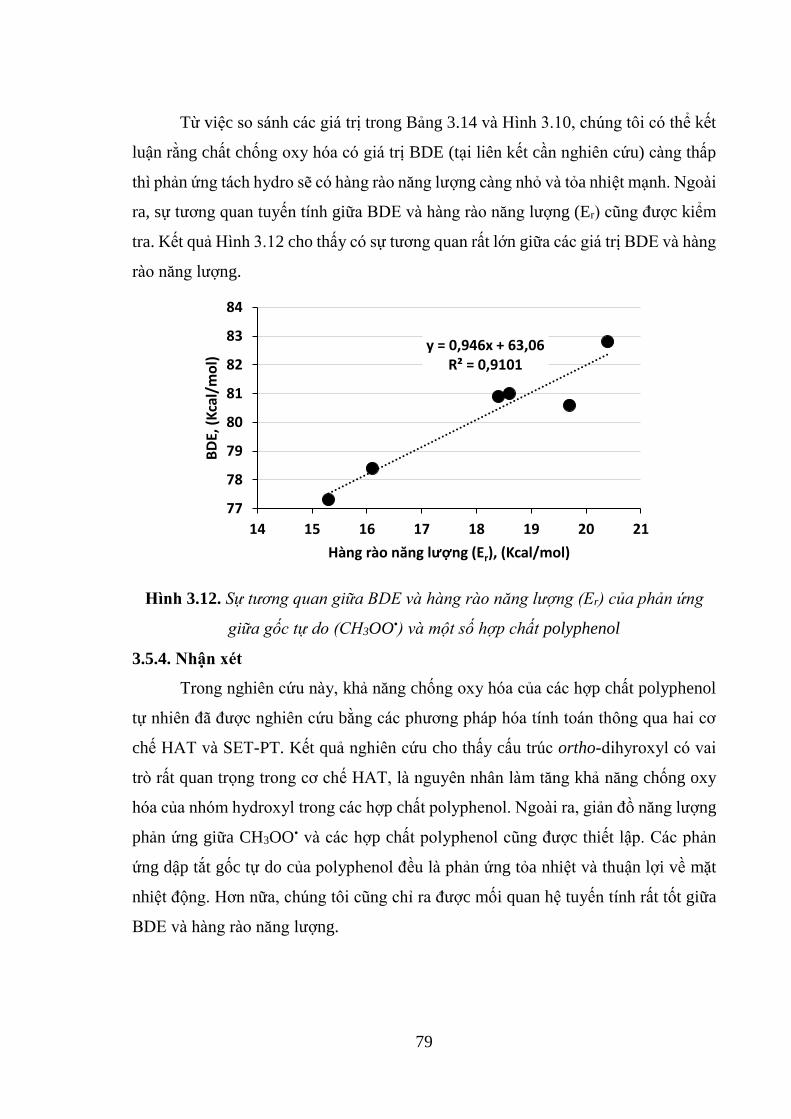
79
Từ việc so sánh các giá trị trong Bảng 3.14 và Hình 3.10, chúng tôi có thể kết
luận rằng chất chống oxy hóa có giá trị BDE (tại liên kết cần nghiên cứu) càng thấp
thì phản ứng tách hydro sẽ có hàng rào năng lượng càng nhỏ và tỏa nhiệt mạnh. Ngoài
ra, sự tương quan tuyến tính giữa BDE và hàng rào năng lượng (Er) cũng được kiểm
tra. Kết quả Hình 3.12 cho thấy có sự tương quan rất lớn giữa các giá trị BDE và hàng
rào năng lượng.
Hình 3.12. Sự tương quan giữa BDE và hàng rào năng lượng (Er) của phản ứng
giữa gốc tự do (CH3OO•) và một số hợp chất polyphenol
3.5.4. Nhận xét
Trong nghiên cứu này, khả năng chống oxy hóa của các hợp chất polyphenol
tự nhiên đã được nghiên cứu bằng các phương pháp hóa tính toán thông qua hai cơ
chế HAT và SET-PT. Kết quả nghiên cứu cho thấy cấu trúc ortho-dihyroxyl có vai
trò rất quan trọng trong cơ chế HAT, là nguyên nhân làm tăng khả năng chống oxy
hóa của nhóm hydroxyl trong các hợp chất polyphenol. Ngoài ra, giản đồ năng lượng
phản ứng giữa CH3OO• và các hợp chất polyphenol cũng được thiết lập. Các phản
ứng dập tắt gốc tự do của polyphenol đều là phản ứng tỏa nhiệt và thuận lợi về mặt
nhiệt động. Hơn nữa, chúng tôi cũng chỉ ra được mối quan hệ tuyến tính rất tốt giữa
BDE và hàng rào năng lượng.
y = 0,946x + 63,06R² = 0,9101
77
78
79
80
81
82
83
84
14 15 16 17 18 19 20 21
BD
E, (
Kca
l/m
ol)
Hàng rào năng lượng (Er), (Kcal/mol)
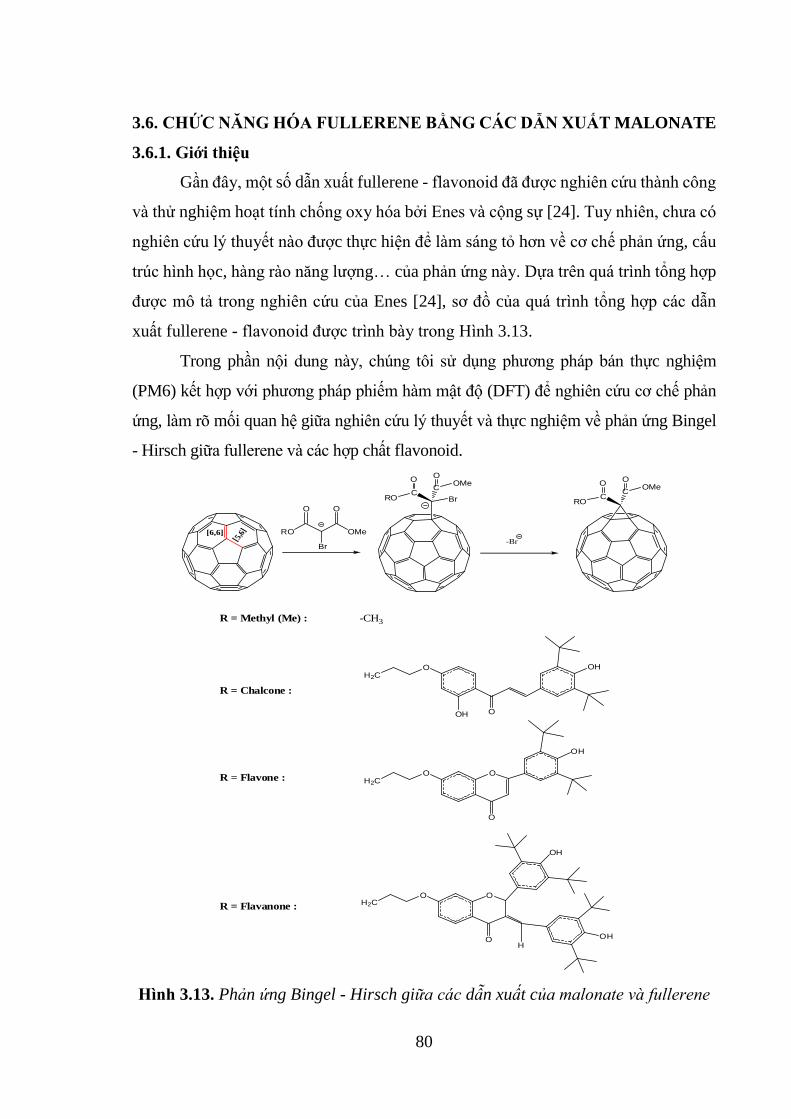
80
3.6. CHỨC NĂNG HÓA FULLERENE BẰNG CÁC DẪN XUẤT MALONATE
3.6.1. Giới thiệu
Gần đây, một số dẫn xuất fullerene - flavonoid đã được nghiên cứu thành công
và thử nghiệm hoạt tính chống oxy hóa bởi Enes và cộng sự [24]. Tuy nhiên, chưa có
nghiên cứu lý thuyết nào được thực hiện để làm sáng tỏ hơn về cơ chế phản ứng, cấu
trúc hình học, hàng rào năng lượng… của phản ứng này. Dựa trên quá trình tổng hợp
được mô tả trong nghiên cứu của Enes [24], sơ đồ của quá trình tổng hợp các dẫn
xuất fullerene - flavonoid được trình bày trong Hình 3.13.
Trong phần nội dung này, chúng tôi sử dụng phương pháp bán thực nghiệm
(PM6) kết hợp với phương pháp phiếm hàm mật độ (DFT) để nghiên cứu cơ chế phản
ứng, làm rõ mối quan hệ giữa nghiên cứu lý thuyết và thực nghiệm về phản ứng Bingel
- Hirsch giữa fullerene và các hợp chất flavonoid.
[6,6]
[5,6
]
CC
Br
O
RO
OOMe
Br
RO OMe
O O
CC
O
RO
OOMe
-Br
R = Methyl (Me) :
R = Flavone :
R = Chalcone :
OH
OH
OOH
OH
R = Flavanone :
OH2C
OH
O
OH2C
O
O
OH2C
O
H
-CH3
Hình 3.13. Phản ứng Bingel - Hirsch giữa các dẫn xuất của malonate và fullerene
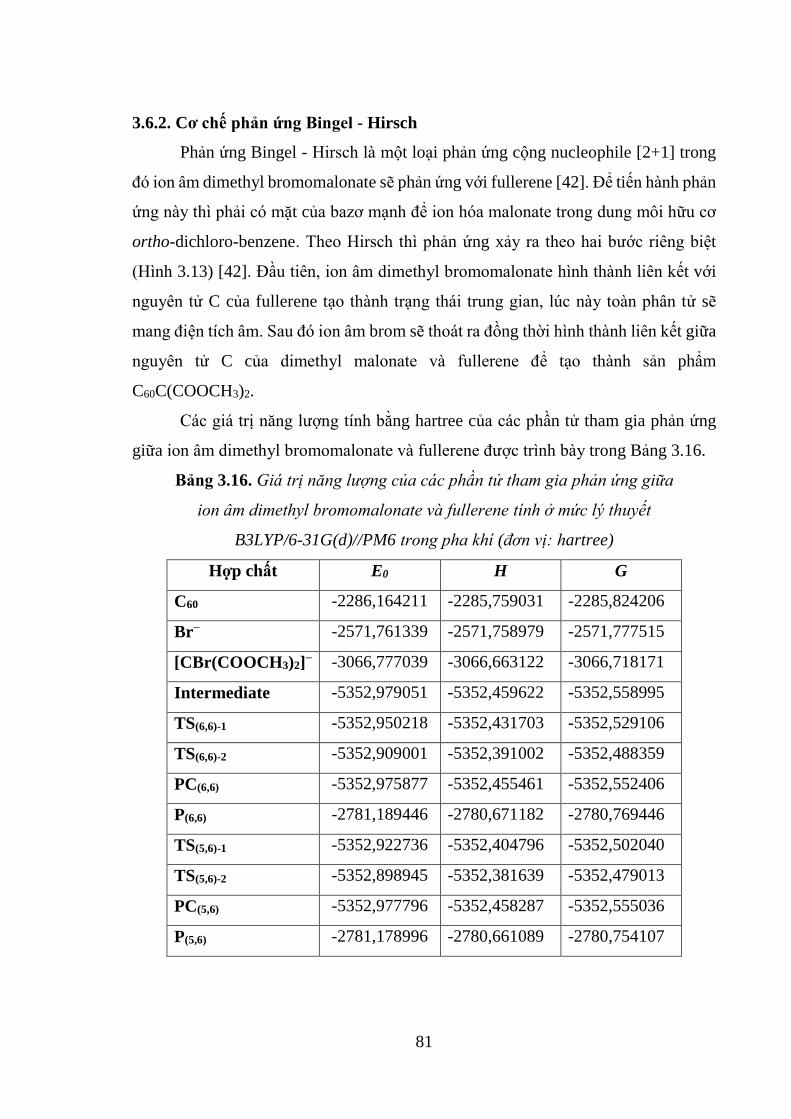
81
3.6.2. Cơ chê phản ứng Bingel - Hirsch
Phản ứng Bingel - Hirsch là một loại phản ứng cộng nucleophile [2+1] trong
đó ion âm dimethyl bromomalonate sẽ phản ứng với fullerene [42]. Để tiến hành phản
ứng này thì phải có mặt của bazơ mạnh để ion hóa malonate trong dung môi hữu cơ
ortho-dichloro-benzene. Theo Hirsch thì phản ứng xảy ra theo hai bước riêng biệt
(Hình 3.13) [42]. Đầu tiên, ion âm dimethyl bromomalonate hình thành liên kết với
nguyên tử C của fullerene tạo thành trạng thái trung gian, lúc này toàn phân tử sẽ
mang điện tích âm. Sau đó ion âm brom sẽ thoát ra đồng thời hình thành liên kết giữa
nguyên tử C của dimethyl malonate và fullerene để tạo thành sản phẩm
C60C(COOCH3)2.
Các giá trị năng lượng tính bằng hartree của các phần tử tham gia phản ứng
giữa ion âm dimethyl bromomalonate và fullerene được trình bày trong Bảng 3.16.
Bảng 3.16. Giá trị năng lượng của các phần tử tham gia phản ứng giữa
ion âm dimethyl bromomalonate và fullerene tính ở mức lý thuyết
B3LYP/6-31G(d)//PM6 trong pha khí (đơn vị: hartree)
Hợp chất E0 H G
C60 -2286,164211 -2285,759031 -2285,824206
Br− -2571,761339 -2571,758979 -2571,777515
[CBr(COOCH3)2]− -3066,777039 -3066,663122 -3066,718171
Intermediate -5352,979051 -5352,459622 -5352,558995
TS(6,6)-1 -5352,950218 -5352,431703 -5352,529106
TS(6,6)-2 -5352,909001 -5352,391002 -5352,488359
PC(6,6) -5352,975877 -5352,455461 -5352,552406
P(6,6) -2781,189446 -2780,671182 -2780,769446
TS(5,6)-1 -5352,922736 -5352,404796 -5352,502040
TS(5,6)-2 -5352,898945 -5352,381639 -5352,479013
PC(5,6) -5352,977796 -5352,458287 -5352,555036
P(5,6) -2781,178996 -2780,661089 -2780,754107

