REDEEMER’S UNIVERSITY COLLEGE OF NATURAL SCIENCES DEPARTMENT OF CHEMICAL SCIENCES INORGANIC CHEMISTRY IV COURSE: CHE421 COORDINATION CHEMISTRY NOTES 2017/18 SESSION SEMESTER 1 LECTURER PROFESSOR G A KOLAWOLE COMPILED BY PROFESSOR GA KOLAWOLE

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

REDEEMER’S UNIVERSITY
COLLEGE OF NATURAL SCIENCES
DEPARTMENT OF CHEMICAL SCIENCES
INORGANIC CHEMISTRY IV
COURSE: CHE421
COORDINATION CHEMISTRY NOTES
2017/18 SESSION
SEMESTER 1
LECTURER
PROFESSOR G A KOLAWOLE
COMPILED BY PROFESSOR GA KOLAWOLE

1. Purpose of the Course
To extend the concept of periodicity to the f-block elements;
To deepen students‟ knowledge of coordination chemistry, introduced from first year;
To introduce students to the new area of Inorganic reaction mechanism
2. Course Outcomes
By the end of the course students should be able to:
• Account for the physical and chemical properties of the lanthanides and actinides in
relation to their electronic configurations highlighting differences and similarities
with the d-block transition metals.
• Identify the reasons for the similarities and differences in the properties of elements in
the two series, and with d-transition metals.
• Account for the prevalence of +3 oxidation state in the lanthanides
• Discuss the basic theories of bonding in Coordination compounds, particularly VBT,
CFT and LFT, bringing out the limitations and strengths of each theory.
• Extract structural information from the physico-chemical analyses of coordination
compounds especially electronic spectra and magnetic susceptibility measurements.
• To be able to extract reaction mechanisms from rate laws in inert octahedral, square
planar and redox reactions.
3. Recommended textbooks:
The under-listed books are some of the books consulted in the preparation of this note and
they are acknowledged.
N N Greenwood and A Earnshaw, Chemistry of the Elements, 2nd
Edition, Elsevier, 2009.
J D Lee, Concise Inorganic Chemistry, 5th Edition, Blackwell, Oxford, 1996.
P Atkins, T Overton, J Rouke, M Weller, F Armstrong, Shriever & Artkins Inorganic
Chemistry, 4th
Edition (or later), Oxford, 2006.
G L Miessler and D A Tarr, Inorganic Chemistry, 3rd
Edition, Pearson, 2004 or later.

4. Course Outline
UNIT Coverage Completion
Week (No.
of lectures)
Unit 1:
Coordination
Chemistry
Synthesis of coordination compounds;
nomenclature;
isomerism;
theories of structure and bonding
Physical methods of structural investigation;
electronic spectra; spectro-chemical series;
Jahn-Teller, tetragonal and trigonal
distortions,
Chelate effect, thermodynamic stability of
complexes,
Magnetochemistry
Weeks 1-5
(15 hours)
ASSESSMENT 1 (60 MINUTES)
Unit 2: The
Chemistry of f-
block elements
Physical and chemical properties related to
electronic structures.
Comparative discussion of the chemistry of
lanthanides and actinides, including
differences and similarities with the d-
transition metals: variable oxidation states,
reactivity, complex formation, electronic and
magnetic properties
Extraction from ores (lanthanides) and
preparation of trans-uranium metals
(actinides), including revision of relevant
areas of radiochemistry required to understand
the preparation of trans-uranium metals..
Electronic properties of lanthanides and
actinides
Magnetic properties of lanthanides and
actinides
6-9
(12 lectures)
MID-SEMESTER ASSESSMENT (90 MINUTES)
Unit 3: Inorganic
reaction
mechanisms
Substitution reactions in inert octahedral
complexes
Substitution reactions in square planar
complexes
Redox reactions: inner sphere and outer
sphere mechanisms
10-12
(9 lectures)
REVISION Week 13
EXAMINATION Week 14
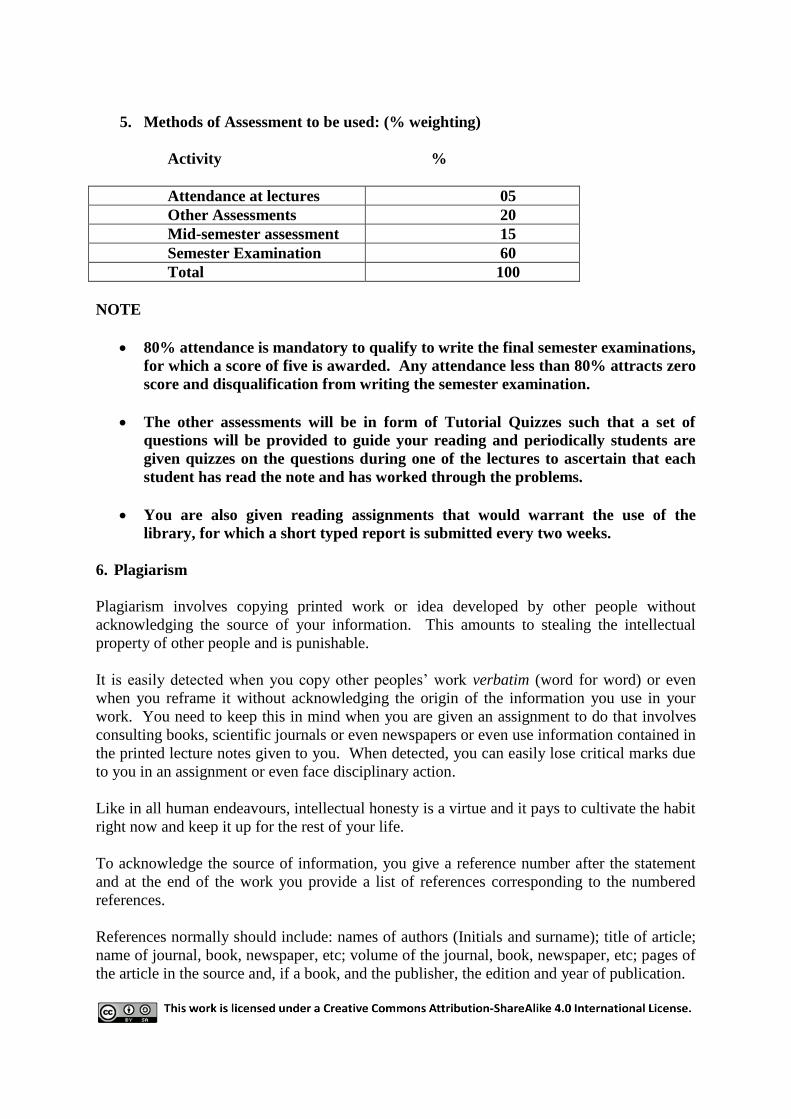
5. Methods of Assessment to be used: (% weighting)
Activity %
Attendance at lectures 05
Other Assessments 20
Mid-semester assessment 15
Semester Examination 60
Total 100
NOTE
80% attendance is mandatory to qualify to write the final semester examinations,
for which a score of five is awarded. Any attendance less than 80% attracts zero
score and disqualification from writing the semester examination.
The other assessments will be in form of Tutorial Quizzes such that a set of
questions will be provided to guide your reading and periodically students are
given quizzes on the questions during one of the lectures to ascertain that each
student has read the note and has worked through the problems.
You are also given reading assignments that would warrant the use of the
library, for which a short typed report is submitted every two weeks.
6. Plagiarism
Plagiarism involves copying printed work or idea developed by other people without
acknowledging the source of your information. This amounts to stealing the intellectual
property of other people and is punishable.
It is easily detected when you copy other peoples‟ work verbatim (word for word) or even
when you reframe it without acknowledging the origin of the information you use in your
work. You need to keep this in mind when you are given an assignment to do that involves
consulting books, scientific journals or even newspapers or even use information contained in
the printed lecture notes given to you. When detected, you can easily lose critical marks due
to you in an assignment or even face disciplinary action.
Like in all human endeavours, intellectual honesty is a virtue and it pays to cultivate the habit
right now and keep it up for the rest of your life.
To acknowledge the source of information, you give a reference number after the statement
and at the end of the work you provide a list of references corresponding to the numbered
references.
References normally should include: names of authors (Initials and surname); title of article;
name of journal, book, newspaper, etc; volume of the journal, book, newspaper, etc; pages of
the article in the source and, if a book, and the publisher, the edition and year of publication.

