DERMAL AND FASCIAL FIBROMATOSIS* JOHN T. PRIOR, M.D., AND BERNARD J. SISSON, M.D. SYRACUSE, NEW YORK DEPARTMENTS OF PATHOLOGY AND SURGERY (PLASTIC SURGERY), STATE UNIVERSITY OF NEW YORK, COLLEGE OF MEDICINE, SYRACUSE, NEW YORK RECENTLY IN OUR routine surgical mate- rial we have encountered an increasing number of cases of an unpredictable type of connective tissue overgrowth. It is our opin- ion that this tumor is not generally recog- nized either by the clinician or by the pathologist, the latter being content to clas- sify it within that maze of fibrous tissue growths variously known as fibromra, sub- epidermal fibroma, dermatofibroma lentic- ulare, extra-abdominal desmoid, fasciitis, etc. The pathologist cannot accept full re- sponsibility for this confusion in nomencla- ture, since the clinician, on the basis of anatomical location alone, has dignified these tumors with such terms as Peyronie's disease when restricted to the dorsal penile fascia, fibromatosis colli referring to con- genital wryneck, Dupuytren's contracture of palmar fascia, fibromatosis of plantar fascia, and desmoid tumor when confined to the musculoaponeurotic tissue of the anterior abdominal wall. Stout,20 in discussing fibrosarcomas, has called attention to the lesion with which this paper is primarily concerned. He states that it is a tumor-like congenital fibrous tis- sue proliferation, concerning which our knowledge is still incomplete, even regard- ing whether or not these peculiar fibroses can produce malignant tumors. Although Stout points out that the lesion has been called progressive myositis fibrosa9 and hereditary polyfibromatosis,23 in a recent communication he has referred to the neo- plasm as infantile fibromatosis.21 Similarly, with intent to emphasize its occurrence in a young age group, a variant of this same process in which calcification is a promi- nent feature has been named calcifying ju- venile aponeurotic fibroma.7 Kapiloff and Prior6 have pointed out that this multiplicity of terms, referring as they do to the same basic pathologic process, has done much to prevent its general recognition. They de- scribed two examples occurring in the ex- tremities of children, both being character- ized by repeated recurrences of histolog- ically benign fibrous tissue growth follow- ing what was judged to be adequate sur- gical excision. In view of the confusion currently exist- ing regarding this type of connective tissue overgrowth, it was felt that a review of a series of cases which have been studied at this center would be of value. The clinical and pathologic characteristics of the lesion will be oontrasted with other tumors of con- nective tissue origin which, although con- sidered benign, are similarly characterized by repeated recurrences and in rare in- stances, distant metastasis. FIBROMATOSIS Some of the pertinent data regarding this series has been arranged in tabulated form (Table I). It will be observed that although the tumor characteristically affects children and young adults, the older age groups are niot spared. It should, however, be empha- 453 * Submitted for publication November, 1953.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

DERMAL AND FASCIAL FIBROMATOSIS*JOHN T. PRIOR, M.D., AND BERNARD J. SISSON, M.D.
SYRACUSE, NEW YORK
DEPARTMENTS OF PATHOLOGY AND SURGERY (PLASTIC SURGERY), STATE UNIVERSITY OF NEW YORK,COLLEGE OF MEDICINE, SYRACUSE, NEW YORK
RECENTLY IN OUR routine surgical mate-rial we have encountered an increasingnumber of cases of an unpredictable type ofconnective tissue overgrowth. It is our opin-ion that this tumor is not generally recog-nized either by the clinician or by thepathologist, the latter being content to clas-sify it within that maze of fibrous tissuegrowths variously known as fibromra, sub-epidermal fibroma, dermatofibroma lentic-ulare, extra-abdominal desmoid, fasciitis,etc. The pathologist cannot accept full re-sponsibility for this confusion in nomencla-ture, since the clinician, on the basis ofanatomical location alone, has dignifiedthese tumors with such terms as Peyronie'sdisease when restricted to the dorsal penilefascia, fibromatosis colli referring to con-genital wryneck, Dupuytren's contractureof palmar fascia, fibromatosis of plantarfascia, and desmoid tumor when confinedto the musculoaponeurotic tissue of theanterior abdominal wall.
Stout,20 in discussing fibrosarcomas, hascalled attention to the lesion with whichthis paper is primarily concerned. He statesthat it is a tumor-like congenital fibrous tis-sue proliferation, concerning which ourknowledge is still incomplete, even regard-ing whether or not these peculiar fibrosescan produce malignant tumors. AlthoughStout points out that the lesion has beencalled progressive myositis fibrosa9 andhereditary polyfibromatosis,23 in a recent
communication he has referred to the neo-plasm as infantile fibromatosis.21 Similarly,with intent to emphasize its occurrence ina young age group, a variant of this sameprocess in which calcification is a promi-nent feature has been named calcifying ju-venile aponeurotic fibroma.7 Kapiloff andPrior6 have pointed out that this multiplicityof terms, referring as they do to the samebasic pathologic process, has done muchto prevent its general recognition. They de-scribed two examples occurring in the ex-tremities of children, both being character-ized by repeated recurrences of histolog-ically benign fibrous tissue growth follow-ing what was judged to be adequate sur-gical excision.
In view of the confusion currently exist-ing regarding this type of connective tissueovergrowth, it was felt that a review of aseries of cases which have been studied atthis center would be of value. The clinicaland pathologic characteristics of the lesionwill be oontrasted with other tumors of con-nective tissue origin which, although con-sidered benign, are similarly characterizedby repeated recurrences and in rare in-stances, distant metastasis.
FIBROMATOSIS
Some of the pertinent data regarding thisseries has been arranged in tabulated form(Table I). It will be observed that althoughthe tumor characteristically affects childrenand young adults, the older age groups areniot spared. It should, however, be empha-
453
* Submitted for publication November, 1953.
PRIOR AND SISSON
sized that particularly in clhildren, the dateof discovery may be far removed from theactual onset, and the probabilities are thatmany of these tumors were present sincebirth. The fact that these neoplasms mayoccur in older age groups is not an original
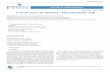
FIG. 1. (Case 3) Recurrent tumor plaques on theinner surfaces of the 3rd and 4th fingers.
observation, since Stout20 states that al-though the process generally starts in thefirst two or three years of life, it may firstappear in adult life. In our series there is nostatistically significant preference for eithersex-a fact of some interest when one con-
siders that a histologically identical con-
nective tissue overgrowth, Dupuytren'scontracture, is said to occur nine times as
frequently in males as in females.22 All buttwo of the present group of cases have in-volved an extremity, and although therehave been repeated recurrences at theoriginal site, we have observed only one
case in which there have been multiple pri-mary foci (Case 3).