82
Bởi vì trong phân tử C60 có 2 loại liên kết C−C bao gồm: liên kết (6,6) và liên
kết (5,6) (Hình 3.13) nên sản phẩm tạo thành sẽ có hai đồng phân tương ứng (6,6)-
C60C(COOCH3)2 và (5,6)-C60C(COOCH3)2. Các đường phản ứng cho sự hình thành
các sản phẩm đồng phân của C60C(COOCH3)2 được minh họa ở Hình 3.14. Mỗi sản
phẩm đồng phân hình thành sẽ đi qua 2 trạng thái chuyển tiếp (TS). Chẳng hạn, phản
ứng đóng vòng qua trạng thái chuyển tiếp TS(6,6)-1 hoặc TS(6,6)-2 sẽ tạo thành sản phẩm
(6,6)-C60C(COOCH3)2. Còn sản phẩm (5,6)-C60C(COOCH3)2 hình thành khi phản ứng
đóng vòng qua trạng thái chuyển tiếp TS(5,6)-1 hoặc TS(5,6)-2 (Hình 3.14).
Vì các đại lượng H và G là các hàm trạng thái nên biến thiên của chúng trong
các phản ứng không phụ thuộc vào đường đi mà chỉ phụ thuộc vào trạng thái đầu và
trạng thái cuối của hệ, nên chúng tôi chỉ quan tâm đến giá trị nhiệt động của các cấu
tử tham gia phản ứng và sản phẩm. Do đó enthalpy và năng lượng tự do Gibbs của
phản ứng hình thành sản phẩm P(5,6) và P(6,6) được tính theo công thức sau:
∆𝐻2980 (P(5,6)) = H(P(5,6)) + H(Br−) − H([CBr(COOCH3)2]−) − H(C60) =
= (-2780,661089) + (-2571,758979) − (-3066,663122) − (-2285,759031) =
= 0,0020855 hartree = 1,31 kcal/mol.
∆𝐺2980 (P(5,6)) = G(P(5,6)) + G(Br−) − G([CBr(COOCH3)2]−) − G(C60) =
= (-2780,754107) + (-2571,777515) − (-3066,718171) − (-2285,824206) =
= 0,0107555 hartree = 6,75 kcal/mol.
∆𝐻2980 (P(6,6)) = H(P(6,6)) + H(Br−) − H([CBr(COOCH3)2]−) − H(C60) =
= (-2780,671182) + (-2571,758979) − (-3066,663122) − (-2285,759031) =
= -0,0080076 hartree = -5,02 kcal/mol
∆𝐺2980 (P(6,6)) = G(P(6,6)) + G(Br−) − G([CBr(COOCH3)2]−) − G(C60) =
= (-2780,769446) + (-2571,777515) − (-3066,718171) − (-2285,824206) =
= -0,0045836 hartree = -2,88 kcal/mol
Vì ∆𝐺2980 (P(6,6)) < ∆𝐺298
0 (P(5,6)) nên phản ứng theo hướng hình thành sản phẩm
P(6,6) được ưu tiên về mặt nhiệt động và phản ứng là tỏa nhiệt vì ∆𝐻2980 (P(6,6)) < 0.
So sánh hàng rào năng lượng của các đường phản ứng này thì sự hình thành
sản phẩm (6,6)-C60C(COOCH3)2 qua trạng thái chuyển tiếp TS(6,6)-1 là thuận lợi nhất,
có giá trị hàng rào năng lượng tướng ứng là 17,6 kcal/mol. Ba con đường phản ứng
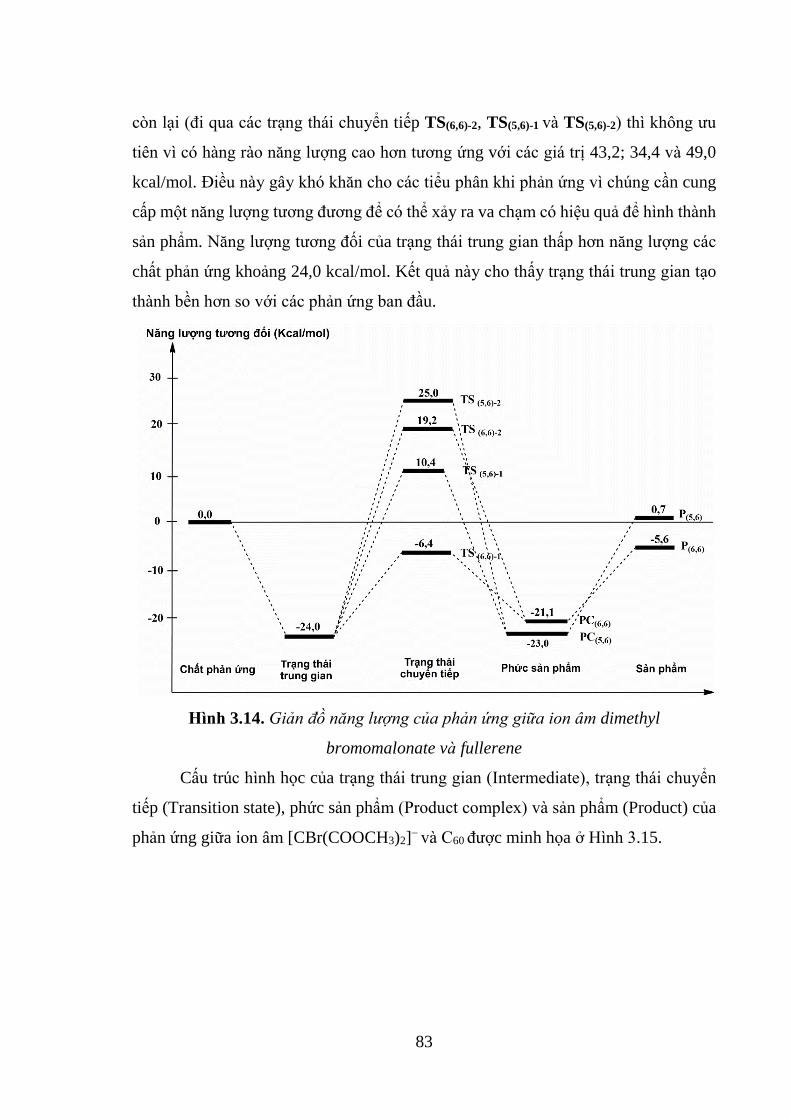
83
còn lại (đi qua các trạng thái chuyển tiếp TS(6,6)-2, TS(5,6)-1 và TS(5,6)-2) thì không ưu
tiên vì có hàng rào năng lượng cao hơn tương ứng với các giá trị 43,2; 34,4 và 49,0
kcal/mol. Điều này gây khó khăn cho các tiểu phân khi phản ứng vì chúng cần cung
cấp một năng lượng tương đương để có thể xảy ra va chạm có hiệu quả để hình thành
sản phẩm. Năng lượng tương đối của trạng thái trung gian thấp hơn năng lượng các
chất phản ứng khoảng 24,0 kcal/mol. Kết quả này cho thấy trạng thái trung gian tạo
thành bền hơn so với các phản ứng ban đầu.
Hình 3.14. Giản đồ năng lượng của phản ứng giữa ion âm dimethyl
bromomalonate và fullerene
Cấu trúc hình học của trạng thái trung gian (Intermediate), trạng thái chuyển
tiếp (Transition state), phức sản phẩm (Product complex) và sản phẩm (Product) của
phản ứng giữa ion âm [CBr(COOCH3)2]− và C60 được minh họa ở Hình 3.15.
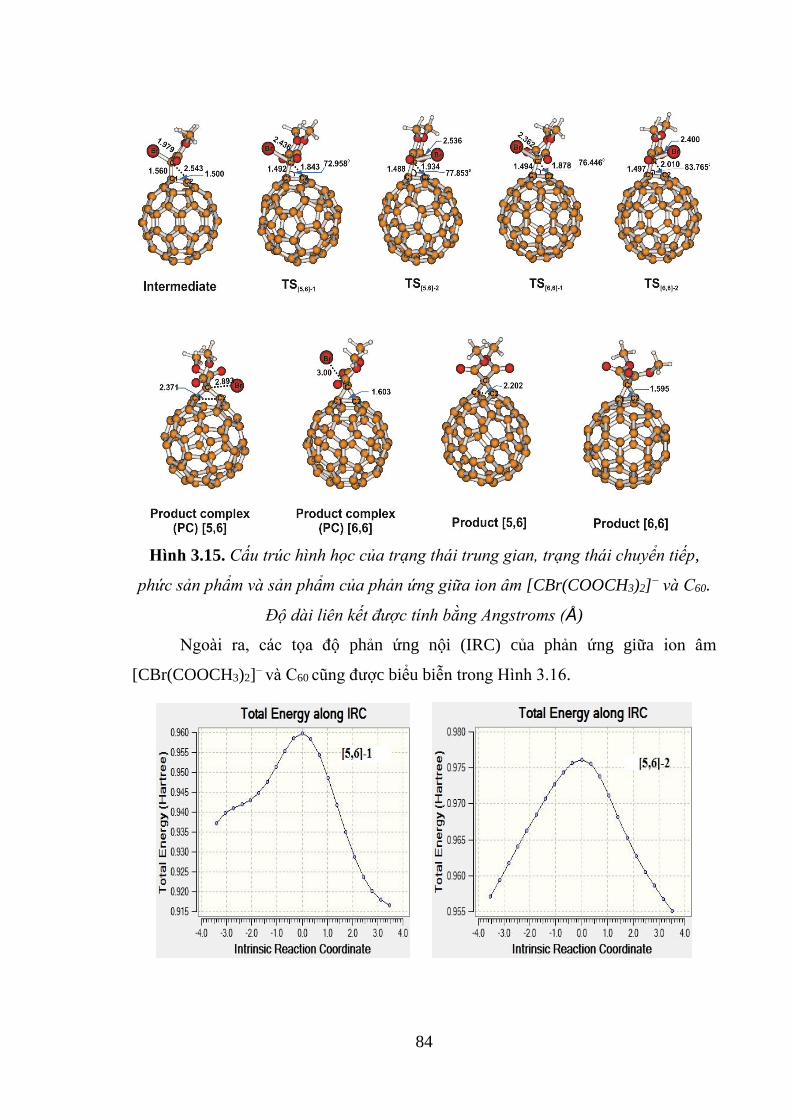
84
Hình 3.15. Cấu trúc hình học của trạng thái trung gian, trạng thái chuyển tiếp,
phức sản phẩm và sản phẩm của phản ứng giữa ion âm [CBr(COOCH3)2]− và C60.
Độ dài liên kết được tính bằng Angstroms (Å)
Ngoài ra, các tọa độ phản ứng nội (IRC) của phản ứng giữa ion âm
[CBr(COOCH3)2]− và C60 cũng được biểu biễn trong Hình 3.16.
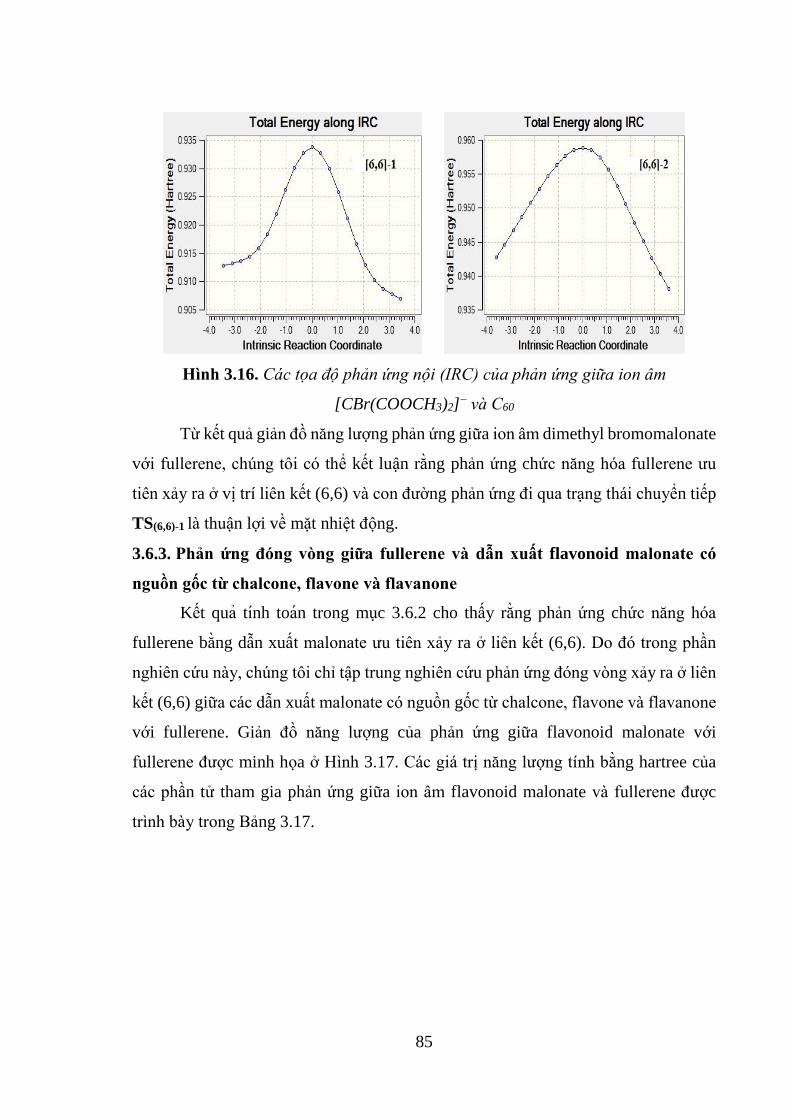
85
Hình 3.16. Các tọa độ phản ứng nội (IRC) của phản ứng giữa ion âm
[CBr(COOCH3)2]− và C60
Từ kết quả giản đồ năng lượng phản ứng giữa ion âm dimethyl bromomalonate
với fullerene, chúng tôi có thể kết luận rằng phản ứng chức năng hóa fullerene ưu
tiên xảy ra ở vị trí liên kết (6,6) và con đường phản ứng đi qua trạng thái chuyển tiếp
TS(6,6)-1 là thuận lợi về mặt nhiệt động.
3.6.3. Phản ứng đóng vòng giữa fullerene và dẫn xuất flavonoid malonate có
nguồn gốc từ chalcone, flavone và flavanone
Kêt qua tinh toan trong mục 3.6.2 cho thấy rằng phản ứng chức năng hóa
fullerene bằng dẫn xuất malonate ưu tiên xảy ra ở liên kết (6,6). Do đó trong phần
nghiên cứu này, chúng tôi chỉ tập trung nghiên cứu phản ứng đóng vòng xảy ra ở liên
kết (6,6) giữa các dẫn xuất malonate có nguồn gốc từ chalcone, flavone và flavanone
với fullerene. Giản đồ năng lượng của phản ứng giữa flavonoid malonate với
fullerene được minh họa ở Hình 3.17. Các giá trị năng lượng tính bằng hartree của
các phần tử tham gia phản ứng giữa ion âm flavonoid malonate và fullerene được
trình bày trong Bảng 3.17.
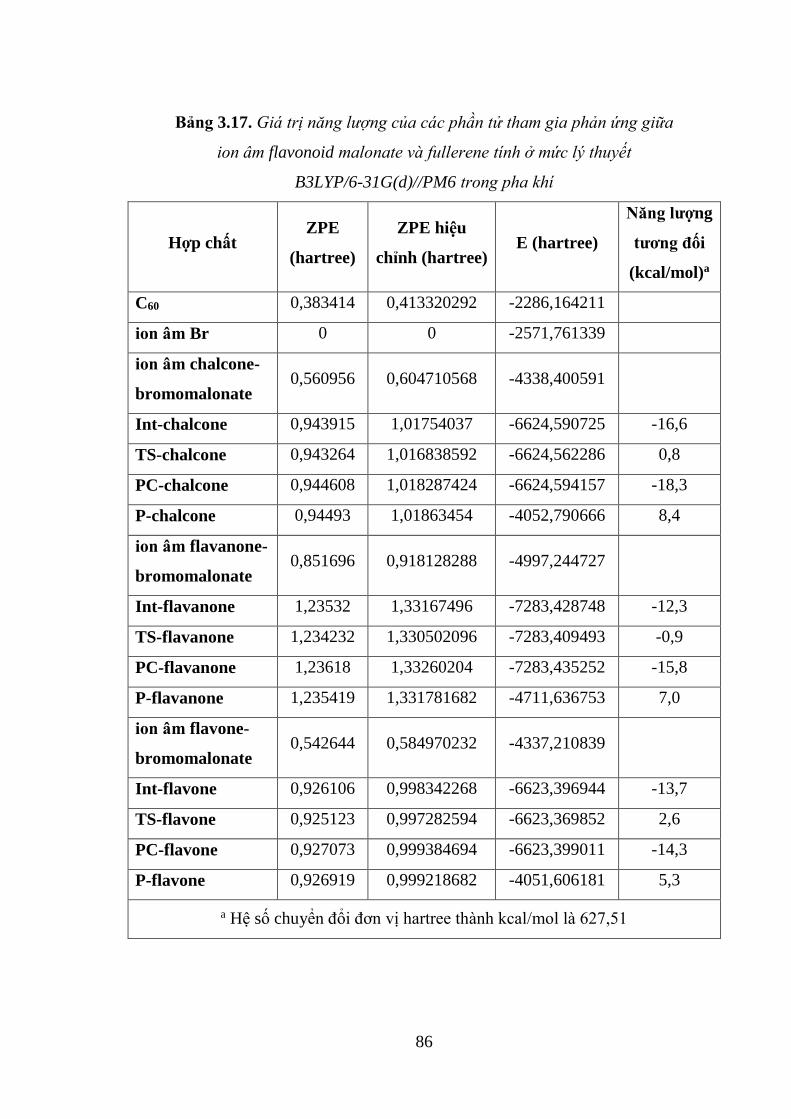
86
Bảng 3.17. Giá trị năng lượng của các phần tử tham gia phản ứng giữa
ion âm flavonoid malonate và fullerene tính ở mức lý thuyết
B3LYP/6-31G(d)//PM6 trong pha khí
Hợp chất ZPE
(hartree)
ZPE hiệu
chỉnh (hartree) E (hartree)
Năng lượng
tương đối
(kcal/mol)a
C60 0,383414 0,413320292 -2286,164211
ion âm Br 0 0 -2571,761339
ion âm chalcone-
bromomalonate 0,560956 0,604710568 -4338,400591
Int-chalcone 0,943915 1,01754037 -6624,590725 -16,6
TS-chalcone 0,943264 1,016838592 -6624,562286 0,8
PC-chalcone 0,944608 1,018287424 -6624,594157 -18,3
P-chalcone 0,94493 1,01863454 -4052,790666 8,4
ion âm flavanone-
bromomalonate 0,851696 0,918128288 -4997,244727
Int-flavanone 1,23532 1,33167496 -7283,428748 -12,3
TS-flavanone 1,234232 1,330502096 -7283,409493 -0,9
PC-flavanone 1,23618 1,33260204 -7283,435252 -15,8
P-flavanone 1,235419 1,331781682 -4711,636753 7,0
ion âm flavone-
bromomalonate 0,542644 0,584970232 -4337,210839
Int-flavone 0,926106 0,998342268 -6623,396944 -13,7
TS-flavone 0,925123 0,997282594 -6623,369852 2,6
PC-flavone 0,927073 0,999384694 -6623,399011 -14,3
P-flavone 0,926919 0,999218682 -4051,606181 5,3
a Hệ số chuyển đổi đơn vị hartree thành kcal/mol là 627,51