If the information is from a website, quote the website and the date you download the
information in addition to the above.
Coordination Chemistry
1.0 Preparation of coordination compounds
Preparation of compounds is the trade mark of every chemist. Research in chemical
industries is largely oriented toward the synthesis of new and useful materials. The chemist
is interested in preparing new compounds because it is an invaluable way of expanding our
knowledge of chemistry.
There are many routes, but related experimental methods to preparing metal complexes. The
method chosen depends upon the metal, the oxidation state of the metal, the ligand and the
electron configuration of the ion. Not all methods can therefore be employed to the synthesis
of a particular compound. Having found a suitable method for making the compound in good
yield one needs to find a suitable way to isolate the product from its reaction mixture.
Some of the commonly used techniques to obtain products from reaction mixtures are:
Evaporation of the solvent to concentrate (could be under reduced pressure usin a
rotary evaporator) and the cooling in an ice-bath (or a refrigerator). Adding a seed
crystal of the desired compound (if available, and often it is not available) or
scratching the inside of the beaker below the liquid surface helps to induce
crystallization.
A slow addition of a solvent that is miscible (but less polar) with the solvent of the
reaction mixture but which does not dissolve the desired product followed by cooling,
(seeding), and scratching.
For a cationic complex an appropriate anion with which it forms an insoluble salt can
be added. A suitable cation may be added to the reaction mixture containing an
anionic complex. E.g. to precipitate [Ni(CN)5]3-
from a solution add a large trivalent
cation like [Cr(en)3]3+
to give [Cr(en)3][Ni(CN)5].
Chromatography can be used to separate and purify complexes.
Other techniques are distillation (could be under reduced pressure if the compound
decomposes before its boiling point) and sublimation (for volatile complexes), and
Soxhlet extraction (of either the complex, if soluble in extractor solvent or of the
impurity if the complex is insoluble).
1.1 General principles of synthesis coordination compounds
There are two important variables that occur in reactions involving transition metals:
Coordination number
Oxidation state.
Either may increase, decrease, or remain unchanged in a reaction. It is, in practice, not
possible to predict either of these variables in a reaction. This is because ligands behave in
peculiar way depending on a number of constraints. E.g. a tetradentate ligand may behave as
a bidentate ligand. It is also possible for a ligand, which should be anionic, to coordinate
neutral or as a radical. Whether a reaction results in a change of oxidation state of the central
metal or not would depend on the mode of coordination of the resulting complex.

In general the following classifications hold:
Addition reaction: Coordination number of an electron acceptor (the metal/metal ion)
increases.
Substitution reaction: Coordination number is unchanged.
Dissociation reaction: Coordination number decreases.
Oxidation – reduction reaction: There is a change in oxidation state.
Coordination compounds are also classified according to the speed at which they undergo
substitution reaction:
Complexes that undergo substitution reaction at room temperature spontaneously are
said to be kinetically labile.
Those where substitution takes hours/days are said to be kinetically inert.
1.3 General rules guiding lability/inertness
1.3.1 Labile complexes
Complexes with central metal atom having d electrons in the eg orbitals, e.g.
[Ga(C2O4)3]3-
, d10
; [Co(NH3)6]2+
, d7+
; [Cu(H2O)6]2+
, d9; [Ni(H2O)6]
2+, d
8 and
[Fe(H2O)6]3+
, d5.
Complexes containing less than 3 electrons in the d orbitals , e.g. [Ti(H2O)6]3+
, d1;
V(phen)3]
3+, d
2 and [Ca(EDTA)]
2+, d
0+.
1.3.2 Inert complexes
Octahedral low-spin d4, d
5 and d
6 complexes, e.g. [Fe(CN)6]
3-; [Co(NH3)6]
3+ and
[PtCl6]2-
, d6.
Octahedral d3 complexes, e.g. [Cr(H2O)6]
3+, d
3.
Crystal field approach helps to see the picture clearly.
1.3.3 Addition reactions
Addition reactions lead to increase in coordination number, usually accompanied by colour
changes.
[ML4] + Y → [ML4Y]
[ML4Y] + Y → [ML4Y2]
Y is an adduct and can be the solvent molecule or another molecule, e.g.
[Cu(acac)2] + py → [Cu(acac)2py]
The product may or may not be isolable but the formation of the product can be detected
because of the change in coordination number.
1.3.4 Substitution reactions
Majority of complexes can be prepared by substitution reactions, in a number of cases,
displacing water. However, the method employed depends on whether the complex being
substituted is labile or inert.
1.3.5 Preparation of labile complexes