Clinically, the gross appearance of thetumor is characterized by a smooth, some-
what rounded, slightly raised, plaque-likelesion which is red in color (Fig. 1). Thetumors have averaged about 1.5 cm. in theirgreatest diamiieter. The examiner is readilvalble to ouitline the limnits of the process,
and the impression of fixation to deeper tis-
sues was present in some of our cases.There is nothing particularly characteristicabout the cut surface of the tumor, the sur-face being described as smooth, grayish-yellow and translucent in appearance, andcutting with increased resistance. The ex-aminer's clinical impression of the lesionhas been of interest, being variously con-
sidered as a scar, keloid, neurofibroma,sclerosing hemangioma, dermatofibrosar-coma protuberans, fibroma, and rarely,fibromatosis.Only rarely did the patient complain of
tenderness in the lesion, and the most fre-quent history obtained was that the tumorarose spontaneously and without pain. Intwo cases (Case 7 and 10) the lesion was
associated with a history of precedingtrauma at the site which was stated to beslight in degree. It will be noted (Table I)that all but two cases of our series affectedan extremity, one case being located withinthe superficial tissue of the breast and theother confined to the lateral aspect of theneck. Multiple tumors were observed inonly one instance (Case 3), although theliterature concerning this type of tumor in-dicates that multiple primary sites are notunusual.The duration of the tumors is particularly
difficult to evaluate, although one gets theimpression that they have been of very longduration and that perhaps in the young
children they were actually present atbirth. The rate of growth is generally very
slow in the adult lesions, while the tumorsin children tend to show a rather rapid in-crease in size following their initial obser-vation. When recurrences appear, theycharacteristically occur between three andsix months following operation.The histopathology of the lesion is highly
characteristic in its appearance and israther simple and orderly in its structure.The more superficial or cutaneous form ofthe tuimor is located within both the papil-lary and reticular layers of the dermis.
454
Antnals of SurgeryApril, 1954
DERMAL AND FASCIAL FIBROMATOSIS
Fl a2 FIG. :3
FIG. 4 FIG. 5
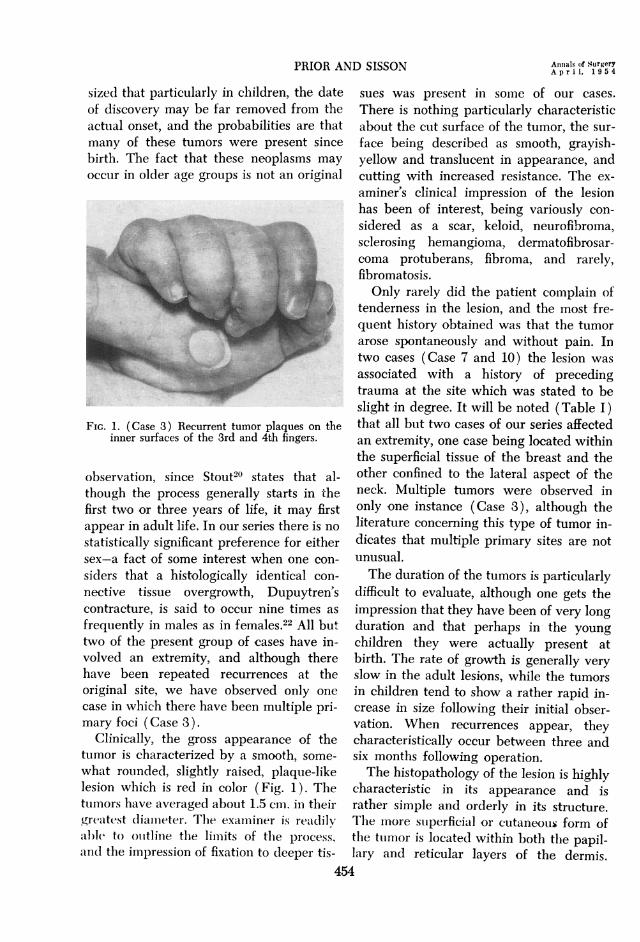
FIG. 2. (Case 3) Replacement of the normal dermal pattern by interlacing connective tissuefascicles extending into the epidermal-dermal junction (x5l).
FIG. 3. (Case 5) Capillary showing the peculiar structure of contiguous tumor cells andthe swollen character of the lining endothelium (x 88).
FIG. 4. (Case 5) Group of vessels showing pronounced so-called "fibrinoid necrosis" (x 100).FIG. 5. (Case 11) Large sheet of deposited calcium replacing tumor in lower right corner
(x71).
In less than half of our cases the process ex-
tended from the junction of the dermis andepidermis into the superficial subcutaneouslayer (Fig. 2). The growth is neither en-
capsulated nor sharply circumscribed, andis composed of thick bundles of connectivetissue, with the individual bundles coursingin opposing directions, reminiscent of thefasciculated, whorled pattern seen in thebanal uiterine leiomyomna. The cellular ele-ments composing the tumor are spindle-shaped fibroblasts, whose vesicular nuclei
and abundant cytoplasm suggest their rel-ative immaturity. Evidence of mitotic activ-ity is extremely difficult to find, and al-though the nuclei may appear somewhathyperchromatic in profile, variation in nu-
clear size and shape was observed only inthe lungs of the patient in whom pulmonarymetastases developed (Case 7). This childdeveloped pulmonary metastatic lesionsfollowing repeated local excisions andeventual amputation of the thumb and, al-
though most of the pulmonary tumor was
455
Volunme 1:39Numtiber 4

PRIOR AND SISSON
histologically identical with the primarylesion, rare foci showed the cellular andnuclear atypism consistent with a rapidlygrowing fibrosarcoma. The Coldner modi-fication of the trichrome Masson stain re-veals the lesion to be very rich in collage-nous tissue, and the Weigert's resorcinfuchsin stain discloses insignificant amountsof elastic fibers to be present. Perl's stainfor iron has been consistently negative.
In areas of maximum growth the neo-plasm tends to suirround, compress and par-tially obliterate the dermal appendages. Allof the cases in our series are characterizedby moderate vascularity, capillary in type,and the lining endothelium frequentlyshows evidence of some hypertrophy (Fig.3). One of the cases studied (Case 3)showed unusual thickening of the capillarywalls, and there was marked so-called fibri-noid change present (Fig. 4). The epithe-lium overlying the tumor characteristicallyshows various degrees of acanthosis, andthe rete pegs may be either narrow andsomewhat pointed distally, or frequentlythey appear as club-like extensions into thetumor area. Hyperkeratosis is always a
prominent feature directly over the neo-
plastic zone. Degenerative changes withinthe tumor were seen in only one instance(Case 11). They took the form of multiplefoci of calcification scattered in an irregularfashion throughout the tumor (Fig. 5).
Other than for the mononuclear andgiant cell reaction this foreign material ex-
cited, the tumor in no way differs from thenon-calcified type. Klasberg7 has attemptedto group these cases in a special category,terming them calcifying juvenile aponeu-rotic fibromas. It is of interest that the fourcases she described were confined to theaponeuroses of either palmar or plantarfascia in children, while Case 11 in thisseries was located in the dermis in the kneeregion in an adult. Repeated recurrences
charaterized Klasberg's cases.