87
Quan sát ở Hình 3.17 cho thấy rằng khuynh hướng phản ứng của các flavonoid
malonate với fullerene là khá giống nhau. Đầu tiên, tạo thành các trạng thái trung
gian Int-chalcone, Int-flavone và Int-flavanone có năng lượng thấp hơn so với các
chất phản ứng tương ứng là −16,0; −12,3 và −13,7 kcal/mol. Điều này chứng tỏ rằng
trạng thái trung gian tạo thành bền hơn so với chất tham gian phản ứng. Hàng rào
năng lượng tương ứng với các trạng thái chuyển tiếp TS-chalcone, TS-flavone và
TS-flavanone có giá trị lần lượt là 16,8; 11,4 và 16,3 kcal/mol. Năng lượng tương
đối của các phức chất sản phẩm PC-chalcone, PC-flavone và PC-flavanone so với
chất phản ứng có giá trị lần lượt là: −18,3; −15,8 và −14,3 kcal/mol. Các sản phẩm
P-chalcone, P-flavone và P-flavanone hình thành sau khi ion âm brom thoát ra mà
không qua trạng thái chuyển tiếp nào, có giá trị năng lượng tương đối cao hơn so với
chất phản ứng tương ứng là: 8,4; 5,3 và 7,0 kcal/mol.
Hình 3.17. Giản đồ năng lượng của phản ứng flavonoid malonate với fullerene
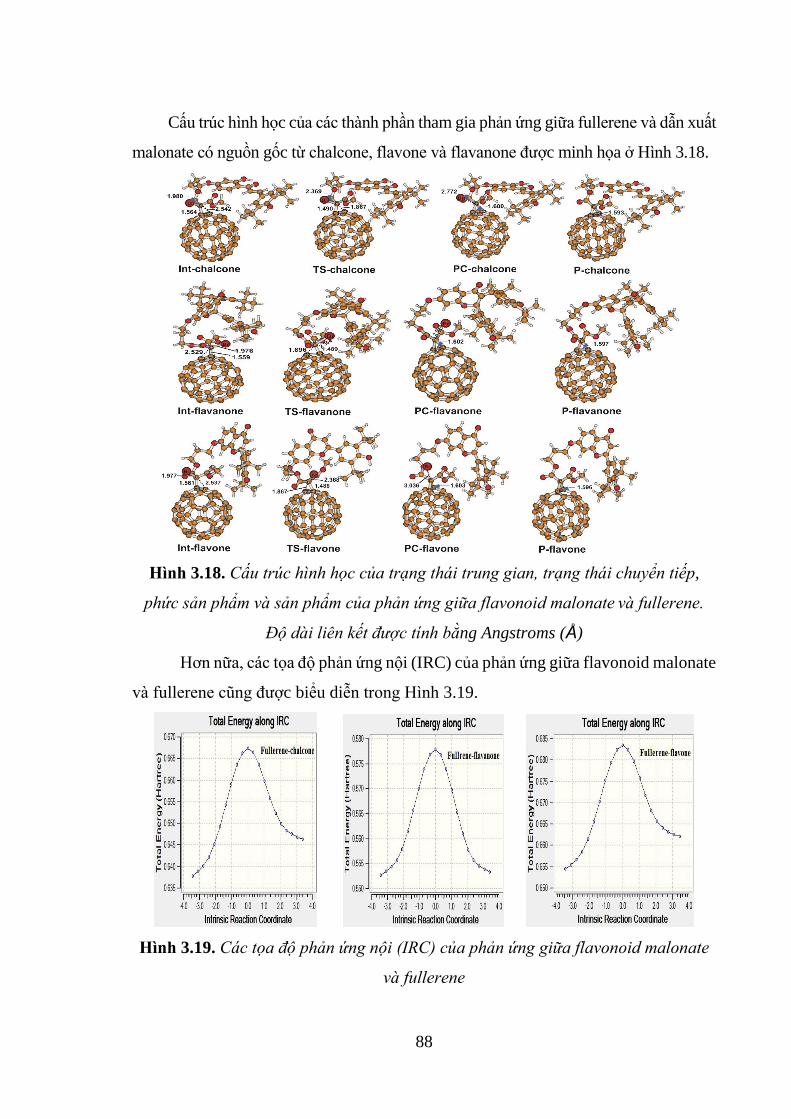
88
Cấu trúc hình học của các thành phần tham gia phản ứng giữa fullerene và dẫn xuất
malonate có nguồn gốc từ chalcone, flavone và flavanone được minh họa ở Hình 3.18.
Hình 3.18. Cấu trúc hình học của trạng thái trung gian, trạng thái chuyển tiếp,
phức sản phẩm và sản phẩm của phản ứng giữa flavonoid malonate và fullerene.
Độ dài liên kết được tính bằng Angstroms (Å)
Hơn nữa, các tọa độ phản ứng nội (IRC) của phản ứng giữa flavonoid malonate
và fullerene cũng được biểu diễn trong Hình 3.19.
Hình 3.19. Các tọa độ phản ứng nội (IRC) của phản ứng giữa flavonoid malonate
và fullerene