Formation of labile complexes is virtually instantaneous upon mixing of the reactants hence
there are few practical difficulties in their preparation, but three points must be remembered:
It is, in practice, difficult to prepare such complexes with several non-ionic ligands
bonded to the same metal atom, although anionic species may be coordinated together
with a neutral ligand.
Although it may be possible to isolate and characterize a solid complex quite a
different complex may be the predominant species in solution.
Some complex ions display incongruent solubility (arising from the second point
above), e. g. if an aqueous solution containing iron(II) sulphate and ammonium
sulphate in 1:1 ratio is allowed to crystallize then [Fe(H2O)6]SO4.(NH4)2SO4 is
formed. The iron(II) ammonium sulphate is said to show congruent solubility.
However, if solutions containing KCl and CuCl2 at ratio 2:1 are allowed to crystallize,
crystals of KCl are obtained first and only later does the complex K2[Cu(H2O)2Cl4]
crystallize. If the complex is re-crystallized there is an initial deposition of KCl again.
The complex is said to display incongruent solubility.
1.3.6 Basic principles for the preparation of metal complexes
Labile complexes are prepared in aqueous medium from hydrated salts.
Inert complexes are prepared from anhydrous complexes if non-aqueous medium is to be
used. Where only hydrated salts are available, salts have to be dehydrated first before use. If
preparation is to be done in aqueous medium then a labile complex of a lower oxidation state
is oxidized or a salt at a higher oxidation state is reduced.
Some ions are unstable to oxygen. Complexes of such ions are prepared in an inert
atmosphere, e.g. under N2 gas.
Examples
Substitution reaction in aqueous solution is the most common method for labile complexes.
The method involves a reaction between a metal salt in water and a coordinating agent.
1. Action of excess ammonia on aqueous solution of copper(II) salts:
[Cu(H2O)6]2+
+ 4 NH3(l) → [Cu(NH3)4]2+
+ 6 H2O
or
[Cu(H2O)6]2+
+ 4 NH3(aq) → [Cu(H2O)2(NH3)4]2+
+ 4 H2O
The instant replacement of water by ammonia at room temperature is shown by a change in
colour. Other species corresponding to stepwise substitution of the water by NH3 exist in
solution, e.g. [Cu(H2O)5(NH3)]2+
, [Cu(H2O)4(NH3)2]2+
, etc.
There are many labile complexes which may be studied readily in solution but which are very
difficult to obtain in the solid state.
1.3.7 Preparation of uncharged complexes
A neutral complex is usually precipitated from either aqueous solution or aqueous alcohol
and, unless highly polymeric, may be re-crystallized from organic solvents. For example,
[Cu(H2O)6]2+
+ 2 Hacac [Cu(acac)2] + 4 H2O + 2 H3O+

For this equilibrium to be displaced to the right (i.e. in favour of the formation of the
complex) the system is usually buffered to about pH 6. Sodium actate is commonly used.
1.3.8 Preparation of inert complexes
As mentioned earlier, substitution is usually slow and preparations can be done in three ways:
1. If water is not being displaced and reaction is being done in water more drastic
experimental conditions are imposed.
For example the preparation of K3[Rh(C2O4)3] is done in boiling concentrated aqueous
solutions of K3[RhCl6] and K2C2O4 for 4 h and then evaporated until product crystallizes
from the solution.
H2O, 4 h
100 oC
K3[RhCl6] + 3 K2C2O4 K3[Rh(C2O4)3] + 6 KClwine red yellow
To prepare [Co(en)3]Cl3 from [CoCl(NH3)5]Cl2, the rection is carried out on a steam bath
because the reaction is slow at room temperature;
[CoCl(NH3)5]Cl2 + 3 en [Co(en)3]Cl3 + 5 NH3
2. If water is being displaced then the water has to be replaced first before he correct
product can be obtained.
For example, Cr(III) complexes cannot be made from aqueous solvents if water is undesirable
in the complexes (alternative routes are available).
For example potassium thiocyanate (m.p. 173o) can be used as a solvent at elevated
temperatures above 173o. Under this condition water is readily displaced from [Cr(H2O)6]
3+.
[Cr(H2O)6]3+
+ 6 NSC-
180o
molten KCNS[Cr(NSC)6]
3-+ 6 H2O
In certain instances the salt is first dehydrated before the product can be obtained.
Dehydration can be effected by use of thionyl chloride or 2,2-dimethoxypropane. Preparation
of anhydrous complexes are best done from organic solvents and starting with, preferably,
anhydrous salts, where they are available.
Examples
1. Consider the preparation of (NEt4)2[NiCl4] from [Ni(H2O)6]Cl2.
[Ni(H2O)6]Cl2 + 6 SOCl2 NiCl2 + 6 SO2 + 12 HCl
NiCl2 + 2 NEt4ClSOCl2
Reflux (NEt4)2[NiCl4] Alternatively,
[Ni(H2O)6]Cl2 + 6 (MeO)2CMe2NiCl2 + 6 Me2CO + 12 MeOH