The cases to be reported in some detailbelow have been selected since we feelthey illustrate the range in clinical behaviorthat the process may follow. The first casedemonstrates the usual course with re-peated local recurrences following whatwas judged to be adequate removal at eachsurgical excision. The second case is pre-sented in some detail since it is believed tobe the first recorded instance of hemato-genous pulmonary metastases from thistype of humor which was originally con-fined to an extremity. This case is partic-ularly important since it has had a profoundinfluence upon our current concept oftherapy.
CASE REPORTS
Case 1. J. K., an 8-months-old male child, wasfirst admitted to Memorial Hospital January 22,1953. The child was one of 7 children, was statedto have hlad a normal birth, an-d all the siblingsare alive and well. Between the age of 2 and 3months the mother noted small, firm masses on thedistal phalanges of the 3rd and 4th fingers of theright hand. These plaques increased steadily insize until at the age of 8 months they involved themiddle phalanx as well as the distal, and com-
pletely surrounded the fingers. The tumor was
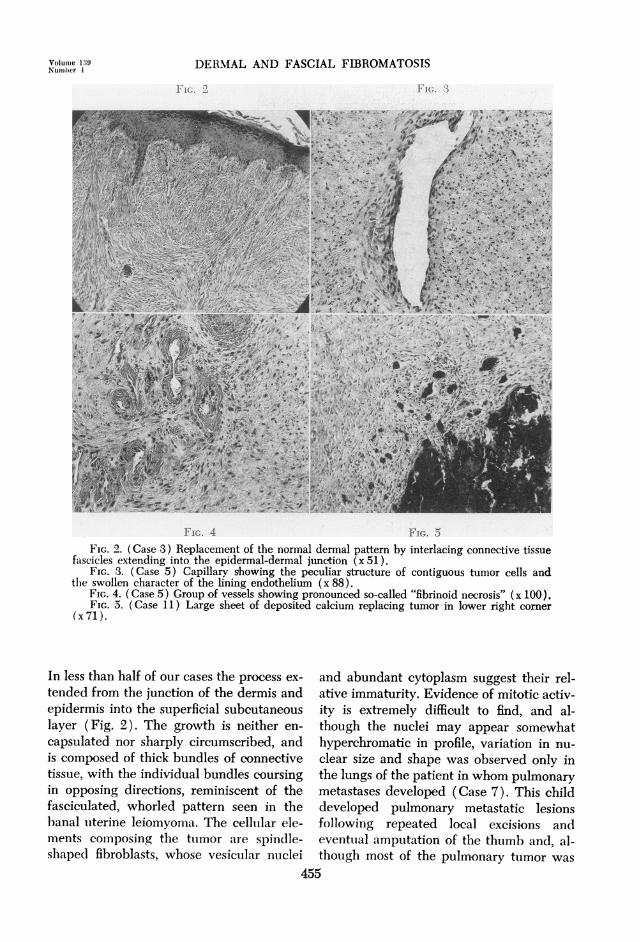
firm, slightly raised, and red in color, and therewere no palpable axillary or epitrochlear lymphglands. A clinical diagnosis of infantile fibroma-tosis was made and both tumors were completelyexcised and a split-thickness skin graft was applied.The microscopic appearance of this tumor is shownin Figure 2. The interlacing bands of connectivetissue extend up to the dermal-epidermal junction,but no evidence of nuclear or cellular atypism isapparent.
Because of the recurrence of the tumor on bothfingers, the child was re-admitted to MemorialHospital May 11, 1953. The extent and clinicalappearance of the tumor at this time is shown inFigure 1. The masses were re-excised and a split-thickness skin graft was re-applied. Histologicalexamination of the tissue showed no changes fromthe original pattern. Evidence of recurrence waspresent in August of 1953, and the decision hasbeen made to re-admit the patient and amputateboth fingers at the metacarpalphalangeal joints.There are no palpable axillary or epitroohlearglands at present.
Annals of SurgeryA p r i 1, 1 9 5 4
DERMAL AND FASCIAL FIBROMATOSIS
FIG. 6 FIG. 7
FIG. 8 FIG. 9FIG. 6. (Case 7) Original biopsy of thumb showing compression of appendages by histolog-
ically benign connective tissue overgrowth (x 82).FIG. 7. (Case 7) Extremely cellular well differentiated tumor within the lung. An invaded
pulmonary artery is shown in the right half of the illustration (x82).FIG. 8. (Case 7) Fibrosarcoma with marked cellular atypism. A small bronchiole is present
in the upper right (x82).FIG. 9. Interlacing connective tissue bundles showing moderate cellularity. Uninvolved
plantar fascia is present in lower right (x80).
Case 2. L. F., an 8-year-old female child,injured the right side of her right thumb in a car
door and the lesion failed to heal. Subsequently,a tender mass, measuring 1 cm. x 0.6 cm., devel-oped in the unhealed portion and the patient was
admitted to Syracuse General Hospital in June,1942, for excision of the mass. The microscopicdiagnosis of the tissue at this time was fibroma(Fig. 6). Review of the sections shows interlacingbands of well-differentiated connective tissue,showing compression of the dermal appendages.In August of 1942, a recurrence was noted in theincision and also another small nodule was presenton the ulnar side of the thumb. The thumb was
swollen to twice its normal size and there was
limited motility and pain on motion. Local excisionwas once more performed, and the microscopic
appearance of the mass showed no change from theprevious examination. Because of a recurrence withan increase in size of the tumor, the thumb wasamputated in October, 1942, and the microscopicpattern was identical with the previous specimens.In November, 1943, the patient was re-admittedto the hospital because of an unhealed wound atthe site of the amputation, and the metacarpal bonestump was removed. No evidence of tumor wasnoted microscopically at this time. In September of1949, during a routine school cheest roentgenogram,a miass in the left middle lung field was noted and,although metastatic tumor was strongly suspected,it was decided to remove the lung. Lymphadeno-pathy was noted in the right axilla and afterbiopsy of the glands disclosed chronic lymph-adenitis, a left pneumonectomy was performed,
457
Volume 159Ntimber 4

PRIOR AND SISSON
The patient developed pulmonary edema imme-diately following operation and expired in January,1950.
Examination of the resected lung disclosedabout a dozen shotty nodules in the anterior por-tion of the upper lobe, the largest of these measur-ing 1.5 cm. in diameter. A similar nodule was notedin the extreme antero-medial inferior portion ofthe lower lobe. Microscopic examination of themajority of the nodules disclosed well differen-tiated, very cellular connective tissue, with evi-dence of blood vessel invasion (Fig. 7). Search ofmany sections did, however, disclose foeci charac-terized by cellular and nuclear atypism andabundant mitoses (Fig. 8). These areas were con-sistent with a high-grade fibrosarcoma. At necropsythere were two nodules, each measuring 0.5 cm. indiameter, located within the right lung, and themicroscopic pattern was similar to that seen inthe resected lung. Marked pulmonary edema waspresent in the right lung. No lymph node involve-ment was noted.