89
Từ các kết quả nghiên cứu ở trên, chúng tôi có thể kết luận rằng nghiên cứu lý
thuyết về quá trình hình thành sản phẩm fullerene-flavonoid ở vị trí liên kết (6,6) là
phù hợp với kết quả thực nghiệm [24].
3.6.4. Nhận xét
Cơ chế của phản ứng Bingel - Hirsch giữa ion âm dimethyl bromomalonate và
fullerene được nghiên cứu chi tiết bằng phương pháp bán thực nghiệm PM6 kết hợp
với phương pháp DFT. Kết quả cho thấy rằng con đường phản ứng đi qua trạng thái
chuyển tiếp TS(6,6)-1 để hình thành sản phẩm (6,6)-C60C(COOCH3)2 là chiếm ưu thế
nhất theo quan điểm của nhiệt động học. Ngoài ra, giản đồ năng lượng của phản ứng
đóng vòng xảy ra ở liên kết (6,6) giữa các dẫn xuất malonate có nguồn gốc từ
chalcone, flavone và flavanone với fullerene cũng được thiết lập. Kết quả nghiên cứu
lý thuyết về quá trình hình thành sản phẩm fullerene-flavonoid ở liên kết (6,6) là phù
hợp với kết quả thực nghiệm [24].
3.7. KHẢ NĂNG CHỐNG OXY HÓA CỦA CÁC DẪN CHẤT LAI HÓA
FULLERENE VÀ CHALCONE, FLAVONE, FLAVANONE
3.7.1. Giới thiệu
Nhằm làm tăng cường khả năng dập tắt gốc tự do từ sự kết hợp hoạt tính chống
oxy hóa cao của flavonoid với khả năng thu nhận gốc tự do của fullerene, một vài
nghiên cứu thực nghiệm về tổng hợp dẫn xuất fullerene - flavonoid có nguồn gốc
thiên nhiên đã được công bố [20, 21, 24]. Cho đến nay, các nghiên cứu về khả năng
chống oxy hóa của hợp chất fullerene - flavonoid đã được thử nghiệm bằng cách đo
năng lượng phân ly liên kết O‒H và hằng số tốc độ phản ứng của chúng với các gốc
peroxyl [24]. Tuy nhiên, cách tiếp cận theo phương pháp hóa tính toán về nghiên cứu
khả năng chống oxy hóa của các hợp chất này chưa được thực hiện. Mục tiêu của
phần nghiên cứu này là đánh giá khả năng chống oxy hóa của hợp chất fullerene -
flavonoid thông qua hai cơ chế chuyển nguyên tử hydro và chuyển electron với các
thông số nhiệt động đặc trưng là BDE và IE. Đồng thời, kết quả nghiên cứu lý thuyết
này là cơ sở khoa học để giải thích và làm sáng tỏ về hoạt tính chống oxy hóa của
dẫn xuất fullerene - flavonoid trong nghiên cứu thực nghiệm.

90
3.7.2. Khả năng chống oxy hóa của các dẫn chất lai hóa fullerene và chalcone,
flavone, flavanone
Như đề cập trong các phần nghiên cứu trước, chúng tôi áp dụng cơ chế HAT
và SET để đánh giá khả năng chống oxy hóa các sản phẩm của phản ứng giữa
flavonoid malonate và fullerene minh họa trong Hình 3.18. Vì vậy, trong phần nghiên
cứu này, BDE và IE là hai thông số nhiệt động quan trọng đánh giá khả năng chống
oxy hóa của các hợp chất này.
Kế thừa các kết quả nghiên cứu trước, chúng tôi sử dụng phương pháp ONIOM
với mô hình ONIOM 1A để tính BDE và phương pháp PM6 để tính IE. Kết quả
BDE(O−H) và IE của các dẫn xuất fullerene được trình bày ở Bảng 3.18.
Bảng 3.18. Giá trị BDE(O−H) và IE của dẫn xuất C60-chalcone, C60-flavone và
C60-flavanone
Hợp chấta BDE(O−H)b - kcal/mol IEc vertical - eV
P-chalcone 66,5 (82,5) 8,97 [8,02]d
P-flavone 70,3 (82,6,) 8,96 [8,19] d
P-flavanone (vòng A) 68,5 (83,7) 8,89 [8,00] d
a Cấu trúc các dẫn xuất C60-flavonoid được hiển thị Hình 3.18.
b Tính bằng phương pháp ONIOM((RO)B3LYP/6-311++G(2df,2p):PM6); Dữ
liệu trong dấu ngoặc đơn là giá trị BDE của chalcone, flavone và flavanone (đơn
vị kcal/mol).
c Tính bằng phương pháp PM6.
d Dữ liệu trong dấu ngoặc vuông là giá trị IE của chalcone, flavone và flavanone.
Kết quả Bảng 3.18 cho thấy rằng fullerene có ảnh hưởng rất lớn đến
BDE(O−H) của các hợp chất flavonoid. Điều này có thể giải thích là do tương tác của
phân tử fullerene lên liên kết O−H. Giá trị BDE(O−H) của dẫn xuất C60-chalcone,
C60-flavone và C60-flavanone lần lượt là: 66,5; 70,3 và 68,5 kcal/mol. So sánh với dữ
liệu BDE của chalcone, flavone và flavanone, ta thấy BDE của dẫn xuất fullerene
giảm rất đáng kể với mức giảm trung bình khoảng 14,5 kcal/mol.

91
Năng lượng ion hóa cũng là một thông số nhiệt động liên quan đến sự đánh
giá khả năng chống oxy hóa của một chất. Đây là một đại lượng xác định độ ổn định
của hợp chất chống oxy hóa trong môi trường giàu oxy. Kết quả Bảng 3.18 cho thấy
giá trị IE tính ở phương pháp PM6 của các dẫn xuất fullerene -flavonoid cao hơn các
flavonoid (chalcone, flavone và flavanone) khoảng 0,87 eV. Do đó, các dẫn xuất
fullerene rất khó cho electron, hiện tượng này xảy ra là vì phân tử fullerene có ái lực
electron rất cao [119].
3.7.3. Nhận xét
Trong phần nghiên cứu này, các giá trị BDE(O−H) của các dẫn xuất fullerene
- flavonoid được tính bằng phương pháp ONIOM 1A dao động trong khoảng 66,5 −
70,3 kcal/mol. Điều này cho thấy rằng đây là những hợp chất này có khả năng chống
oxy hóa mạnh theo cơ chế HAT. Ngược lại với xu hướng của BDE, các giá trị IE của
dẫn xuất fullerene - flavonoid cao hơn so với các flavonoid. Vì vậy, trong cơ chế
chuyển electron (SET) các hợp chất fullerene - flavonoid không được ưu tiên, khó
cho electron hơn so với các hợp chất flavonoid. Nguyên nhân của hiện tượng này có
thể được giải thích là do sự ái lực electron rất cao của phân tử fullerene.
3.8. THIẾT KẾ CÁC HỢP CHẤT CHỐNG OXY HÓA CÓ NGUỒN GỐC TỪ
CÂY SA KÊ VÀ MĂNG CỤT TRÊN NỀN FULLERENE
3.8.1. Giới thiệu
Bên cạnh chất chống oxy hóa tự nhiên, chất chống oxy hóa tổng hợp cũng đã
được nghiên cứu một cách rộng rãi. Một số nhóm nghiên cứu đã tập trung vào việc
thiết kế và tổng hợp các chất chống oxy hóa mới với hoạt tính cao và độc tính thấp.
Trong nhom chât này, các dẫn xuất fullerene - polyphenol được xem là các chất chống
oxy hóa tiềm năng [20, 21, 24], bởi vì phân tử fullerene cũng như các dẫn xuất của
nó có ái lực electron rất cao nên có thể thu nhận nhiều gốc tự do trên mỗi phân tử
[119]. Nhiều dẫn xuất fullerene - flavonoid đươc tao ra tư phản ứng đóng vòng giữa
fullerene và các dẫn xuất malonate có nguồn gốc từ chalcone, flavone và flavonoid
theo cơ chế Bingel-Hirsch [24]. Trên cơ sở các thông tin thu được từ nghiên cứu thực
nghiệm [24] và nghiên cứu lý thuyết [114] về phản ứng chức năng hóa fullerene bằng
các dẫn xuất malonate, chúng tôi đã thiết kế chất chống oxy hóa mới có nguồn gốc

92
từ cây sa kê và măng cụt trên nền fullerene (C60) bằng phương pháp hóa tính toán. Sơ
đồ của quá trình tổng hợp được minh họa ở Hình 3.20.
Trong nghiên cứu này, chúng tôi chọn hai hợp chất altilisin J và mangostin có
nguồn gốc từ cây sa kê và măng cụt để đính lên bề mặt fullerene bởi vì chúng đã được
chứng minh là có khả năng chống oxy hóa mạnh trong các nghiên cứu trước [115,
116]. Ngoài ra, giản đồ năng lượng của phản ứng chức năng hóa fullerene và các
thông số nhiệt động đặc trưng cho khả năng chống oxy hóa thông qua hai cơ chế
chuyển nguyên tử hydro (HAT) và chuyển một electron (SET) bao gôm: năng lượng
phân ly liên kết (BDE), năng lượng ion hóa (IE) và ái lực electron (EA) cũng được
làm rõ trong nghiên cứu này.
OH3CO
O O
+C60(CBr4, DBU, rt)
OH3CO
O O
OH3CO
O O
+C60(CBr4, DBU, rt)
OH3CO
O O
+C60(CBr4, DBU, rt)
OH3CO
O O
C60-Altilisin J
OH3CO
O O
OO
OH O
OCH3
OH
C60-Mangosteen-1
OO
OOH
OH
1
2
41
2
34
5
6
A
B 3
OO
OOH
OH
1
2
41
2
34
5
6
A
B 3
O
OH
O
O
H3CO OH
O
OH
O
O
H3CO OH
OO
OH O
OCH3
OH
C60-Mangosteen-2(1) (2)
(3)
B
B
B
B
1 2
3
1 2
3
1
2 3
1
2 3
Hình 3.20. Phản ứng Bingel - Hirsch giữa các dẫn xuất của malonate và fullerene
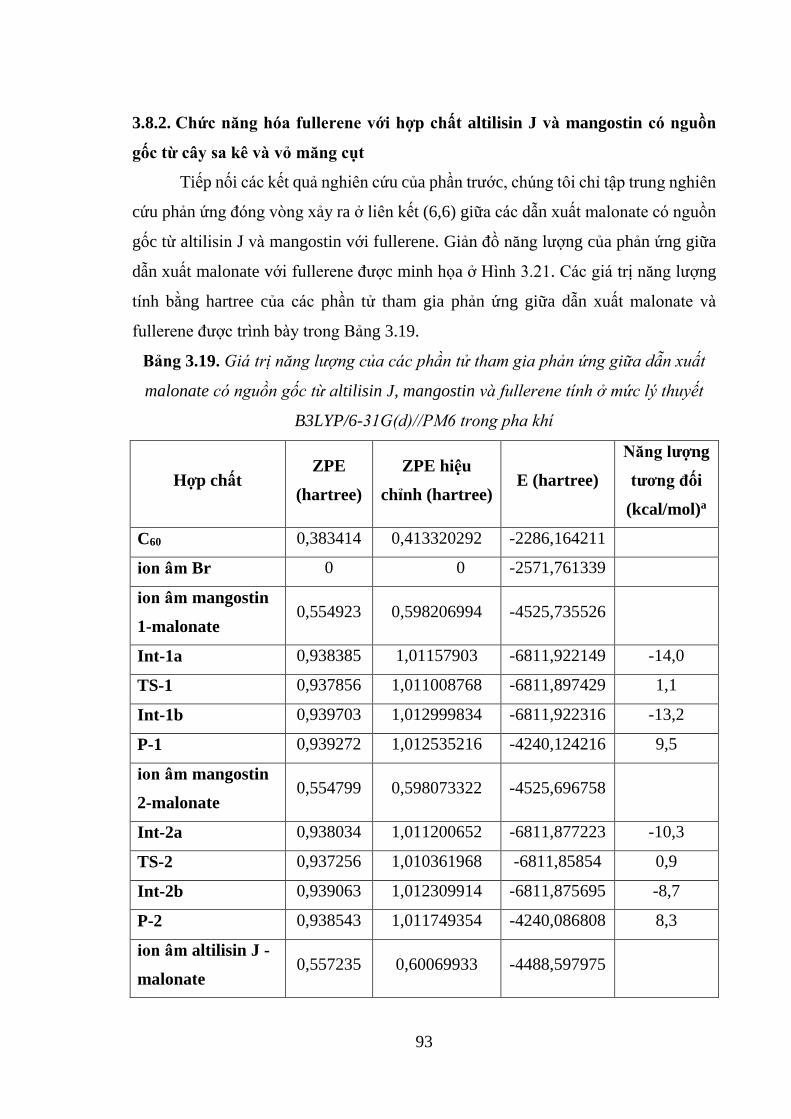
93
3.8.2. Chức năng hóa fullerene với hợp chất altilisin J và mangostin có nguồn
gốc từ cây sa kê và vỏ măng cụt
Tiếp nối các kết quả nghiên cứu của phần trước, chúng tôi chỉ tập trung nghiên
cứu phản ứng đóng vòng xảy ra ở liên kết (6,6) giữa các dẫn xuất malonate có nguồn
gốc từ altilisin J và mangostin với fullerene. Giản đồ năng lượng của phản ứng giữa
dẫn xuất malonate với fullerene được minh họa ở Hình 3.21. Các giá trị năng lượng
tính bằng hartree của các phần tử tham gia phản ứng giữa dẫn xuất malonate và
fullerene được trình bày trong Bảng 3.19.
Bảng 3.19. Giá trị năng lượng của các phần tử tham gia phản ứng giữa dẫn xuất
malonate có nguồn gốc từ altilisin J, mangostin và fullerene tính ở mức lý thuyết
B3LYP/6-31G(d)//PM6 trong pha khí
Hợp chất ZPE
(hartree)
ZPE hiệu
chỉnh (hartree) E (hartree)
Năng lượng
tương đối
(kcal/mol)a
C60 0,383414 0,413320292 -2286,164211
ion âm Br 0 0 -2571,761339
ion âm mangostin
1-malonate 0,554923 0,598206994 -4525,735526
Int-1a 0,938385 1,01157903 -6811,922149 -14,0
TS-1 0,937856 1,011008768 -6811,897429 1,1
Int-1b 0,939703 1,012999834 -6811,922316 -13,2
P-1 0,939272 1,012535216 -4240,124216 9,5
ion âm mangostin
2-malonate 0,554799 0,598073322 -4525,696758
Int-2a 0,938034 1,011200652 -6811,877223 -10,3
TS-2 0,937256 1,010361968 -6811,85854 0,9
Int-2b 0,939063 1,012309914 -6811,875695 -8,7
P-2 0,938543 1,011749354 -4240,086808 8,3
ion âm altilisin J -
malonate 0,557235 0,60069933 -4488,597975
94
Hợp chất ZPE
(hartree)
ZPE hiệu
chỉnh (hartree) E (hartree)
Năng lượng
tương đối
(kcal/mol)a
Int-3a 0,94031 1,01365418 -6774,790547 -18
TS-3 0,939781 1,013083918 -6774,76537 -2,6
Int-3b 0,941908 1,015376824 -6774,793854 -19
P-3 0,941112 1,014518736 -4202,989751 7,3
a Hệ số chuyển đổi đơn vị hartree thành kcal/mol là 627,51
Hình 3.21. Giản đồ năng lượng của phản ứng giữa ion âm dimethyl
bromomalonate có nguồn gốc từ cây sa kê và măng cụt với fullerene
Quan sát Hình 3.21 cho thấy khuynh hướng phản ứng của các dẫn xuất
malonate với fullerene là khá giống nhau. Đầu tiên, cac phan ưng tạo thành các trạng
thái trung gian Int-1a, Int-2a và Int-3a có năng lượng thấp hơn so với các chất phản
ứng ban đâu tương ứng là −14,0; −10,3 và −18,0 kcal/mol. Điều này chứng tỏ rằng
trạng thái trung gian tạo thành bền hơn so với chất tham gian phản ứng. Hàng rào