NiCl2 + 2 NEt4Cl (NEt4)2[NiCl4]
2. Preparation of [Cr(en)3]Cl3 from CrCl3.6H2O
[Cr(H2O)6]Cl3 + 6 (MeO)2CMe6 CrCl3 + 6 Me2CO + 12 MeOH CrCl3 + 3 en Cr(en)3]Cl3
1.3.9 Oxidation-reduction reaction in the preparation of inert complexes
Oxidation
Co(II) salts are usually used as starting materials for the preparation of Co(III) complexes;
CoCl2 + 2NH4Cl + 10 NH3 + H2O2 2 [Co(NH3)6]Cl3 + 2 H2OCharcoal
Charcoal acts as a catalyst. In its absence the product is mostly [Co(NH3)5X] complexes, X =
H2O or Cl.
If it is only hydrated salts of an inert complex that is available it is best to dehydrate the salt
first before use.
Reduction
The preparation of K3[Cr(C2O4)3] from K2Cr2O7
Here the dichromate is reduced by an aqueous solution of oxalic acid and potassium oxalate:
K2Cr2O7 + 7 H2C2O4 + 2 K2C2O42 K3[Cr(C2O4)3] + 6 CO2 + 7H2O
Note that in the instances where oxidation-reduction reactions are used the starting
compounds are labile. Cr(III) complexes can also be prepared from Cr(II). However, Cr(II)
compounds are rather unstable and can only be stored under inert atmosphere.
Thermal dissociation reactions
In certain instances a new complex can form by controlled heating of another complex,
usually with evolution of a volatile compound.
For example the preparation of anhydrous copper(II) sulphate from the hydrated salt is done
by controlled heating of the latter.
CuSO4.5H2O CuSO4.4H2O CuSO4.H2OCuSO4.3H2O
CuSO4
96.5o 102
o115
o
220o
The blue hydrated copper(II) sulphate loses water stepwise until all the water of
crystallization is lost to give the off-white anhydrous salt. Controlled heating under vacuum
is therefore a viable method for making a number of complexes.

Examples
The conversion of [Cr(en)3]Cl3 to cis-[Cr(en)2Cl2]Cl is done by controlled heating:
[Cr(en)3]Cl3 → cis-[Cr(en)3Cl2]Cl
1.3.10 Some methods employed in characterizing coordination compounds
Before embarking on characterization, compounds must be ascertained to be pure. To
establish purity the following could be done:
Determine the melting point. The melting point of a pure compound is expected to be
sharp. However, a sharp melting point does not necessarily refer to the melting point
of the compound anticipated. Where the melting point of the compound has been
reported, the melting point of the compound could be compared with the literature
value.
Microanalysis. The percentages of all the elements present in the compound are
determined and compared with the values calculated theoretically.
When the purity of the compound has been established the compound could then be
characterized using some of the following techniques:
Conductivity measurement. The molar ionic conductance of a compound (the
conductance of 1 mole of ions from the compound at infinite dilution) is determined.
The contribution to the molar ionic conductance of an ion Ix±
is about 60 ohm-1
cm2.
For ions Mm+
and Xn-
in the salt MnXm the contribution will be 60m ohm-1
(from Mm+
)
and 60n ohm-1
(from Xn-
). Multiplying by the number of ions of each sort and adding
leads to the conclusion that a salt MnXm will have a molar conductivity of about
120nm ohm-1
at 20 oC .
Example: The molar conductance of CoCl3.5NH3 is 261 ohm-1
cm2, hence
120nm = 261; nm = 2;
n = 1 and m = 2; n + m = 3.
That is, the number of ions is 3, hence the structure is [Co(NH3)5Cl]Cl2.
Generally m 120nm
Chemical reactions (already mentioned under introduction)
Infrared spectroscopy (i.r.): Normally the infrared spectrum of the ligands and the
complexes are required for meaningful comparison. The differences between the
spectra fall into four categories:
1. Band positions may change
2. Relative band intensities may change. Usually new, often weak, bands may
appear.
3. Single peaks in the free ligands may split into several, closely spaced, bands in
the complex.
4. Some peaks in the ligand may disappear while new ones, particularly in the region
due to Metal – Ligand bond (below 600 cm-1
may appear. Evidence for coordination and