PLANTAR FIBROMATOSIS
Attention has recently been directedtowards this lesion by Pickren, et al.,17 whoreviewed 104 previously reported instancesand added 16 new cases to the literature.This process is histologically identical withthe lesions described above, and it is our
feeling that there is little value in attempt-ing to classify this group separately. Bothdermal and plantar tumors are character-ized by repeated recurrences, insidiousgrowth qualities and frequent occurrences
in younger age groups. It is of interest thatthe plantar lesion has been found in asso-
ciation with a similar process in the palmarfascia (Dupuytren's contracture) in a sig-nificant number of cases. That this plantarlesion may also have malignant potential-ities is attested to by the case reported byCollins and Anspach,5 in which groin andpulmonary metastases developed over a
four-year period. The process in the plantarfascia appears to differ from its homologuein the palm only in the fact that the formerrarely produces flexion deformities.The following case illustrates the clinical
course usually observed where the process
is located within the plantar fascia:
Case 3. D. S., a 20-year-old woman, had asmall ttmior remiioved from the plantar surface ofthe right foot in 1946. At this time the diagnosis ofneurofibroma was made on the tissue examined.In 1949 the patient noted a hard mass about theprevious sear, although the scar itself did notseem to be involved. No glands were palpable inthe inguinal area. The mass was re-excised andthe microscopic opinion was again neurofibroma.In November of 1950, 2 tender masses were notedon the plantar surface, one toward the middle ofthe foot and the other beside the previous scar.The mass was excised and the microscopic diag-nosis was fibromatosis of the plantar fascia (Fig.9). Review of the slides from the lesion in 1946and 1949 disclosed the same process to be pres-ent, and there was no evidence of increased activ-ity in the latest lesion.
DESMOID TUMORS
These tumors have been defined as "be-nign fibrous neoplasms which arise from themusculo-aponeurotic structures throughoutthe body, and which have the peculiar char-acteristics of locally invading adjacent mus-
cle tissue and engulfing the striped musclefibers."12 The term, by popular usage, hascome to be restricted to those tumors con-
fined to the musculo-aponeurotic tissue ofthe anterior abdominal wall. Musgrove andMcDonald,'2 however, under the name
"extra-abdominal desmoid," have reported34 examples of this type of growth, involv-ing a variety of locations, including theface, chest, back, buttocks, arms and legs.When the tumor occurs on the anterior ab-dominal wall it is characteristically foundin women in the younger age groups whohave borne children. This rule is not with-out exception, however, in view of the largenumber of cases Booker and Pack have re-
ported occurring in children.2Not only is there disagreement as to the
malignant capabilities of the desmoid, butthere are those who seriously questionwhether the process should be considereda true neoplasm. Stout20 attempts to sepa-
rate desmoids from what he terms fibrosar-comas of the abdominal wall, the latter pur-
suing a course similar to fibrosarcoma else-458
Annals of SurgeryA p r i 1, 1 9 5 4
DERMAL AND FASCIAL FIBROMATOSIS
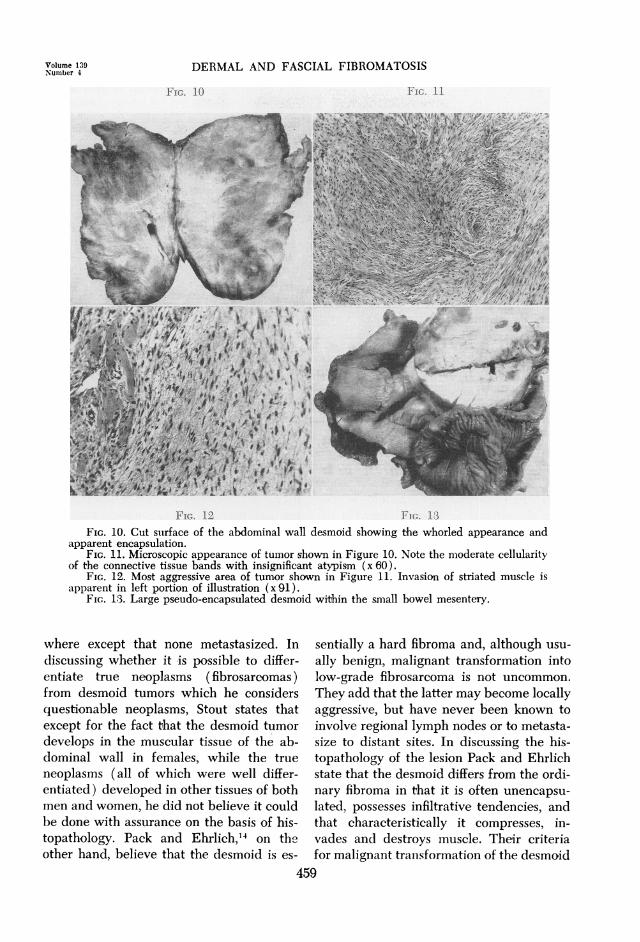
FIG. 10 FIG. 11
FIG. 12 FIG. 13FIG. 10. Cut suirface of the abdominal wall desmoid showing the whorled appearance and
apparent encapsulation.FIG. 11. Microscopic appearance of tumor shown in Figure 10. Note the moderate cellularity
of the connective tissue bands with insignificant atypism (x 60).FIG. 12. Most aggressive area of tumor shown in Figure 11. Invasion of striated muscle is
apparent in left portion of illustration (x 91).FIG. 13. Large pseudo-encapsulated desmoid within the small bowel mesentery.
where except that none metastasized. Indiscussing whether it is possible to differ-entiate true neoplasms (fibrosarcomas)from desmoid tumors which he considersquestionable neoplasms, Stout states thatexcept for the fact that the desmoid tumordevelops in the muscular tissue of the ab-dominal wall in females, while the trueneoplasms (all of which were well differ-entiated) developed in other tissues of bothmen and women, he did not believe it couldbe done with assurance on the basis of his-topathology. Pack and Ehrlich,14 on theother hand, believe that the desmoid is es-
sentially a hard fibroma and, although usu-
ally benign, malignant transformation intolow-grade fibrosarcoma is not uncommon.
They add that the latter may become locallyaggressive, but have never been known toinvolve regional lymph nodes or to metasta-size to distant sites. In discussing the his-topathology of the lesion Pack and Ehrlichstate that the desmoid differs from the ordi-nary fibroma in that it is often unencapsu-
lated, possesses infiltrative tendencies, andthat characteristically it compresses, in-vades and destroys muscle. Their criteriafor malignant transformation of the desmoid
459
Volume 139Numitber 4

PRIOR AND SISSON
are not clearly defined, and they simplystate that the histological picture may varyfrom an acelluluar fibroma to a cellular low-grade fibrosarcoma, and that it is not un-common to encounter areas of fibrosarco-matous and myosarcomatous transformationin this group of neoplasms.