95
năng lượng tương ứng với các trạng thái chuyển tiếp TS-1, TS-2 và TS-3 có giá trị lần
lượt là 15,1; 11,2 và 15,4 kcal/mol. Năng lượng tương đối của các phức chất sản phẩm
Int-1b, Int-2b và Int-3b so với chất phản ứng ban đâu có giá trị lần lượt là: −13,2;
−8,7 và −19,0 kcal/mol. Các sản phẩm P-1, P-2 và P-3 được hình thành sau khi ion âm
brom thoát ra mà không qua trạng thái chuyển tiếp nào, có giá trị năng lượng tương đối
cao hơn so với chất phản ứng tương ứng là: 9,5; 8,3 và 7,3 kcal/mol. Cấu trúc hình học
tối ưu của các thành phần tham gia phản ứng giữa fullerene và dẫn xuất malonate có
nguồn gốc từ altilisin J và -mangostin được minh họa ở Hình 3.22.
Hình 3.22. Cấu trúc tối ưu của trạng thái trung gian, trạng thái chuyển tiếp, phức
sản phẩm và sản phẩm của phản ứng giữa polyphenol malonate và fullerene
96
Hơn nữa, các tọa độ phản ứng nội (IRC) của phản ứng giữa dimethyl
bromomalonate có nguồn gốc từ cây sa kê và măng cụt với fullerene cũng được biểu
diễn trong Hình 3.23.
Hình 3.23. Tọa độ phản ứng (IRC) của phản ứng giữa dimethyl bromomalonate có
nguồn gốc từ cây sa kê và măng cụt với fullerene
3.8.3. Khả năng chống oxy hóa của các dẫn xuất fullerene-altilisin J và fullerene-
mangostin
Trong phần nghiên cứu này, chúng tôi tiếp tục sử dụng phương pháp
ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) với mô hình 1A để tính BDE(OH)
của các hợp chất nghiên cứu. Độ tin cậy và tính chính xác của phương pháp này đã
được chứng minh trong các nghiên cứu trước của chúng tôi [85, 115]. Ngoài ra, câu
truc các phân tử trong nghiên cứu này chứa một số lượng lớn các nguyên tử. Do vậy
để tiết kiệm thời gian tính toán, chúng tôi sử dụng phương pháp PM6 để tính các
thông số IE và EA, và đây là những thông số quan trọng để đánh giá khả năng chống
oxy hóa của các chất nghiên cứu theo cơ chế SET [76, 77].
Các thông số đặc trưng cho hai cơ chế dập tắt gốc tự do như: BDE, IE và EA
của các hợp chất nghiên cứu được liệt kê trong Bảng 3.20. Dữ liệu trong Bảng 3.20
cho thấy fullerene có ảnh hưởng rất lớn đến BDE(OH) của mangostin và altilisin J
với độ giảm trung bình 13,5 kcal/mol. Điều này có thể giải thích do tương tác của
phân tử fullerene lên liên kết OH. Giá trị BDE(OH) của các hợp chất C60-
mangostin-1, C60-mangostin-2 và C60-altilisin J lần lượt là: 69,3; 67,7 và 65,3
kcal/mol.

97
Khi thảo luận về cơ chế chuyển electron thì giá trị IE và EA là các thông số
quan trọng cần được xem xét khi nghiên cứu khả năng chống oxy hóa của các hợp
chất. Giá trị IE càng thấp thì càng dễ cho electron trong khi đó giá trị EA càng cao
thì càng dễ nhận electron. Từ kết quả Bảng 3.20 cho thấy giá trị IE và EA của các
dẫn xuất fullerene lớn hơn đáng kể so với mangostin và altilisin J. Do đó, các dẫn
xuất fullerene rất khó cho electron nhưng ngược lại sự nhận electron thì dễ dàng hơn
so với hợp chất ban đầu. Hiện tượng này xảy ra là vì phân tử fullerene có ái lực
electron rất cao [119].
Kết quả nghiên cứu trên cho thấy các dẫn xuất fullerene đã kết hợp được cả
tính chất của fullerene và hợp chất phenolic nên vừa co kha năng chống oxy hóa theo
cơ chế ngắt mạch oxy hoa vừa là “chiếc lồng thu nhặt gốc tự do”.
Bảng 3.20. Giá trị BDE(O−H), EA và IE của mangostin, altilisin J,
C60-mangostin-1, C60-mangostin-2 và C60-altilisin J
Hợp chất BDE(O−H)a
kcal/mol IEb vertical, eV EAb vertical, eV
Mangostin 82,8 8,31 1,47
Altilisin J 77,3 8,08 2,14
C60-mangostin-1 69,3 8,90 3,53
C60-mangostin-2 67,7 8,91 3,56
C60-altilisin J 65,3 8,92 3,61
a Sử dụng mô hình 1A và tính ở mức ONIOM(ROB3LYP/6-
311++G(2df,2p):PM6).
b Tính bằng phương pháp PM6.
3.8.4. Nhận xét
Các tính toán lý thuyết đã được tiên hành để dự đoán sự hình thành sản phẩm
chống oxy hóa tiềm năng giữa fullerene và các dẫn xuất malonate có nguồn gốc từ
cây sa kê và măng cụt thông qua phản ứng Bingel - Hirsch. Giản đồ năng lượng của
phản ứng đóng vòng giữa fullerene và dẫn xuất malonate ở vị trí liên kết (6,6) cũng

98
được thiết lập. Năng lượng phản ứng tương đối giữa fullerene với các dẫn xuất
malonat mangostin-1, mangostin-2 và altilisin J lần lượt là: 9,5; 8,5 và 7,3 kcal/mol.
Các dẫn xuất fullerene mới được đánh giá là có khả năng chống oxy hóa cao thông
qua các thông số đặc trưng như: BDE, IE và EA. Câu truc phân tử fullerene ảnh
hưởng rất lớn đến khả năng chống oxy hóa cua các hợp chất fullerene-altilisin J và
fullerene-mangostin: lam giảm giá trị BDE nhưng lại tăng IE và EA.

99
NHỮNG KẾT LUẬN CHÍNH CỦA LUẬN ÁN
Trong luận án này, chúng tôi đã tiến hành nghiên cứu một cách hệ thống khả
năng chống oxy hóa của các hợp chất polyphenol có nguồn gốc từ cây sa kê, măng
cụt và dẫn xuất fullerene - flavonoid bằng phương pháp hóa tính toán. Thông qua đó,
chúng tôi đã thiết kế các hợp chất chống oxy hóa có nguồn gốc từ thiên nhiên trên
nền fullerene. Qua quá trình nghiên cứu, chúng tôi rút ra một số kết luận sau:
1. Đã chứng minh được tính chính xác và độ tin cậy của phương pháp
ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) thông qua 7 mô hình khác nhau.
Trong đó, phương pháp ONIOM 1A là sự lựa chọn tốt nhất dùng để tính
BDE(O−H) của các hợp chất phenolic.
2. Từ sự phân tích và so sánh giữa giá trị tính toán và giá trị thực nghiệm, chúng tôi
có thể kết luận rằng phương pháp PM6 được xem là phương pháp thích hợp để
tính năng lượng ion hóa của các hợp chất là dẫn xuất phenol.
3. Đã nghiên cứu khả năng chống oxy hóa các hợp chất polyphenol có nguồn gốc từ
cây sa kê và măng cụt thông qua 3 cơ chế HAT, SET-PT và SPLET. Dựa vào giá
trị BDE(O−H), hợp chất S12, M10 và M11 được xem như là một chất chống oxy
hóa tiềm năng với giá trị BDE tính trong pha khí lần lượt là 77,3; 82,3 và 82,8
kcal/mol.
4. Đã nghiên cứu khả năng dập tắt gốc tự do CH3OO• của một số hợp chất polyphenol
bằng phương pháp hóa tính toán. Hàng rào năng lượng của phản ứng giữa gốc tự
do CH3OO• và altilisin J, -mangostin, flavanone, chalcone, flavone và
quecertin có giá trị lần lượt là 15,3; 20,4; 18,4; 18,6; 19,7 và 16,1 kcal/mol.
5. Đã làm sáng tỏ cơ chế của phản ứng Bingel - Hirsch giữa ion âm dimethyl
bromomalonate và fullerene (C60). Kết quả nghiên cứu cho thấy rằng sản phẩm
tạo thành ở liên kết (6,6) là ưu tiên hơn so với liên kết (5,6), điều này cũng phù
hợp với kết quả thực nghiệm [24] và con đường phản ứng đi qua trạng thái chuyển
tiếp TS(6,6)-1 là thuận lợi về mặt nhiệt động.
6. Đã tính được giá trị BDE(O−H) của dẫn xuất C60-chalcone, C60-flavone và C60-
flavanone lần lượt là 66,5; 70,3 và 68,5 kcal/mol, thấp hơn rất nhiều so với giá trị

100
BDE(O−H) của các hợp chất flavonoid với độ giảm trung bình 14,5 kcal/mol. Tuy
nhiên, giá trị IE của các dẫn xuất fullerene này thì cao hơn so với các flavonoid
(chalcone, flavone và flavanone) khoảng 0,87 eV. Vì vậy, các dẫn xuất fullerene
- flavonoid dễ cho nguyên tử hydro nhưng khó cho electron.
7. Đã thiết kế được các hợp chất chống oxy hóa mới từ fullerene và hợp chất phenolic
co nguôn gôc thiên nhiên bằng phản ứng Bingel-Hirsch. Khả năng chống oxy hóa
của các hợp chất này được khao sat thông qua các thông số đặc trưng là BDE, IE
và EA. Kết quả nghiên cứu trên cho thấy các dẫn xuất fullerene mới có khả năng
chống oxy hóa rất cao vì đã kết hợp được cả tính chất của fullerene và hợp chất
phenolic.