atoms in coordination can be established from i.r. spectra. The mode of coordination of some
anionic ligands can also be detected.
Examples
CO32-
can be monodentate or bidentate. Free CO32-
absorbs at 890 cm-1
; coordinated
CO32-
absorbs at ~850 cm-1
(if monodentate) and at ~830 cm-1
(if bidentate).
SCN- can be S-bonded, where (C-S) is at ~700 cm
-1 or N-bonded where (C-S) is at
~820 cm-1
.
NO2- could be N-bonded [(N-O) is at ~1310 cm
-1] or O-bonded [(N-O) is at ~1065 cm
-
1].
Stretching frequency in metal-oxygen double bond, as in V=O occurs at 960±50 cm-1
.
Metal-ligand absorptions are generally weak and occur at 600 – 100 cm-1
region, which
may present instrumental problems. Usually any i.r. spectrometer that does not record to
200 cm-1
is of limited use in coordination chemistry. This is the region to concentrate on
for M-N, M-O, and M-X frequencies.
Infrared spectra for coordination compounds are usually recorded preferably in the solid
(KBr or CsBr pellets). There are overlaps when run in Nujol and one cannot go below
600 cm-1
in Nujol.
Other techniques that can be used in the characterization of coordination compounds include:
UV-Visible spectroscopy
Photoluminescence
Magnetochemistry
Thermogravimetric analysis
Differential thermal analysis
Cyclic Voltammetry (for oxidation-reduction properties of complexes)
Mass spectrometry
Nuclear magnetic resonance spectrometry
Mössbauer Spectrometry
Optical rotatory dispersion and circular dichroism (for optically active complexes)
X-ray diffraction (which gives the ultimate structure of the compound unequivocally)
2. Nomenclature
The International Union of Pure and Applied Chemistry (IUPAC) system will be discussed.
2.1 Naming ligands
Ligands can be anionic or neutral. Both anionic and neutral ligands can
be monodentate or polydentate.
2.1.1 Anionic ligands
Anionic ligands end in “-o”
Monodentate anionic ligands:
Ligand Name Ligand Name
Cl-
Chloro O2-
Oxo
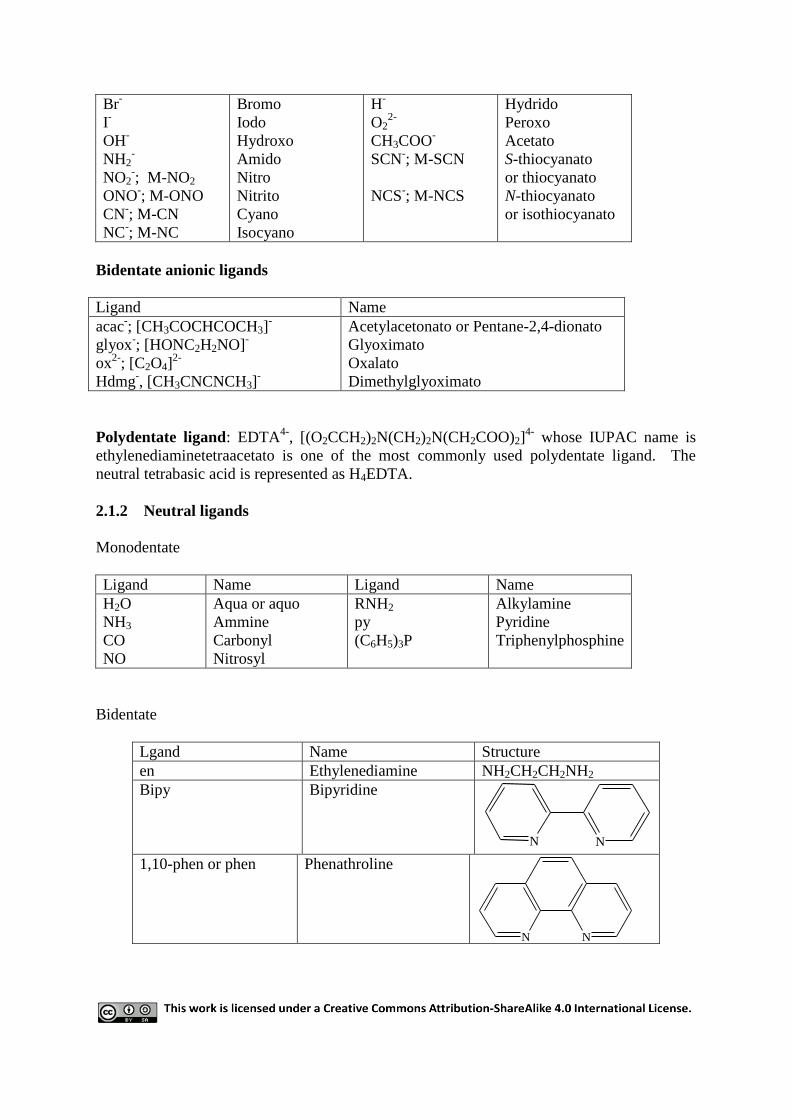
Br-
I-
OH-
NH2-
NO2-; M-NO2
ONO-; M-ONO
CN-; M-CN
NC-; M-NC
Bromo
Iodo
Hydroxo
Amido
Nitro
Nitrito
Cyano
Isocyano
H-
O22-
CH3COO-
SCN-; M-SCN
NCS-; M-NCS
Hydrido
Peroxo
Acetato
S-thiocyanato
or thiocyanato
N-thiocyanato
or isothiocyanato
Bidentate anionic ligands
Ligand Name
acac-; [CH3COCHCOCH3]
-
glyox-; [HONC2H2NO]
-
ox2-
; [C2O4]2-
Hdmg-, [CH3CNCNCH3]
-
Acetylacetonato or Pentane-2,4-dionato
Glyoximato
Oxalato
Dimethylglyoximato
Polydentate ligand: EDTA4-
, [(O2CCH2)2N(CH2)2N(CH2COO)2]4-
whose IUPAC name is
ethylenediaminetetraacetato is one of the most commonly used polydentate ligand. The
neutral tetrabasic acid is represented as H4EDTA.
2.1.2 Neutral ligands
Monodentate
Ligand Name Ligand Name
H2O
NH3
CO
NO
Aqua or aquo
Ammine
Carbonyl
Nitrosyl
RNH2
py
(C6H5)3P
Alkylamine
Pyridine
Triphenylphosphine
Bidentate
Lgand Name Structure
en Ethylenediamine NH2CH2CH2NH2
Bipy
Bipyridine
N N
1,10-phen or phen Phenathroline
N N

2.2 Naming complexes
2.2.1 If it is a salt:
Name cation first and then the anions, like in all salts. There is a space between the
cation and the anion.
Within a complex:
Name negative ligands.
Name the neutral ligands.
Name the metal (with oxidation state, in Roman numeral, in brackets).
Where there is more than one type of ligand in a complex, name in alphabetical order or in
order of complexity where they start with the same alphabet.
For the number of ligand (of the same type) use di-, tri-, tetra-, penta- and hexa- for 2, 3, 4,
5, and 6 respectively.
If the ligands are multi-syllabic put the name of the ligand in parenthesis. The numerical
prefixes are changed to bis-, tris-, tetrakis-, pentakis, and hexakis- for 2, 3… 6.
In anionic complexes the name of the metal ends “-ate”. In some cases the Latin name is
used; e.g. iron becomes ferrate.
In cationic complexes the metal retains its English name.
Examples
[Cr(NH3)6](NO3)3 hexaamminechromium(III) nitrate
K2[PtCl6] potassium hexachloroplatinate(IV)
K3[Fe(ox)3].3H2O potassium trioxalatoferrate(III), trihydrate or 3-water
Na[Co(CO)4] sodium tetracarbonylcobaltate(-I)
K4(Ni(CN)4] potassium tetracyanonickelate(0)
[Co(en)2Cl2]Cldichlorobis(ethylenediamine)cobalt(III) chloride
[Co(NO2)3(NH3)3] triamminetrinitrocobalt(III)
In a neutral complex, the ligands are named first adopting the rules above followed by the
metal.
Example
[Ni(Hdmg)2] bis(dimethylglyoximato)nickel(II)
2.2.2 Bridging complexes
Ligands that bridge two centres of coordination (polynuclear) are preceded by the Greek
letter, , which is repeated before the name of each different kind of bridging group.

[(H2O)4Fe
HO
OH
Fe(H2O)4](SO4)2
[(en)2Co
-dihyroxobis[tetraaquairon(III)] sulphate or tetraaquairon(III)--dihydroxotetraaquairon(III) sulphate
HN
OH
Co(en)2]Cl4
bis(ethylenediamine)--imido-m-hydroxo-bis(ethylenediamine)cobalt(III) chlorideor hydroxo--imidobis[bis(ethylenediamine)cobalt(III)] chloride
[(NH3)4Co
NO2
HN
Co(NH3)4] (NO3)4
-amido--nitrobis[tetraamminecobalt(III)] nitrate
2.2.3 Point of attachment
Whenever necessary the point of attachment of a ligand is designated by placing the symbol
(in italics) of the element attached after the name of the group is separated by hyphen.
(NH3)3[Cr(NCS)6] ammonium hexathiocyanato-N –chromate(III) or
hexaisothiocynatochromate(III)
(NH3)2[Pt(SCN)6] ammonium hexathiocyanato-S-platinate(IV)
2.2.4 Naming geometric isomers
Geometric isomers are generally named by the use of the terms cis to designate adjacent (90o
apart) positions and trans for the opposite (180o apart) positions. It is occasionally necessary
to use a number system to designate the position of each ligand. For square-planar
complexes, groups 1-3 and 2-4 are in trans positions. Note that only two of the
transpositions need be numbered in the name of the second complex below. This is because
in a square-planar complex the other two ligands must then be in trans positions. Since
positions 2 and 4 are equivalent these numbers need not be mentioned.