-:: ~ ::.
FIG. 14. The raised nodular character of thetumor with central ulceration is obvious in the lefthalf of the illustration.
Our own experience with the desmoidtumor leads us to conclude that it is an in-filtrative tumor of connective tissue whichcannot be separated on an histologic basisfrom the dermal and fascial growths de-scribed above. The following case is pre-sented to illustrate the fact that despite an
innocuous microscopic appearance, the des-moid may bring about a fatal terminationby reason of recurrent growth and uncon-
trollable local extension.
Case 4. J. C., a 21-year-old multiparous whitewoman, first entered Memorial Hospital on May18, 1949, complaining of intermittent bloody stoolsof 2 years' duration. Significant in the family his-tory was the fact that the patient's mother andone sister died at ages 32 and 27 years, respec-
tively, of cancer of the bowel. A diagnosis ofbenign familial polyposis of the colon was estab-lished, and a resection of the large bowel fromthe cecum to the sigmoid colon was performed.She returned to the hospital 3 months after her
discharge, for -the removal of the sigmoid and rec-
tum. Microscopic examination of the resected sig-moid at this time disclosed Grade II adenocarci-noma within a large pedunculated polyp withoutinvasion of the stalk. In November of 1951, thepatient was again admitted to the hospital because
of a discrete area of progressive swelling of theanterior abdominal wall to the left of, but attachedto the left rectus scar. The mass measured 3 cm.in diameter and had been present for 7 months.At operation the tumor was found to invade bothrectus muscles and to extend to the iliac crest onthe right side and upwards to a point just belowthe costal margin. The mass completely surroundedthe ileostomy opening and had also invaded themesentery. Following a frozen section report ofdesmoid tumor, much of the abdominal mass wasremoved, the ileostomy opening revised, and about35 cm. of ileum was removed with the mesentericmass. The abdominal wall tumor was pinkish-grayin color and cut with a grating sensation. Its cutsurface disclosed a whorled appearance and therewas pseudo-encapsulation of the mass (Fig. 10).Microscopically, the tumor consisted of interlacingfascicles of well-differentiated connective tissuewithout evidence of atypism or unusual mitoticactivity (Fig. 11). The most aggressive area ofthe tumor is shown in Figure 12 as it is invadingstriated muscle. The tumor within the mesenterymeasured 7 x 7 x 4 cm., and was generally similarin appearance (Fig. 13). The bowel mucosa was
dimpled and there was marked compression of thelumen, and the tumor had extended into the serosaland muscular coats.Two months following discharge the patient
developed recurrent abdominal tumor just abovethe ileostomy site. Two masses of tissue, 4 x 2 x 2cm. and 6 x 4 x 2 cm., were excised andthe microscopic examination revealed desmoidtumor with no change from the previous cyto-logic pattern. The patient was again admittedin March of 1952, complaining of ulceration, witha scar superior to the ileostomy opening. At oper-ation there was a recurrence within the abdominalwall measuring 6 cm. in diameter, with extensioninto the mesentery of the terminal ileum. Themass in the latter area measured 8 cm. in diameter,and 40 cm. of ileum was resected at the time ofits removal. No change in the microscopic patternof the desmoid tumor was observed. Roentgen raytherapy to the abdominal wall was instituted atthis time in an effort to suppress the growth, butno beneficial results were noted. Five months latera recurrent abdominal wall mass measuring 4 x 4x 1 cm. was excised and at this time peritonealexploration revealed no evidence of tumor. InJanuary, 1953, however, an abdominal mass meas-
uring 5 cm. in diameter was palpated and this wasfound at surgery to be embedded within themesentery of the jejunum. Thirty centimeters ofsmall bowel were removed at this time and a
jejunostomy was performed. The remaining unin-volved ibowel was measured and. was estimated to
460
Annals of SurgeryApril. 1954

DERMAL AND FASCIAL FIBROMATOSIS
be less than 28 cm. in length. At the present time,4 months since her last hospital admission, thepatient shows no tumor recurrence, but has becomea very severe nutritional as well as psyohiatricproblem.
DERMATOFIBROSARCOMA PROTUBERANS
Another connective tissue tumor whichis dermal in location and which may clin-ically and microscopically simulate thefibromatoses, is a neoplasm known as der-matofibrosarcoma protuberans. This tumorhas a peculiar predilection for the trunk,and tends to spare the extremities. Althoughit may present as an ill-defined, fibroticplaque, it more frequently tends to occur as
a hemispherical, mushroom-shaped, sessiletumor, with bluish-red overlying slin andfrequent ulceration (Fig. 14). This tumoris also characterized by long durati-on andvery slow growth. According to Pack andTabah,15 the tumor is notorious for its ten-dency to recur locally following excision,and the erroneous interpretation of its mi-croscopic features has been responsiible forits awe-inspiring name. In none of the 39patients reported by Pack and Tabah was
there evidence of metastases to regionallymph nodes or viscera. There have been,however, rare instances of tumors of thistype which have metastasized, both cases
following multiple local excisions.1 16 It isregrettable to note that the term "primary
fibrosarcoma of skin" has recently been sug-gested for the lesion, notwithstanding thefact that the cases reported did not metas-tasize."