101
NHỮNG ĐỊNH HƯỚNG NGHIÊN CỨU TIẾP THEO
Những kết quả đạt được trong luận án đã mở ra những định hướng nghiên cứu
triển vọng có thể tiếp cận trong thời gian đến để đề tài này hoàn chỉnh. Cụ thể:
1. Tiếp tục nghiên cứu về mặt động hóa học khả năng chống oxy hóa của các
hợp chất polyphenol và dẫn xuất fullerene.
2. Tiến hành tổng hợp các dẫn xuất fullerene - polyphenol và thử hoạt tính sinh
học trong in vivo và in vitro.
3. Kết hợp phương pháp lý thuyết với thực nghiệm để đánh giá và làm rõ hoạt
tính và cơ chế chống oxy hóa của các hợp chất có nguồn gốc thiên nhiên. Trên cơ sở
đó có thể thiết kế và tổng hợp các hợp chất mới có khả năng chống oxy hóa cao.

102
DANH MỤC CÁC CÔNG TRÌNH LIÊN QUAN ĐẾN LUẬN ÁN
1. Nguyen Minh Thong, Thi Chinh Ngo, Duy Quang Dao, Tran Duong, Quoc
Tri Tran, Pham Cam Nam (2016), “Functionalization of fullerene via the
Bingel reaction with α-chlorocarbanions: an ONIOM approach”, Journal of
Molecular Modeling, 22, pp.113-121.
2. Nguyen Minh Thong, Duy Quang Dao, Thi Chinh Ngo, Trinh Le Huyen,
Pham Cam Nam (2016), “Antioxidant activities of [60]fullerene derivatives
from chalcone, flavone and flavanone: A ONIOM approach via H-atom and
electron transfer mechanism”, Chem. Phys. Lett., 652, pp.56–61.
3. Nguyen Minh Thong, Thi Chinh Ngo, Truc Xuyen Nguyen Phan, Duy Quang
Dao, Truong Van Nam, Phan Thi Tuyet Trinh, Pham Cam Nam (2015) “A
Study of the Substituent Effects on the O−H Bond Dissociation Enthalpies of
Phenol Derivatives Using the ONIOM Method”, American Journal of
Chemistry, 5(4), pp.91-95.
4. Nguyen Minh Thong and Pham Cam Nam (2015) “Theoretical investigation
on antioxidant activity of phenolic compounds extracted from Artocarpus
altilis”, Springer International Publishing, 46, pp. 464-469.
5. Nguyen Minh Thong, Duong Tuan Quang, Ngoc Hoa Thi Bui, Duy Quang
Dao, Pham Cam Nam (2015), “Antioxidant properties of xanthones extracted
from the pericarp of Garcinia mangostana (Mangosteen): A theoretical study”,
Chem. Phys. Lett., 625, pp. 30–35.
6. Nguyen Minh Thong, Tran Duong, Linh Thuy Pham, and Pham Cam Nam
(2014), “Theoretical investigation on the bond dissociation enthalpies of
phenolic compounds extracted from Artocarpus altilis using
ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) method”, Chem. Phys. Lett.,
613, pp. 139–145.
7. Nguyễn Minh Thông và Phạm Cẩm Nam (2016), “Nghiên cứu phản ứng chức
năng hóa fullerene (C60) với các dẫn xuất flavonoid malonate bằng phương
pháp hóa tính toán”, Tạp chí KHCN - ĐHĐN, Số 5(102), trang 95-99.

103
8. Nguyên Minh Thông, Đào Duy Quang, Ngô Thị Chinh, Trần Dương, Phạm
Cẩm Nam (2016), “Nghiên cứu khả năng dập tắt gốc tự do (ROO•) của các
hợp chất polyphenol tự nhiên bằng phương pháp hóa tính toán”, Tạp chí Khoa
học Đại học Huế, Số 3(117). (Đã chấp nhận đăng)
9. Nguyên Minh Thông, Phạm Lê Minh Thông, Đinh Tuấn (2016), “Thiết kế các
hợp chất chống oxy hóa từ một số dẫn xuất malonate của Altilisin J và Mangostin
trên nền Fullerene (C60) bằng phương pháp hóa tính toán”, Tạp chí Khoa học và
Công nghệ (Viện Hàn lâm KH & CN Việt Nam), số 54(2B), trang 149-155.

104
TÀI LIỆU THAM KHẢO
1. Alecu I.M., Zheng J., Zhao Y., Truhlar D.G. (2010), "Computational
Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational
Frequencies Obtained from Electronic Model Chemistries", J. Chem. Theory.
Comput., 6(9), pp. 2872-2887.
2. Anilkumar P., Lu F., Cao L., Luo P.G., Liu J.H., Sahu S., Tackett Ii N.K., Wang
Y., Sun Y.P. (2011), "Fullerenes for Applications in Biology and Medicine",
Current Medicinal Chemistry, 18(14), pp. 2045-2059.
3. Balch A.L., Catalano V.J., Lee J.W., Olmstead M.M., Parkin S.R. (1991),
"(.eta.2-C70)Ir(CO)Cl(PPh3)2: the synthesis and structure of an iridium
organometallic derivative of a higher fullerene", J. Am. Chem. Soc., 113(23),
pp. 8953-8955.
4. Beck R.D., St. John P., Alvarez M.M., Diederich F., Whetten R.L. (1991),
"Resilience of all-carbon molecules C60, C70, and C84: a surface-scattering time-
of-flight investigation", J. Phys. Chem., 95(21), pp. 8402-8409.
5. Beckhaus H.-D., Rüchardt C., Kao M., Diederich F., Foote C.S. (1992), "The
Stability of Buckminsterfullerene(C60): Experimental Determination of the Heat
of Formation", Angew. Chem. Int. Ed. Engl., 31(1), pp. 63-64.
6. Bensasson R.V., Brettreich M., Frederiksen J., Göttinger H., Hirsch A., Land
E.J., Leach S., Mcgarvey D.J., Schönberger H. (2000), "Reactions of e−aq, CO2
•−,
HO•, O2•− and O2(1δg) with a dendro[60]fullerene and C60[C(COOH)2]n (n = 2–
6)1", Free Radical Biology and Medicine, 29(1), pp. 26-33.
7. Bingel C. (1993), "Cyclopropanierung von Fullerenen", Chemische Berichte,
126(8), pp. 1957-1959.
8. Bingel C., Schiffer H. (1995), "Biscyclopropanation of C70", Liebigs Annalen,
1995(8), pp. 1551-1553.
9. Brewer M.S. (2011), "Natural Antioxidants: Sources, Compounds, Mechanisms
of Action, and Potential Applications", Comprehensive Reviews in Food Science
and Food Safety, 10(4), pp. 221-247.

105
10. Bühl M., Hirsch A. (2001), "Spherical Aromaticity of Fullerenes", Chem. Rev.,
101(5), pp. 1153-1184.
11. Bürgi H.-B., Blanc E., Schwarzenbach D., Liu S., Lu Y., Kappes M.M., Ibers
J.A. (1992), "The Structure of C60: Orientational Disorder in the Low-
Temperature Modification of C60", Angew. Chem. Int. Ed. Engl., 31(5), pp. 640-
643.
12. Camps X., Hirsch A. (1997), "Efficient cyclopropanation of C60 starting from
malonates", J. Chem. Soc., Perkin Trans. 1, 11, pp. 1595-1596.
13. Cancès E., Mennucci B. (1998), "New applications of integral equations
methods for solvation continuum models: ionic solutions and liquid crystals",
Journal of Mathematical Chemistry, 23(3/4), pp. 309-326.
14. CanceS E., Mennucci B., Tomasi J. (1997), "A new integral equation formalism
for the polarizable continuum model: Theoretical background and applications
to isotropic and anisotropic dielectrics", The Journal of Chemical Physics,
107(8), pp. 3032-3041.
15. Chen S.X., Wan M., Loh B.N. (1996), "Active constituents against HIV-1
protease from Garcinia mangostana", Planta. Med., 62(4), pp. 381-382.
16. Chung L.W., Sameera W.M., Ramozzi R., Page A.J., Hatanaka M., Petrova
G.P., Harris T.V., Li X., Ke Z., Liu F., Li H.B., Ding L., Morokuma K. (2015),
"The ONIOM Method and Its Applications", Chem. Rev., 115(12), pp. 5678-
5796.
17. Conforti F., Sosa S., Marrelli M., Menichini F., Statti G.A., Uzunov D., Tubaro
A., Menichini F., Loggia R.D. (2008), "In vivo anti-inflammatory and in vitro
antioxidant activities of Mediterranean dietary plants", J. Ethnopharmacol.,
116(1), pp. 144-151.
18. Da Ros T., Prato M. (1999), "Medicinal chemistry with fullerenes and fullerene
derivatives", Chem. Commun., 8, pp. 663-669.
19. Dapprich S., Komáromi I., Byun K.S., Morokuma K., Frisch M.J. (1999), "A
new ONIOM implementation in Gaussian98. Part I. The calculation of energies,

106
gradients, vibrational frequencies and electric field derivatives", Journal of
Molecular Structure: THEOCHEM, 461-462, pp. 1-21.
20. De La Torre M.D.L., Rodrigues A.G.P., Tomé A.C., Silva A.M.S., Cavaleiro
J.A.S. (2004), "[60]Fullerene–flavonoid dyads", Tetrahedron, 60(16), pp.
3581-3592.
21. De La Torre M.D.L., Tomé A.C., Silva A.M.S., Cavaleiro J.A.S. (2002),
"Synthesis of [60]fullerene–quercetin dyads", Tetrahedron Letters, 43(26), pp.
4617-4620.
22. Dugan L.L., Lovett E.G., Quick K.L., Lotharius J., Lin T.T., O'malley K.L.
(2001), "Fullerene-based antioxidants and neurodegenerative disorders",
Parkinsonism & Related Disorders, 7(3), pp. 243-246.
23. Echegoyen L., Echegoyen L.E. (1998), "Electrochemistry of Fullerenes and
Their Derivatives", Accounts of Chemical Research, 31(9), pp. 593-601.
24. Enes R.F., Farinha A.S.F., Tomé A.C., Cavaleiro J.A.S., Amorati R., Petrucci
S., Pedulli G.F. (2009), "Synthesis and antioxidant activity of [60]fullerene–
flavonoid conjugates", Tetrahedron, 65(1), pp. 253-262.
25. Enes R.F., Tomé A.C., Cavaleiro J.A., Amorati R., Fumo M.G., Pedulli G.F.,
Valgimigli L. (2006), "Synthesis and antioxidant activity of [60]fullerene-BHT
conjugates", Chemistry–A European Journal, 12(17), pp. 4646-4653.
26. Fang J.J., Ye G., Chen W.L., Zhao W.M. (2008), "Antibacterial phenolic
components from Eriocaulon buergerianum", Phytochemistry, 69(5), pp. 1279-
1286.
27. Fiorucci S., Golebiowski J., Cabrol-Bass D., Antonczak S. (2007), "DFT study
of quercetin activated forms involved in antiradical, antioxidant, and prooxidant
biological processes", J. Agric. Food. Chem., 55(3), pp. 903-911.
28. Foresman J.B., Frisch Æ. (2016), Exploring chemistry with electronic structure
methods, 3rd Edition, Gaussian, Inc., Wallingford, CT USA.
29. Foti M.C., Daquino C.,Geraci C. (2004), "Electron-transfer reaction of cinnamic
acids and their methyl esters with the DPPH(*) radical in alcoholic solutions",
J. Org. Chem., 69(7), pp. 2309-2314.