M
1
2
3
4
Pt
Cl
NH3
NO2
NH3 Pt
NH3
Cl NO2
Br
trans-diamminechloronitroplatinum(II) 1-ammine-3-bromochloronitroplatinum(II) ion
-
Number system in square-planar complexes
The number system for octahedral complexes has the trans positions numbered 1-6, 2-4, and
3-5.
M
12
3
4
5
6
Rh
NH3
NH3
NH3
NH3 Br
Br
Pt
Cl
NH3
py NO2
Br
I
cis-tetraamminedibromorhodium(III) ion1-ammine-2-bromo-4-chloro-6-iodonitro(pyridine)platinum(IV)or trans-ammineiodo-trans-bromochloronitro(pyridine)platinum(IV)
+
2.2.5 Naming optical isomers
If a solution rotates plane-polarised yellow light (the NaD line) to the right, the solute is
designated a (+) isomer; if to the left, a (-) isomer.
(+) K3[Ir(C2O4)3] potassium (+)trioxalatoiridate(III)
(-) [Cr(en)3]Cl3 (-)tris(ethylenediamine)chromium(III) chloride
3 Isomerism in metal complexes
Isomerism can be divided into two broad divisions: structural isomerism and
stereoisomerism.
Structural isomerism: Ionization isomerism, hydration isomerism, coordination isomerism,
linkage isomerism, ligand isomerism, and polymerization isomerism. Stereoisomerism:
Geometric isom

3.1 Structural isomerismerism, conformational isomerism, and optical isomerism.
3.1.1 Structural isomerism: ionization isomerism
Ionization isomerism results from the interchange of negative ligand within the first
coordination sphere of a complex that has an anion outside the coordination sphere. Such
isomers yield different ions in solution.
Examples:
[Co(NH3)4ClNO2]I and [Co(NH3)4ICl]NO2: When both are dissolved in water the first gives a
complex as cation and I- as anion whereas the second gives a complex as cation and NO2
- as
anion. Other examples are [Co(NCS)2(en)2]Cl and [Co(NCS)Cl(en)2]NCS; Pt(NH3)3Br]NO2
and [Pt(NH3)3NO2]Br. The isomers can be readily distinguished by appropriate qualitative
analysis.
[Co(NH3)5Br]SO4 and [Co(NH3)5SO4]Br: These can also be distinguished by appropriate
qualitative analysis.
Develop qualitative analysis schemes to distinguish between each pair of the compounds
above.
3.1.2 Structural isomerism: hydration isomerism/solvate isomerism
Hydration isomerism results from the interchange of H2O and another ligand between the
first coordination sphere and the ligand outside. Here H2O can be a ligand or water of
crystallization.
Most common example is CrCl3.6H2O, which can give three possible isomers,
distinguishable by their colours:
[Cr(H2O)6]Cl3 Violet
[Cr(H2O)5Cl]Cl2.H2O Blue green
[Cr(H2O)4Cl2]Cl.2H2O Dark green
Other examples are [CoCl(en)2H2O]Cl2 and [CoCl2(en)2]Cl.H2O; [CrCl2(py)2(H2O)2]Cl and
[CrCl3(py)2H2O].H2O.
The isomers can be distinguished by quantitative precipitation of free chloride using silver
nitrate.
Describe how you would carry out the quantitative precipitation of free chlorides in the
isomers in 5.2. Write appropriate equations and show how you would use your results to
distinguish the isomers.
3.1.3 Structural isomerism: coordination isomerism
Coordination isomerism occurs in salts in which both cation and anion are complex ions.
Isomerism arises from interchange of ligands between the two complex ions.
Examples:
[Co(NH3)6][Cr(ox)3] and [Cr(NH3)6][Co(ox)3]
[PtII(NH3)4][Pt
IVCl6] and [Pt
IV(NH3)4Cl2][Pt
II(NH3)4]
[Co(NH3)6][Cr(CN)6] and [Cr(NH3)6][Co(CN)6]
[Co(NH3)6][Co(NO2)6] and [Co(NH3)4(NO2)2][Co(NH3)2(NO2)4]
3.1.4 Structural isomerism: linkage isomerism
Linkage isomerism occurs when one or more of the ligands can coordinate to the metal ion in
more than one way. Linkages with two or more possible sites of attachment to a metal are
known as ambidentate ligands. In principle they include NO2-, SCN
-, CN
-, S2O3
2-, CO,