Despite its dermal origin and its ten-dency to recur and metastasize, it is our
belief that the dermatofibrosarcoma protu-berans can be readily separated on a histo-logic basis from the other dermal connec-
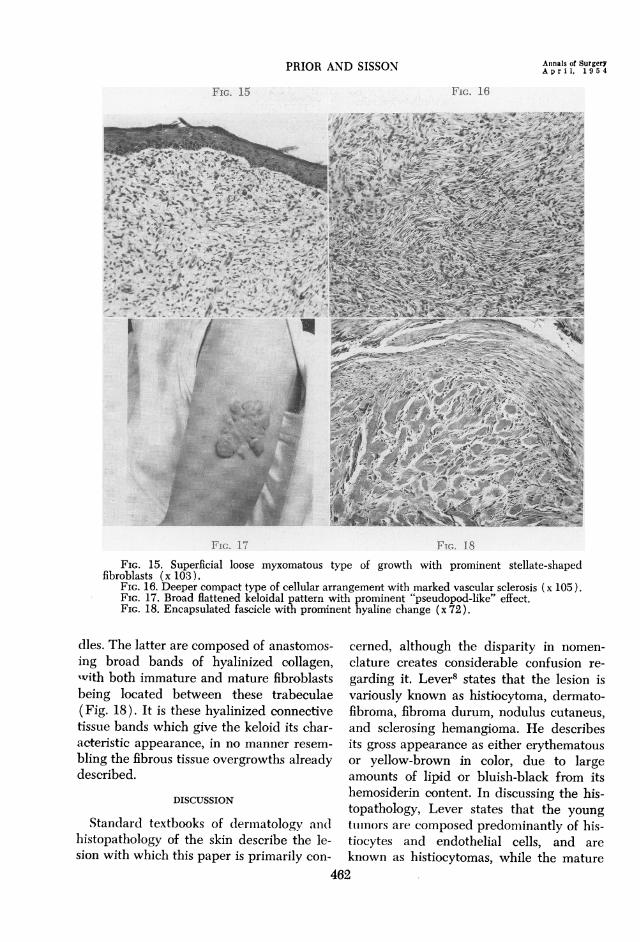
tive tissue growths referred to above. Thetypical dermatofibrosarcoma protuberansreveals two distinctive microscopic patternswithin the dermal connective tissue. Justbeneath and contiguous with a compressedepidermis, the tumor has a pale, loose myxo-matous appearance (Fig. 15). The cellular
elements in this zone are chiefly fibroblastsand rare mononuclear cells, the formerbeing characterized by abundant cytoplasmextending as stellate processes from the cellmargins. These cells are not, however, ar-ranged in fascicles, but tend to be scattereddiffusely throughout the edematous retic-ular type of stroma. Collagen fibers andelastic tissue is virtually absent. This zoneis relatively avascular and the connectivetissue cells have no constant relationshipto the few capillaries which are present.The second pattern is usually confined
to the reticular layer of the dermis, but mayextend into the subcutaneous adipose tissue(Fig. 16). The fibroblasts in this area are
more compactly arranged, spindle-shaped,and there is abundant collagenous tissuepresent. With low-power magnification one
gets the impression of attempted palisadingof these fibroblasts, an appearance some-
what suggestive of the structure seen inthe neurilemmoma. Many of the fibroblastsare concentrically arranged about minutecapillaries, the latter being present in largenumbers. It is this architecture which givesthe deeper portion of the lesion an appear-
ance not unlike that seen in sclerosinghemangiomas. Mitotic figures and cellularatypism are lacking, but it is the extremecellularity of the lesion that may cause itto be mistaken for a fibrosarcoma.
KELOID
The fact that some of the cases of fibro-matosis have been associated with a historyof preceding trauma is responsible for thefact that many of these tumors have beenclassified as keloids. Keloids tend to occur
in predisposed subjects, particularlyNegroes, and present as a broad, flat thick-ening of the skin in areas which have beenpreviously exposed to trauma (Fig. 17).Microscopically, the typical keloid consistsof broad connective tissue fascicles locatedwithin the dermis, and with a tendencytoward encapsulation of the individual bun-
461
Volume 139Number 4
PRIOR AND SISSON
FIG. 15
Annals of SurgeryApril, 1954
FIG. 16
FIG. 17 FIG. 18FIG. 15. Superficial loose myxomatous type of growth with prominent stellate-shaped
fibroblasts (x 103 ).FIG. 16. Deeper compact type of cellular arrangement with marked vascular sclerosis (x 105).FIG. 17. Broad flattened keloidal pattern with prominent "pseudopod-like" effect.FIG. 18. Encapsulated fascicle with prominent hyaline change (x 72).
dles. The latter are composed of anastomos-ing broad bands of hyalinized collagen,xvith both immature and mature fibroblastsbeing located between these trabeculae(Fig. 18). It is these hyalinized connectivetissue bands which give the keloid its char-acteristic appearance, in no manner resem-bling the fibrous tissue overgrowths alreadydescribed.
DISCUSSION
Standard textbooks of dermi-atology andhistopathology of the skin describe the le-sion with which this paper is primarily con-
cerned, although the disparity in nomen-clature creates considerable confusion re-garding it. Lever8 states that the lesion isvariously known as histiocytoma, dermato-fibroma, fibroma durum, nodulus cutaneus,and sclerosing hemangioma. He describesits gross appearance as either erythematousor yellow-brown in color, due to largeamounts of lipid or bluish-black from itshemosiderin content. In discussing the his-topathology, Lever states that the youngttumors are composed predominantly of his-tiocytes and endothelial cells, and areknown as histiocytomas, while the mature
462

DERMAL AND FASCIAL FIBROMATOSIS
tumors contain only fibroblasts and arecalled dermatofibromas. The description ofthe latter lesion appears consistent with thetumors we have described above, and Leveralso stresses its circumscribed nature andlack of mitotic figures and nuclear atypism.Ormsby and Montgomery13 refer to thegrowth as fibroma durum, obviously con-
sidering it a synonym for dermatofibroma,since they trace the historical backgroundof the latter lesion. It is of interest, how-ever, that their illustration of the micro-scopic features of this lesion is not at allconsistent with dermatofibroma, their pho-
tomicrograph being quite typical of derma-tofibrosarcoma protuberans. Furthermore,certain statements are found in their dis-cussion of fibroma durum which do not ap-pear to be in harmony with our observa-tions. Ormsby and Montgomery state thatthe tumor attains its size, remains station-ary, and that there is no need for treatmentexcept for cosmetic purposes or when thereis doubt as to the diagnosis. They add thatthere is general agreement that a definiteclinical differentiation between dermato-fibroma and histiocytoma cannot be made,but that the histiocytoma tends to be more
superficial, larger, elevated above the levelof the skin, and simulate solitary xanthomain oolor. Finally, they state that both con-
ditions (dermatofibroma and histiocytoma)can be distinguished from keloids by theiruniform size, contour, and lack of recur-
rence after excision.Becker and Obermayer2 make a distinc-
tion between fibroma durum and fibro-xan-thoma (histiocytoma). In their opinion theformer is only of cosmetic significance andwhen treatment is requested, the lesionmay be excised, electrocoagulated or cau-
terized. They state that the fibro-xanthomais characterized by cells containing rela-tively less lipid material than the ordinaryxanthomata. The histiocytes are not massed
together, but rather distributed uniformlythroughout the tumor, and they feel that
this tumor has a definite tendency to recur-
rence, unless properly treated.It would appear that the small space
devoted to the lesion in standard dermato-logical textbooks is but a reflection of thepaucity of reported cases, particularly inthe American literature. Traub and Mo-nash24 described two cases which clinicallyresembled Boeck's sarcoid. The lesions tookthe form of multiple tumors occurring inadults, and these had been present a longperiod of time, with itching being the onlysubjective symptom. No mention of recur-
rence is to be found in this report. Michel-son'0 described the lesion under the term"nodular subepidermal fibrosis," and statedthat the lesions are observed more fre-quently than the scanty literature on thesubject would indicate. He felt that this isthe same lesion that Unna25 named "fibromasimplex" or "fibroma durum," and statedthat it was characterized by an insidiousorigin, with possible trivial injury at thesite, a round or spherical shape, smooth redsurface, and lack of symptomatology. Indiscussing the histology of the tumorMichelson observed that the collagen bun-dles seemed to lose their normal form, andarrangement had tended to run in wavy
lines. In the more recent lesions, the con-
nective tissue cells were greatly increasedin number and gave the tumor a fibrosar-comatous appearance. Michelson's opinionwas that excision of the lesion was final andthat the tumors did not recur. In the dis-cussion that followed the presentation ofMichelson's paper, Ormsby echoed thesame sentiments, stating that the lesionsare always benign and that he had not seen
recurrences after excision. Perhaps the mostcomplete review in the literature is to befound in a paper by Senear and Caro en-titled "Histiocytoma Cutis."'I8 These authorspresent a detailed historical review of thelesion, with a study of 25 cases from theirown files. The writers stimulate additionalinterest, since they suggest a new concept
463
'oltziiie 139Number 4

PRIOR AND SISSON Annals of SurgeryA p r i l, 9 5 4
TABLE I. Summary of Cases.