107
30. Friedman S.H., Decamp D.L., Sijbesma R.P., Srdanov G., Wudl F., Kenyon
G.L. (1993), "Inhibition of the HIV-1 protease by fullerene derivatives: model
building studies and experimental verification", J. Am. Chem. Soc., 115(15), pp.
6506-6509.
31. Frisch Æ., Hratchian H.P., Dennington Ii R.D., Keith T.A., Millam J., Nielsen
A.B., Holder A.J., Hiscocks J. (2009), GaussView 5 Reference,Gaussian. Inc.,
Wallingford.
32. Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A.,
Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A.,
Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J.,
Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R.,
Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T.,
Montgomery J.A.Jr., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers
E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K.,
Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam
J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J.,
Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C.,
Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A.,
Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas Ö., Foresman
J.B., Ortiz J.V., Cioslowski J., Fox D.J. (2009), Gaussian 09, Gaussian, Inc.:
Wallingford, CT, USA.
33. Froese R.D.J., Morokuma K. (1999), "Accurate calculations of bond-breaking
energies in C60 using the three-layered ONIOM method", Chem. Phys. Lett.,
305(5-6), pp. 419-424.
34. Galano A., Mazzone G., Alvarez-Diduk R., Marino T., Alvarez-Idaboy J.R.,
Russo N. (2016), "Food Antioxidants: Chemical Insights at the Molecular
Level", Annu. Rev. Food Sci. Technol., 7, pp. 335-352.
35. Haddon R.C. (1993), "Chemistry of the fullerenes: the manifestation of strain in
a class of continuous aromatic molecules", Science, 261(5128), pp. 1545-1550.

108
36. Haddon R.C., Brus L.E., Raghavachari K. (1986), "Rehybridization and π-
orbital alignment: the key to the existence of spheroidal carbon clusters", Chem.
Phys. Lett., 131(3), pp. 165-169.
37. Haddon R.C., Brus L.E., Raghavachari K. (1986), "Electronic structure and
bonding in icosahedral C60", Chem. Phys. Lett., 125(5-6), pp. 459-464.
38. Hale P.D. (1986), "Discrete-variational-X.alpha. electronic structure studies of
the spherical C60 cluster: prediction of ionization potential and electronic
transition energy", J. Am. Chem. Soc., 108(19), pp. 6087-6088.
39. Hedberg K., Hedberg L., Bethune D.S., Brown C.A., Dorn H.C., Johnson R.D.,
De Vries M. (1991), "Bond lengths in free molecules of buckminsterfullerene,
C60, from gas-phase electron diffraction", Science, 254(5030), pp. 410-412.
40. Hileman E.O., Liu J., Albitar M., Keating M.J., Huang P. (2004), "Intrinsic
oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity",
Cancer Chemother. Pharmacol., 53(3), pp. 209-219.
41. Hiroshi I., Kiyoshi H., Tsuyoshi A., Masanori A., Seiji T., Tadashi O., Masahiro
S., Yoshiteru S. (1996), "The small reorganization energy of C60 in electron
transfer", Chem. Phys. Lett., 263(3-4), pp. 545-550.
42. Hirsch A., Brettreich M. (2005), Fullerenes: Chemistry and Reactions,Wiley-
VCH Verlag GmbH & Co. KGaA, Weinheim, Germany.
43. Hou C. (2014), "Theoretical study of antioxidative ability and antioxidative
mechanism of norathyriol in solution", Computational and Theoretical
Chemistry, 1028, pp. 87-91.
44. Jabbari M., Mir H., Kanaani A., Ajloo D. (2014), "Kinetic solvent effects on the
reaction between flavonoid naringenin and 2,2-diphenyl-1-picrylhydrazyl
radical in different aqueous solutions of ethanol: An experimental and
theoretical study", Journal of Molecular Liquids, 196, pp. 381-391.
45. Jagtap U.B., Bapat V.A. (2010), "Artocarpus: a review of its traditional uses,
phytochemistry and pharmacology", J. Ethnopharmacol., 129(2), pp. 142-166.
46. Jensen A.W., Wilson S.R., Schuster D.I. (1996), "Biological applications of
fullerenes", Bioorganic & Medicinal Chemistry, 4(6), pp. 767-779.

109
47. Jovanovic S.V., Steenken S., Hara Y., Simic M.G. (1996), "Reduction potentials
of flavonoid and model phenoxyl radicals. Which ring in flavonoids is
responsible for antioxidant activity?", J. Chem. Soc., Perkin Trans. 2, 11, pp.
2497.
48. Jovanovic S.V., Steenken S., Tosic M., Marjanovic B., Simic M.G. (1994),
"Flavonoids as Antioxidants", J. Am. Chem. Soc., 116(11), pp. 4846-4851.
49. Jung H.A., Su B.N., Keller W.J., Mehta R.G., Kinghorn A.D. (2006),
"Antioxidant xanthones from the pericarp of Garcinia mangostana
(Mangosteen)", J. Agric. Food Chem., 54(6), pp. 2077-2082.
50. Kim S.K., Joo T.H., Suh S.W., Kim M. S. (1986), "Surface-enhanced Raman
scattering (SERS) of nucleic acid components in silver sol: Adenine series",
Journal of Raman Spectroscopy, 17(5), pp. 381-386.
51. Kozlowski D., Marsal P., Steel M., Mokrini R., Duroux J.L., Lazzaroni R.,
Trouillas P. (2007), "Theoretical investigation of the formation of a new series
of antioxidant depsides from the radiolysis of flavonoid compounds", Radiat.
Res., 168(2), pp. 243-252.
52. Kozlowski D., Trouillas P., Calliste C., Marsal P., Lazzaroni R., Duroux J.L.
(2007), "Density functional theory study of the conformational, electronic, and
antioxidant properties of natural chalcones", J. Phys. Chem. A, 111(6), pp. 1138-
1145.
53. Kräutler B., Maynollo J. (1996), "Diels-Alder reactions of the [60]fullerene
functionalizing a carbon sphere with flexibly and with rigidly bound addends",
Tetrahedron, 52(14), pp. 5033-5042.
54. Kroto H.W. (1987), "The stability of the fullerenes Cn, with n = 24, 28, 32, 36,
50, 60 and 70", Nature, 329(6139), pp. 529-531.
55. Kroto H.W., Heath J.R., O'brien S.C., Curl R.F., Smalley R.E. (1985), "C60:
Buckminsterfullerene", Nature, 318(6042), pp. 162-163.
56. Krusic P.J., Wasserman E., Keizer P.N., Morton J.R., Preston K.F. (1991),
"Radical reactions of C60", Science, 254(5035), pp. 1183-1185.

110
57. Lan W.C., Tzeng C.W., Lin C.C., Yen F.L., Ko H.H. (2013), "Prenylated
flavonoids from Artocarpus altilis: antioxidant activities and inhibitory effects
on melanin production", Phytochemistry, 89, pp. 78-88.
58. Langa F., Nierengarten J.F. (2007), Fullerenes: Principles and Applications,
The Royal Society of Chemistry.
59. Lengyel J., Rimarcik J., Vaganek A., Klein E. (2013), "On the radical
scavenging activity of isoflavones: thermodynamics of O-H bond cleavage",
Phys. Chem. Chem. Phys., 15(26), pp. 10895-10903.
60. Leopoldini M., Marino T., Russo N., Toscano M. (2004), "Antioxidant
Properties of Phenolic Compounds: H-Atom versus Electron Transfer
Mechanism", J. Phys. Chem. A, 108(22), pp. 4916-4922.
61. Leopoldini M., Marino T., Russo N., Toscano M. (2004), "Density functional
computations of the energetic and spectroscopic parameters of quercetin and its
radicals in the gas phase and in solvent", Theoretical Chemistry Accounts:
Theory, Computation, and Modeling (Theoretica Chimica Acta), 111(2-6), pp.
210-216.
62. Leopoldini M., Pitarch I.P., Russo N., Toscano M. (2004), "Structure,
Conformation, and Electronic Properties of Apigenin, Luteolin, and Taxifolin
Antioxidants. A First Principle Theoretical Study", J. Phys. Chem. A, 108(1),
pp. 92-96.
63. Leopoldini M., Rondinelli F., Russo N., Toscano M. (2010),
"Pyranoanthocyanins: a theoretical investigation on their antioxidant activity",
J. Agric. Food Chem., 58(15), pp. 8862-8871.
64. Lespade L., Bercion S. (2012), "Theoretical investigation of the effect of sugar
substitution on the antioxidant properties of flavonoids", Free Radic. Res.,
46(3), pp. 346-358.
65. Li M.-J., Liu L., Fu Y., Guo Q.-X. (2007), "Accurate bond dissociation
enthalpies of popular antioxidants predicted by the ONIOM-G3B3 method",
Journal of Molecular Structure: THEOCHEM, 815(1-3), pp. 1-9.

111
66. Li M., Liu W., Peng C., Ren Q., Lu W., Deng W. (2013), "A DFT study on
reaction of eupatilin with hydroxyl radical in solution", International Journal of
Quantum Chemistry, 113(7), pp. 966-974.
67. Li X., Chung L.W., Paneth P., Morokuma K. (2009), "DFT and
ONIOM(DFT:MM) studies on Co-C bond cleavage and hydrogen transfer in
B12-dependent methylmalonyl-CoA mutase. Stepwise or concerted
mechanism?", J. Am. Chem. Soc., 131(14), pp. 5115-5125.
68. Litwinienko G., Ingold K.U. (2003), "Abnormal solvent effects on hydrogen
atom abstractions. 1. The reactions of phenols with 2,2-diphenyl-1-
picrylhydrazyl (dpph*) in alcohols", J. Org. Chem., 68(9), pp. 3433-3438.
69. Lu L., Qiang M., Li F., Zhang H., Zhang S. (2014), "Theoretical investigation
on the antioxidative activity of anthocyanidins: A DFT/B3LYP study", Dyes
and Pigments, 103, pp. 175-182.
70. Lucarini M., Pedrielli P., Pedulli G.F., Valgimigli L., Gigmes D., Tordo P.
(1999), "Bond Dissociation Energies of the N−H Bond and Rate Constants for
the Reaction with Alkyl, Alkoxyl, and Peroxyl Radicals of Phenothiazines and
Related Compounds", J. Am. Chem. Soc., 121(49), pp. 11546-11553.
71. Luo Y.-R. (2003), Handbook of Bond Dissociation Energies in Organic
Compounds, CRC Press.
72. Mai N.T.T., Hai N.X., Phu D.H., Trong P.N.H., Nhan N.T. (2012), "Three new
geranyl aurones from the leaves of Artocarpus altilis", Phytochemistry Letters,
5(3), pp. 647-650.
73. Marcorin G.L., Da Ros T., Castellano S., Stefancich G., Bonin I., Miertus S.,
Prato M. (2000), "Design and Synthesis of Novel [60]Fullerene Derivatives as
Potential HIV Aspartic Protease Inhibitors", Organic Letters, 2(25), pp. 3955-
3958.
74. Marcus Y., Smith A.L., Korobov M.V., Mirakyan A.L., Avramenko N.V.,
Stukalin E.B. (2001), "Solubility of C60 Fullerene", J. Phys. Chem. B, 105(13),
pp. 2499-2506.

112
75. Martinez A., Galano A.,Vargas R. (2011), "Free radical scavenger properties of
alpha-mangostin: thermodynamics and kinetics of HAT and RAF mechanisms",
J. Phys. Chem. B, 115(43), pp. 12591-12598.
76. Martinez A., Hernandez-Marin E., Galano A. (2012), "Xanthones as
antioxidants: a theoretical study on the thermodynamics and kinetics of the
single electron transfer mechanism", Food Funct., 3(4), pp. 442-450.
77. Martinez A., Vargas R., Galano A. (2009), "What is important to prevent
oxidative stress? A theoretical study on electron-transfer reactions between
carotenoids and free radicals", J. Phys. Chem. B, 113(35), pp. 12113-12120.
78. Mcewen C.N., Mckay R.G., Larsen B.S. (1992), "C60 as a radical sponge", J.
Am. Chem. Soc., 114(11), pp. 4412-4414.
79. Mendoza-Wilson A.M., Glossman-Mitnik D. (2006), "Theoretical study of the
molecular properties and chemical reactivity of (+)-catechin and (−)-epicatechin
related to their antioxidant ability", Journal of Molecular Structure:
THEOCHEM, 761(1-3), pp. 97-106.
80. Mendoza-Wilson A.M., Lardizabal-Gutiérrez D., Torres-Moye E., Fuentes-
Cobas L., Balandrán-Quintana R.R., Camacho-Dávila A., Quintero-Ramos A.,
Glossman-Mitnik D. (2007), "Optimized structure and thermochemical
properties of flavonoids determined by the CHIH(medium)–DFT model
chemistry versus experimental techniques", Journal of Molecular Structure,
871(1-3), pp. 114-130.
81. Mikulski D., Molski M. (2012), "A quantum chemical study on the antioxidant
activity of bioactive polyphenols from peanut (Arachis hypogaea) and the major
metabolites of trans-resveratrol", Computational and Theoretical Chemistry,
981, pp. 38-46.
82. Mohajeri A., Asemani S.S. (2009), "Theoretical investigation on antioxidant
activity of vitamins and phenolic acids for designing a novel antioxidant",
Journal of Molecular Structure, 930(1-3), pp. 15-20.
83. Monajjemi M., Heshmat M., Haeri H.H., Kaveh F. (2006), "Theoretical study
of vitamin properties from combined QM-MM methods: Comparison of