CONH2, CS(NH2)2, and (CH3)2SO but only the first four have been shown to form linkage
isomers.
Examples
[Co(NH3)5NO2]2+
and [Co(NH3)5ONO)]2+
are isomers . In the first one NO2- bonds to Co by
N (a nitro complex) whereas in the second it bonds by O (a nitrito complex). The complexes
can be distinguished by using IR spectroscopy. For the O-bonded ligand, characteristic
absorption bands at 1065 and 1470 cm-1
are observed whereas corresponding bands for the N-
bonded ligand are at 1310 and 1430 cm-1
. The above complexes can be written as
[Co(NH3)5(NO2-N)]2+
and [Co(NH3)5(NO2-O)]2+
respectively.
[Co(NH3)5SCN]2+
has two isomers, [Co(NH3)5SCN-S]2+
and [Co(NH3)5SCN-N]2+
, where
SCN- bonds via S and N respectively. Other examples can be found in complexes of CN
-
where the ligand can bond by C (cyno complexes) or N (isocyano complexes).
Other examples: [dipyPd(SCN)2] and [dipyPd(NCS)2]; [Mn(CO)5SCN] and [Mn(CO)5NCS].
All linkage isomers can be readily identified by IR spectroscopy.
e.g. M-N=C=S has a characteristic band at 780 – 860 cm-1
whereas in M-S-CN it occurs at
690 – 720 cm-1
.
3.1.5 Structural isomerism: polymerization isomerism
Polymerization isomerism refers to complexes, which have the same empirical formulae but
different molecular masses.
Examples:
[PtCl2(NH3)2] and [Pt(NH3)4][PtCl4] and [Co(NH3)3(NO2)3] and [Co(NH3)6][Co(NO2)6]
3.1.6 Structural isomerism: Ligand isomerism
Ligand isomerism is due to isomerism in the organic compounds that constitute the ligands.
Since many ligands are organic compounds and the latter have a large number of possibilities
for isomerism, the resulting complexes can show isomerism from this source.
Examples of isomeric ligands are 1, 2-diaminopropane (pn) and 1,3-diaminopropane (tn) or
o-, m- and p-toluidine (CH3C6H4NH2). When these compounds are used as ligands the
resulting complexes are also isomeric.
e.g. [Co(pn)3 and [Co(tn)3 are isomers.
3.2 Stereoisomerism
3.2.1 Stereoisomerism: geometrical isomerism
Geometrical isomerism occurs when a metal complex of the same formula and same basic
geometry has a different arrangement of ligands. This occurs in 4-coordinate square-planar
and 6-coordinate octahedral complexes.
3.2.2 Geometric isomerism in square planar complexes
cis and trans isomerism occurs when ligands are arranged adjacent (90o) and opposite (180
o)
respectively. Common geometric isomers are found in complexes of the type MA2B2. They
are more common in Pt and Pd complexes. Only two isomers are known in square-planar
complexes of this type. If the compound is tetrahedral only one isomer is possible..
A second type is of the form MABCD. Three square-planar complexes of this type is
possible. If it is tetrahedral, then only one isomer (which is also potentially optically active,
but not resolvable) is possible. The first type is illustrated below:

Pt Pt Pt
Cl
Cl
Cl
Cl
Cl
Cl
NH3 NH3 NH3 NH3NH3 NH3
The trans-isomer isnon-polar
Asymmetric stretchIR active
Symmetric stretchIR inactive
Cl
Cl
Cl
Cl ClPt Pt Pt
Cl
NH3
NH3
NH3
NH3
NH3
NH3
The cis-isomer is polar
Asymmetric stretchIR active
Symmetric stretchIR active
The cis- and trans-isomers of [Pt(NH3)2Cl2] can be distinguished by IR spectroscopy. An IR
active vibration leads to a change in molecular dipole moment.
The three isomers of the second type are given below.
M M M
A
B
C
D
A
C
B
D
A
B
D
C
3.2.3 Geometric isomerism in octahedral complexes
Two types of geometric isomerism are recognized in octahedral complexes: the simple types,
which exist in complexes of the type MA2B4, in which A may be adjacent to each other or
opposite and the second for complexes of the type MA3B2
MA2B4 type
An example is [Co(NH3)4Cl2]+

Cl
NH3
NH3
NH3NH3
Cl
Co
Cl
NH3
NH3NH3
Co
Cl
NH3
cis-isomer trans-isomer
MA3B3 type
Two types are possible: (1) here the ligands of one type forms an equilateral triangle on one
of the faces (the facial isomer, abbreviated as fac) and (2) the ligands span three positions
such that two are opposite, ortrans, to each other (the meridionalisomer, abbreviated as mer).
The following complexes can display the fac-mer isomerism: [Ru(H2O)3Cl3]; [Pt(NH3)3Br3]+;
[Pt(NH3)3I3]; [Ir(H2O)3Cl3]; [Rh(CH3CN)3Cl3]; [Co(NH3)3(NO2)3]; and [M(CO)3PR3], (M =
Cr, Mo, W).
Cl
H2O
Cl
ClH2O
H2O
Ru
OH2
Cl
ClCl
Ru
H2O
H2O
fac-isomermer-isomer
3.2.4 Stereoisomerism: conformational isomerism
Conformational isomerism occurs if a metal complex can exist in two totally different
geometric forms. For example, [NiCl2(Ph2PCH2Ph)2] can exist as square-planar in the solid
state and tetrahedral in solution.

Ni
P
P
Cl
Cl
Square-planar
Ni
Cl
PCl
P
Tetrahedral
P = PH2PCH2Ph
Properties of geometric isomers:
One isomer is usually stable in solid. In solution it often isomerizes to a mixture or
the other isomer. For example, green trans-[Co(en)2Cl2]+ isomerizes to a mixture of
cis and trans. The purple cis-isomer also isomerises to cis and trans-isomers.
Isomers usually have different colours.
Their chemical properties are usually different; for example they react at different
rates.
3.2.5 Stereoisomerism: optical isomerism
Optical isomerism is concerned with chirality.
A molecule is chiral if it possesses a non-superimposable mirror image. Octahedral
complexes, like [Co(acac)3], which has three bidentate chelating ligands also possesses non-
superimposable mirror images. Chiral molecules (enantiomorphs) rotate plane polarized
light in opposite directions. This property is known as optical activity and the two mirror
images are known as optical isomers or enantiomers.
Enantiomers rotate plane polarized light to equal extents in opposite directions, the
dextrorotatory (dor+) enantiomorphs to the right and the laevorotatory (lor-) to the left at a
particular wavelength. The observation of optical activity depends upon the chemical
properties of the chiral molecules; if the two enantiomorphs interconvert rapidly to give an
equilibrium mixture containing equal amounts of the two forms, there will not be any overall
rotation. A mixture of equal amounts of enantiomorphs is called a racemic mixture.
A polarimeter is used to measure the rotation, , of an enantiomorph. The amount of rotation
depends on the wavelength of the light, temperature, and the concentration of the compound.
The specific rotation, [], for a chiral compound in solution is given by:
[] =
c x l where = observed rotation, l = path length of solution in the polarimeter (in dm) and c =
concentration (in g cm-1
). Light of a single frequency is used for specific rotation
measurements and a common choice is sodium D-line in the emission spectrum of atomic
sodium; the specific rotation at this wavelength is denoted as []D. The importance of
chirality has been found in the dramatic differences in the activities of different enantiomers
of chiral drugs.
Read: E Thall (1996) Journal of Chemical Education, Vol. 73, p.481 – “When drug
molecules look in the mirror”.