Case No. Surgical No. Age First noted Sex Location Recurrences
I...................... 51-4206 7 yrs. M Left side of neck Yes (3)2. .52-717 2 yrs. F Right middle finger Yes (2)3. .................. 53-286 3 mos. M Right 3rd and 4th fingers Yes (2)4................ 53-675 27 yrs. F Left breast No5.................... CIH 53-386 66 yrs. F Base of right toe No6. .................. 52-270 8 yrs. M Left plantar fascia Yes (4)7...................... 43-1788 2 yrs. F Right thumb Yes (3)
Pulmonary metastasis8. .................. 52-3276 65 yrs. F Right plantar fascia Yes (1)9......... ..... 53-2489 29 yrs. F Right deltoid area No
10 .CIH 53-1365 26 yrs. M Right thumb No11 ............53-2468 41 yrs. F Right knee No
12 ......... ...........49-222516 yrs. F Right plantar fascia Yes (2)
of pathogenesis of the tumor in which therole of the histiocyte becomes paramount.With the use of the Sudan III stain, theywere able to demonstrate the presence ofintracellular lipoids in 15 cases. In three ofthe remaining ten in which they were ableto carry out vital staining, the histiocyticnature of the tumor cells was shown by thepresence of intracellular iron granules.They differentiated this lesion from thexanthoma in which the histiocytes are sodistended with lipoid that they form typicalfoam and giant cells. Furthermore, they dis-tinguish the histiocytoma from the fibroma,stating that the latter is more frequentlymultiple, softer than the histiocytoma clin-ically, and that the histologic differentiationcan be easily made. They cite Sezary andLevy Coblentz,19 who gave a detailed de-scription on the morphologic characteristicsof fibromas contrasted with histiocytomas.These French investigators observed thatin the fibroma the tumor cell is the fibro-blast, a fusiform or branching well withfibrillar or clear cytoplasm, and containinga spindle-shaped nucleus. There is practi-cally no variation in the nuclear and cyto-plasmic structure of the fibroblasts through-out the tumor, and the cells were separatedby fine collagenous lamellae. In the histio-cytoma, on the other hand, while the basiccell is also fusiform and is likely to anasto-mrose and forn whorls, the spindle is short
and thick and the cell is rich in cytoplasm.The nuclei are larger and less regular thanin the cells of a fibroma. Finally, theseauthors state that within the histiocytomathe cells sometimes form irregular clustersor are packed one against another withoutregular interposition of collagenous lamel-lae. In view of the close interrelationship,and in tissue cultures, actual transformationof histiocytes to fibroblasts, it may beas Senear and Caro8' suggest, that in abroad sense the histiocytoma is a fibromaarrested in evolution. In our experience, wehave not been able to distinguish eitherclinically or microscopically a lesion whichcorresponds with the so-called pure histio-cytoma-the lesion richest in histiocytes isfrequently the sclerosing hemangiomawhich should not be confused histologicallywith the fibromatoses. The cells comprisingthe subepidermal tumors we have studiedare morphologically fibroblastic, and theuse of iron stains and Sudan preparationshave been of no assistance to us in recog-nizing cells of supposed histiocytic naturein these tumors. In this connection it is per-tinent that Senear and Caro'8 deny thestatement that only histiocytes can storelipoids, and add that they have demon-strated the presence of subanophilic lipoidsand doubly refractile crystals within thetumor cells in cases of dermatofibrosarcomaprotuberans and of fibrosarcoma.
464

DERMAL AND FASCIAL FIBROMATOSIS
Should this group of dermal and fascialconnective tissue overgrowths be classifiedas low grade fibrosarcomas? As Stout20 haspointed out, a perusal of the literaturemakes it obvious that there are many differ-ent and oonflicting conceptions regardingwhat actually constitutes a fibrosarcoma.He states that a variety of both benign andmalignant lesions have been included underthe term, all of which serves to confuse our
understanding and makes its recognitionone of the most baffling problems of oncol-ogy. Stout divides tumors of fibroblasticorigin into three categories: non-neoplastichyperplasias, fibrous growth which may or
may not be neoplastic, and true fibrosar-comas. Hereditary polyfibromatosis is in-cluded in the first group, fibrous prolifera-tion of plantar and palmar fascia in thesecond, while the true fibrosarcomas haveas their outstanding feature, infiltrativegrowth. The latter may extend over a
period of many years, with static periodsintervening between times of more rapidproliferation. It has been pointed out thatwhile the microscopic diagnosis of thehigher grade tumor is relatively easy, theslower growing, well differentiated tumorsare frequently underestimated, particularlyif the number of mitotic figures alone istaken into consideration. Broders et al.4state that in the fibrous tumors of low ma-
lignancy the cellular elements closely re-
semble fibroblasts and show a moderate de-gree of variation in size and form. Thenuclei are oval, slightly lobulated or spin-dle-shaped, and the nuclear chromatinforms a delicate reticular network, thewhole structure not appearing very chro-
matic. One or more prominent basophilic or
achromatic nucleoli are frequently present.The cytoplasm is abundant, acidophilic,and drawn out into long processes at theends of the cells. In this type of tumor,cellular division occurs by mitosis and isnot usually very prolific. As the tumorascends the scale of malignancy, one notes
an increase in the cytoplasmic and nuclearvolumes, an increase in nuclear chromatin,gigantic cells with multiple nuclei andatypical multiple mitotic figures. Theseauthors state that recurrent tumors nearlyalways show the same structure as the pri-mary, and in persistently recurring tumorsthe structure and degree of malignancy are
essentially the same for each recurrence.