113
chemical shifts and energy", Russian Journal of Physical Chemistry, 80(7), pp.
1061-1068.
84. Morton J.R., Negri F.,Preston K.F. (1998), "Addition of Free Radicals to C60",
Accounts of Chemical Research, 31(2), pp. 63-69.
85. Nam P.C., Chandra A.K.,Nguyen M.T. (2013), "Performance of an integrated
approach for prediction of bond dissociation enthalpies of phenols extracted
from ginger and tea", Chem. Phys. Lett., 555, pp. 44-50.
86. Navarrete M., Rangel C., Espinosa-García J., Corchado J.C. (2005),
"Theoretical Study of the Antioxidant Activity of Vitamin E: Reactions of α-
Tocopherol with the Hydroperoxy Radical", Journal of Chemical Theory and
Computation, 1(2), pp. 337-344.
87. Nenadis N., Sigalas M.P. (2011), "A DFT study on the radical scavenging
potential of selected natural 3′,4′-dihydroxy aurones", Food Research
International, 44(1), pp. 114-120.
88. Nierengarten J.-F., Gramlich V., Cardullo F., Diederich F. (1996), "Regio- und
diastereoselektive Bisfunktionalisierung von C60-Fulleren und enantioselektive
Synthese eines C60-Fullerenderivates mit chiralem Additionsmuster", Angew.
Chem. Int. Ed. Engl., 108(18), pp. 2242-2244.
89. Okonogi S., Duangrat C., Anuchpreeda S., Tachakittirungrod S.,
Chowwanapoonpohn S. (2007), "Comparison of antioxidant capacities and
cytotoxicities of certain fruit peels", Food Chem., 103(3), pp. 839-846.
90. Otto V., Andreas H. (2006), "Heterofullerenes", Chem. Rev., 106(12), pp. 5191-
5207.
91. Park K.H., Park Y.D., Han J.M., Im K.R., Lee B.W., Jeong I.Y., Jeong T.S., Lee
W.S. (2006), "Anti-atherosclerotic and anti-inflammatory activities of
catecholic xanthones and flavonoids isolated from Cudrania tricuspidata",
Bioorg. Med. Chem. Lett., 16(21), pp. 5580-5583.
92. Peres V., Nagem T.J., De Oliveira F.F. (2000), "Tetraoxygenated naturally
occurring xanthones", Phytochemistry, 55(7), pp. 683-710.

114
93. Perez-Gonzalez A., Galano A. (2011), "OH radical scavenging activity of
Edaravone: mechanism and kinetics", J. Phys. Chem. B, 115(5), pp. 1306-1314.
94. Pérez-González A., Galano A. (2012), "On the •OH and •OOH scavenging
activity of 3-methyl-1-pyridin-2-yl-5-pyrazolone: Comparisons with its parent
compound, edaravone", International Journal of Quantum Chemistry, 112(21),
pp. 3441-3448.
95. Pokorný J. (2007), "Are natural antioxidants better – and safer – than synthetic
antioxidants?", European Journal of Lipid Science and Technology, 109(6), pp.
629-642.
96. Prato M.,Maggini M. (1998), "Fulleropyrrolidines: A Family of Full-Fledged
Fullerene Derivatives", Accounts of Chemical Research, 31(9), pp. 519-526.
97. Pratt D.A., Dilabio G.A., Brigati G., Pedulli G.F., Valgimigli L. (2001), "5-
Pyrimidinols: Novel Chain-Breaking Antioxidants More Effective than
Phenols", J. Am. Chem. Soc., 123(19), pp. 4625-4626.
98. Ramachandran K.I., Deepa G., Namboori K. (2008), Computational Chemistry
and Molecular Modeling: Principles and Applications, Springer-Verlag Berlin
Heidelberg.
99. Rimarčík J., Lukeš V., Klein E., Ilčin M. (2010), "Study of the solvent effect on
the enthalpies of homolytic and heterolytic N–H bond cleavage in p-
phenylenediamine and tetracyano-p-phenylenediamine", Journal of Molecular
Structure: THEOCHEM, 952(1-3), pp. 25-30.
100. Rong Y., Wang Z., Wu J., Zhao B. (2012), "A theoretical study on cellular
antioxidant activity of selected flavonoids", Spectrochim. Acta A Mol. Biomol.
Spectrosc., 93, pp. 235-239.
101. Ros T.D., Prato M., Lucchini V. (2000), "Additions of Azomethine Ylides to
Fullerene C60 Assisted by a Removable Anchor", J. Org. Chem., 65(14), pp.
4289-4297.
102. Ruoff R.S., Tse D.S., Malhotra R., Lorents D.C. (1993), "Solubility of fullerene
(C60) in a variety of solvents", The Journal of Physical Chemistry, 97(13), pp.
3379-3383.

115
103. Sadasivam K., Kumaresan R. (2011), "Theoretical investigation on the
antioxidant behavior of chrysoeriol and hispidulin flavonoid compounds – A
DFT study", Computational and Theoretical Chemistry, 963(1), pp. 227-235.
104. Sadasivam K., Kumaresan R. (2011), "A comparative DFT study on the
antioxidant activity of apigenin and scutellarein flavonoid compounds",
Molecular Physics, 109(6), pp. 839-852.
105. Schramm D.D., Karim M., Schrader H.R., Holt R.R., Cardetti M., Keen C.L.
(2003), "Honey with high levels of antioxidants can provide protection to
healthy human subjects", J. Agric. Food Chem., 51(6), pp. 1732-1735.
106. Scrivens W.A.,Tour J.M. (1993), "Potent solvents for C60 and their utility for
the rapid acquisition of 13C NMR data for fullerenes", J. Chem. Soc., Chem.
Commun., 15), pp. 1207.
107. Sijbesma R., Srdanov G., Wudl F., Castoro J.A., Wilkins C., Friedman S.H.,
Decamp D.L., Kenyon G.L. (1993), "Synthesis of a fullerene derivative for the
inhibition of HIV enzymes", J. Am. Chem. Soc., 115(15), pp. 6510-6512
108. Sivaraman N., Dhamodaran R., Kaliappan I., Srinivasan T.G., Rao P.R.V.,
Mathews C.K. (1992), "Solubility of C60 in organic solvents", J. Org. Chem.,
57(22), pp. 6077-6079.
109. Śliwa W. (1997), "Diels-Alder Reactions of Fullerenes", Fullerene Science and
Technology, 5(6), pp. 1133-1175.
110. Tabata Y., Ikada Y. (1999), "Biological functions of fullerene", Pure and
Applied Chemistry, 71(11).
111. Tachakittirungrod S., Okonogi S., Chowwanapoonpohn S. (2007), "Study on
antioxidant activity of certain plants in Thailand: Mechanism of antioxidant
action of guava leaf extract", Food Chem., 103(2), pp. 381-388.
112. Taylor R. (1999), Lecture Notes on Fullerene Chemistry: A Handbook for
Chemists, London, Imperial College Press.
113. Taylor R., Walton D.R.M. (1993), "The chemistry of fullerenes", Nature,
363(6431), pp. 685-693.

116
114. Thong N.M., Dao D.Q., Ngo T.C., Huyen T.L., Nam P.C. (2016), "Antioxidant
activities of [60]fullerene derivatives from chalcone, flavone and flavanone: A
ONIOM approach via H-atom and electron transfer mechanism", Chem. Phys.
Lett., 652, pp. 56-61.
115. Thong N.M., Duong T., Thuy P.T., Nam P.C. (2014), "Theoretical investigation
on the bond dissociation enthalpies of phenolic compounds extracted from
Artocarpus altilis using ONIOM(ROB3LYP/6-311++G(2df,2p):PM6) method",
Chem. Phys. Lett., 613, pp. 139-145.
116. Thong N.M., Quang D.T., Bui N.H.T., Dao D.Q., Nam P.C. (2015),
"Antioxidant properties of xanthones extracted from the pericarp of Garcinia
mangostana (Mangosteen): A theoretical study", Chem. Phys. Lett., 625, pp.
30-35.
117. Trouillas P., Marsal P., Siri D., Lazzaroni R., Duroux J.-L. (2006), "A DFT
study of the reactivity of OH groups in quercetin and taxifolin antioxidants: The
specificity of the 3-OH site", Food Chemistry, 97(4), pp. 679-688.
118. Tsuneda T. (2014), Density Functional Theory in Quantum Chemistry, Springer.
119. Tzirakis M.D., Orfanopoulos M. (2013), "Radical reactions of fullerenes: from
synthetic organic chemistry to materials science and biology", Chem. Rev.,
113(7), pp. 5262-5321.
120. Vagánek A., Rimarčík J., Lukeš V., Klein E. (2012), "On the energetics of
homolytic and heterolytic OH bond cleavage in flavonoids", Computational and
Theoretical Chemistry, 991, pp. 192-200.
121. Vreven T., Byun K.S., Komaromi I., Dapprich S., Montgomery J.A., Morokuma
K., Frisch M.J. (2006), "Combining Quantum Mechanics Methods with
Molecular Mechanics Methods in ONIOM", .J Chem. Theory. Comput., 2(3),
pp. 815-826.
122. Vreven T., Morokuma K. (1999), "The accurate calculation and prediction of
the bond dissociation energies in a series of hydrocarbons using the IMOMO
(integrated molecular orbital+molecular orbital) methods", The Journal of
Chemical Physics, 111(19), pp. 8799.

117
123. Vreven T., Morokuma K. (2002), "Prediction of the Dissociation Energy of
Hexaphenylethane Using the ONIOM(MO:MO:MO) Method", The Journal of
Physical Chemistry A, 106(25), pp. 6167-6170.
124. Wang G., Xue Y., An L., Zheng Y., Dou Y., Zhang L., Liu Y. (2015),
"Theoretical study on the structural and antioxidant properties of some recently
synthesised 2,4,5-trimethoxy chalcones", Food Chem., 171, pp. 89-97.
125. Wang I.C., Tai L.A., Lee D.D., Kanakamma P.P., Shen C.K.F., Luh T.-Y.,
Cheng C.H., Hwang K.C. (1999), "C60 and Water-Soluble Fullerene Derivatives
as Antioxidants Against Radical-Initiated Lipid Peroxidation", Journal of
Medicinal Chemistry, 42(22), pp. 4614-4620.
126. Wang Y., Xu K., Lin L., Pan Y., Zheng X. (2007), "Geranyl flavonoids from
the leaves of Artocarpus altilis", Phytochemistry, 68(9), pp. 1300-1306.
127. Wilson S.R., Kaprinidis N., Wu Y., Schuster D.I. (1993), "A new reaction of
fullerenes: [2+2]-photocycloaddition of enones", J. Am. Chem. Soc., 115(18),
pp. 8495-8496.
128. Wilson S.R., Wu Y., Kaprinidis N.A., Schuster D.I., Welch C.J. (1993),
"Resolution of enantiomers of cis- and trans-fused C60-enone [2+2]
photoadducts", J. Org. Chem., 58(24), pp. 6548-6549.
129. Wright J.S., Johnson E.R., Dilabio G.A. (2001), "Predicting the activity of
phenolic antioxidants: theoretical method, analysis of substituent effects, and
application to major families of antioxidants", J. Am. Chem. Soc., 123(6), pp.
1173-1183.
130. Xue Y., Zhang L., Li Y., Yu D., Zheng Y., An L., Gong X., Liu Y. (2013), "A
DFT study on the structure and radical scavenging activity of newly synthesized
hydroxychalcones", J. Phys. Org. Chem., 26(3), pp. 240-248.
131. Xue Y., Zheng Y., An L., Dou Y., Liu Y. (2014), "Density functional theory
study of the structure-antioxidant activity of polyphenolic deoxybenzoins",
Food Chem., 151, pp. 198-206.

118
132. Xue Y., Zheng Y., An L., Zhang L., Qian Y., Yu D., Gong X., Liu Y. (2012),
"A theoretical study of the structure–radical scavenging activity of
hydroxychalcones", Computational and Theoretical Chemistry, 982, pp. 74-83.
133. Xue Y., Zheng Y., Zhang L., Wu W., Yu D., Liu Y. (2013), "Theoretical study
on the antioxidant properties of 2'-hydroxychalcones: H-atom vs. electron
transfer mechanism", J. Mol. Model., 19(9), pp. 3851-3862.
134. Yang S.H., Pettiette C.L., Conceicao J., Cheshnovsky O., Smalley R.E. (1987),
"Ups of buckminsterfullerene and other large clusters of carbon", Chem. Phys.
Lett., 139(3-4), pp. 233-238.
135. Zadernowski R., Czaplicki S.,Naczk M. (2009), "Phenolic acid profiles of
mangosteen fruits (Garcinia mangostana)", Food Chem., 112(3), pp. 685-689.
136. Zhang H.Y., Ji H.F. (2006), "How vitamin E scavenges DPPH radicals in polar
protic media", New Journal of Chemistry, 30(4), pp. 503-504.

Related Documents