Likewise, Wilson26 has emphasized that a
clearcut differentiation between cellularfibromas and low-grade fibrosarcomas maybe impossible because of the lack of defi-nite criteria. He states that every cellularfibroma, no matter how casual its clinical or
histological appearance, should be consid-ered malignant until its subsequent behav-ior is established. In discussing the differen-tial diagnosis, Wilson points out that fibro-sarcomas may be differentiated fromfibromas by their size, more rapid expan-
sible type of growth, characteristic locationabout the thigh, shoulder, knee and elbow,and by their tendency to invade and de-stroy skin and bone. He adds that fibrosar-comas arising in the abdominal wall are
particularly difficult to differentiate grosslyfrom desmoid tumors, but he does not say
whether this can be done microscopicallyand, if so, what criteria are employed.Wide surgical excision, as employed in
the treatment of any potential malignancy,has been the treatment of choice in our
cases, although, particularly in children, re-
currences have occurred despite what was
felt to be adequate surgical removal. Theremay be, however, a certain understandableamount of reluctance to employ wide exci-sion in children, particularly where there isphalangeal involvement, and this may inpart explain their notorious tendency torecur. With repeated recurrences in a dig-ital extremity one should seriously con-
sider amputation with the realization, ofcourse, that the individual may developadditional similar primary tumors in otherlocations, Case 7, in which pulmonary
465
Volume 139Number 4

PRIOR AND SISSON ^Annals of SurgeryPRIORANDSISSON ~~~~~~~~~~Apr il, 1 9 54
metastases were discovered seven years fol-lowing amputation of the thumb, wouldindicate that even amputation may fail tocontrol the growth and that it may pursuethe characteristic course of a fibrosarcomain the very rare instance. The use of irra-diation in these cases in place of surgery ispredicated on its reported beneficial effectsin controlling the growth of keloids. In ourexperience it has been of no value either toobviate surgery or as pre- or postoperativetherapy, and furthermore there is someevidence to suggest that it may actually beharmful. Stout20 classifies one type offibrous hyperplasia and neoplasia as irradia-tion induced, and cites cases of psoriasisand hypertrichosis which, following irradia-tion therapy, developed connective tissueproliferation which he considered question-able fibrosarcomas in nature.
SUMMAY
A general review of dermal and fascialfibromatosis has been presented. Many ofthese connective tissue overgrowths, de-spite a benign histologic appearance, havebeen characterized by an insidious progres-sive course, repeated recurrences, and inone case by distant metastases. Our inves-tigations have suggested that the entity"infantile fibromatosis" is the same lesionvariously known as dermatofibroma andfibroma durum, although the lesion appearsto have considerably greater growth poten-tial when it occurs in the younger agegroups.The superficial or dermal form of the
lesion cannot be histologically distinguishedfrom the fascial and aponeurotic connectivetissue overgrowths variously known as pal-mar (Dupuytren's) and plantar fibroma-tosis, and from desmoid tumors, and theoccasional clinically malignant course ofthese latter neoplasms has been pointedouit. Dermatofibrosarcoma protuiberans andthe so-called keloid, although sometimesfollowing a clinical course similar to these
fibromatoses, can be readily distinguishedfrom them on a histologic basis. The stateof confusion regarding what constitutes alow-grade fibrosarcoma has been noted andthe impossibility of differentiating thesedermal and fascial overgrowths from fibro-sarcoma on a histologic basis has beenpointed out.
BIBLIOGRAPHY1 Binkley, G. W.: Dermatofibrosarcoma pro-
tuberans: Report of Six Cases. Arch. Dermat.and Syph., 40: 578, 1939.
2 Becker, S. W., and M. E. Obermayer: ModemDermatology and Syphilology. J. B. Lippin-cott Co., Philadelphia, 1947, ed. 2, pp. 672and 685.
3 Booker, R. J., and G. T. Pack: Desmomas of theAbdominal Wall in Children. Cancer, 4:1052, 1951.
4 Broders, A. C., R. Hargrave and H. W. Meyer-ding: Pathological Features of Soft TissueFibrosarcoma with Special Reference to theGrading of its Malignancy. Surg., Gynec.,and Obst., 69: 267, 1939.
5 Collins, N. C., and W. E. Anspach: Fibrosar-coma of Plantar Tissues. Am. J. Cancer, 40:465, 1940.
6 Kapiloff, B., and J. T. Prior: Fibromatosis inChildren. Plastic and Reconstructive Surgery,10: 276, 1952.
7 Keasbey, L. E.: Juvenile Aponeurotic Fibroma(Calcifying Fibroma). Cancer, 6: 338, 1953.
8 Lever, W.: Histopathology of the Skin. J. B.Lippincott Co., Philadelphia, 1949, ed. 1,p. 350.
9 Meyenburg, H., von.: In Henke and Lubarsch:Handbuch der speziellen pathologischenAnatomie und Histologie. Berlin. J. Springer,1929; vol. IX/I, p. 386.
10 Michelson, H. E.: Nodular Subepidermal Fibro-sis. Arch. Dermat. and Syph., 27: 813, 1933.
Mopper, C.: Primary Fibrosarcoma of the Skin.J. A. M. A., 152: 570, 1953.
12 Musgrove, J. E., and J. R. McDonald: Extra-abdominal Desmoid Tumors; Their Differen-tial Diagnosis and Treatment. Arch. Path.,45: 513, 1948.
13 Ormsbv, 0. S., and H. Montgomery: Diseasesof the Skin. Lea and Febiger, Philadelphia,1953, ed. 6, p. 704.
14 Pack, 0. T., and H. E. Ehrlich: Neoplasms ofthe Anterior Abdominal Wall with Snecial
466

Volume 139 DERMAL AND FASCIAL FIBROMATOSISNumber 4
Consideration of Desmoid Tumors. InternalAbst. Surg., 79: 177, 1944.
15 Pack, G. T., and E. J. Tabah: Dermatofibrosar-coma Protuberans: A Report of Thirty-nineCases. Arch. Surg., 62: 391, 1951.
16 Penner, D. W.: Metastasizing Dermatofibrosar-coma Protuberans: A Case Report. Cancer,4: 1083, 1951.
17 Pickren, J. W., A. G. Smith, T. W. Stevenson,Jr., and A. P. Stout: Fibromatosis of thePlantar Fascia. Cancer, 4: 846, 1951.
18 Senear, F. E., and M. R. Caro: HistiocytomaCutis. Arch. Dermat. and Syph., 33: 209,1936.
19 Sezary, A., and G. Levy-Coblentz: Fibroma enPastille and Histiocytoma. Bull. Soc. franc dedermat. et syph., 40: 1269, 1933.
20 Stout, A. P.: Fibrosarcoma; the MalignantTumor of Fibroblasts. Cancer, 1: 30, 1948.
21 --- Personal communication.22 Tanzer, R. C.: Dupuytren's Contracture. The
New England Journal of Medicine, 246: 807,1952.
23 Touraine, A., and H. Ruel: Hereditary Polyfibro-matosis. Ann. de dermat. et syph., 5: 1, 1945.
24 Traub, E. F., and S. Monash: Dermatofibroma.Arch. Dermat. and Syph., 26: 250, 1932.
25 Unna, P. G.: The Histopathology of the Dis-eases of the Skin, Translated by NormanWalker, Edinburg, W. F. Clay Co., 1896,p. 836.
26 Wilson, D. A.: Tumors of Subcutaneous Tissueand Fascia. Surg., Gynec. and Obst., 80:500, 1945.
467
Related Documents