3.3 Nomenclature of chiral molecules
Nomenclature of chiral molecule is complicated.
In terms of the sign of the rotation of plane-polarized light:
- the rotation is denoted (+) or d for dextrorotatory and (-) or lfor laevorotatory
- the sign and magnitude of rotation could be incorporated ; (-)589 or (-)D (where D is
sodium D-line at a wavelength of 589 nm)
This method of naming defined in terms of an observable (the rotation) does not bear any
direct relationship with the absolute configuration of the molecule.
IUPAC recommended and Δ system
3.3.1 Definitions and notation of chiral complexes
Enantiomers are a pair of stereoisomers that are non-superimposable mirror images.
Diastereomersare stereoisomers that are not an enantiomeric pairs.
(+) and (-)prefixes: the specific rotation of enantiomers is equal and opposite. Enantiomers
are distinguished by the sign of []D. Two enantiomers of a compound A with []D values of
+12 and -12 are denoted as (+)-A and (-)-A.
d and lprefixes: sometimes (+) and (-) are denoted by dextro- andlaevo- for right and left
rotations respectively.
The +/- or d/l notation, as mentioned above, has nothing to do with the absolute configuration
of an enantiomer (the arrangement of the substituents or ligands). The following prefixes are
used for describing absolute configuration.
R and S prefixes: this convention is used for labeling chiral carbon atoms (tetrahedral with
four different groups attached) and is based on the Cahn-Ingold-Prelog notation.
The four groups attached to the chiral carbon atom are prioritized according to the atomic
number of the attached atoms, highest priority being assigned to highest atomic number, and
the molecule then viewed down the C-X vector, where X has the lowest priority. The R- and
S-labels for the enantiomers refer to a clockwise (rectus) and anticlockwise (sinister)
sequence of the prioritized atoms, working from high to low. Example: CHClBrI, view down
the C-H bond:
C
I
H
BrCl
C
I
H
ClBr
1
23
1
2 3
R S
This notation is used for chiral organic ligands, and also for tetrahedral complexes.
Δ and prefixes: The enantiomers of octahedral complexes containing three equivalent
bidentate ligands (tris-chelate complexes) are among those which are distinguished using Δ

(delta) and (lambda) prefixes. The octahedron is viewed down a 3-fold axis, and the
chelates then define either a right- or a left-handed helix. The enantiomer with right-
handedness is labeled Δ, and that with left-handedness is .
=
=
Depending on the metal ion and the number and nature of chelate rings enantiomers can be
separated.
3.3.2 Rules for optical activity
An optically active compound must not have:
A centre of inversion
A plane of symmetry
An improper axis S
The mirror image must not be superimposable.
Examples of stereoisomers
[Co(en)2Cl2] can exist as cis- and trans-isomers. The cis isomer is potentially optically active
because its mirror image is not superimposable.
N Cl
N Cl
Co
N
N
NCl
NCl
Co
N
N
cis-[Co(en)2Cl2]
But the trans-isomer has a plane of symmetry and a centre of inversion. It is therefore not
optically active.

N N
N N
Co
Cl
Cl
trans-[Co(en)2Cl2]
The first compound to be resolved was cis-[Co(en)2(NH3)Cl]2+
. It was resolved with
(+)bromo-π-camphor sulphate. In the solid state, the enantiomorphs are stable. However,
they often racemize in solution and therefore may not be possible to isolate isomers even if
you know they exist.
The best complexes to isolate and keep in solution are those where
M is inert (undergoes slow substitution); e.g. CoIII
(d6) and Cr
III (d
3) complexes. For
example [Co(en)3]Cl3 is potentially active and boiling for hours will not convert one isomer
to the other. On the other hand [Zn(en)3]2+
racemises very rapidly because Zn2+
is a d10
ion
and is very labile.
Ligands are bidentate or polydentate. Such complexes are stabilized by chelate effect.
Good examples are found in EDTA complexes.
The first purely inorganic complex to be resolved into its optical isomers was [CoL3]6+
,
where L+
ligand = cis-[Co(NH3)4(OH)2]+ complex. L
+ chelates through the two O-donor
atoms.
Co
HO
OH
Co(NH3)4
3
6+
3.3.3 Methods for resolution of isomers
Most common methods used for least soluble diastereoisomers (cations and anions)
only, involve reacting the diastereoisomer with an optically active organic anion.
For example,
(±) C+ + optically active organic anion (+)A
- → (+)C
+.(+)A
- and (-)C
+.(+)A
-
Useful for diastereoisomeric salts (not mirror images).

Employing differences in solubility of the two diastereoisomeric salts
Example,
(±)[Co(en)3]3+
+ (+)tartrate2-
→ (+)[Co(en)3].(+)tartCl.5H2O precipitates but
(-)[Co(en)3].(+)tart stays in solution. This is a practical method.
The separated diastereoisomers must now be reconverted:
Example,
(-)[Co(en)3].(+)tart + NaI → (-)[Co(en)3]I3 (precipitated)
(+)[Co(en)3].(+)tart + xss KI → (+)[Co(en)3]I3
Other methods
Partial asymmetric synthesis
Example
(±)[Fe(phen)3]2+
+ (+)[SbO tartrate] → (-)[Fe(phen)3].(+)[SbOtart] ppt
(+) form still remains in solution.
This method only works if the metal complex has a certain amount of mobility. The
precipitate is fairly labile and on heating, the (+) form goes to (-) form, which is insoluble.
For anions the same technique is used except that organic cations are used as resolving
agents.
A metal complex which is optically active can itself be used as a resolving agent for
other metal complexes.
Examples,
Using (+)[Co(en)2(NO2)2]+ can be used to resolve [Co(en(ox)2]
- with 100% yield and
[Co(edta)]-. Once resolved these metal complexes can be used to resolve optically active
metal cations.
3.3.4 Resolving neutral metal complexes
The complex can be adsorbed on an optically active column like quartz, (+) lactose, or starch.
Example,
[Co(acac)3]0 on (+)lactose can be eluted with C6H6 or hexane. One isomer is preferentially
adsorbed and one is eluted.
4 Stereochemistry
Stereochemistry is that branch of chemistry that concerns with the structures of compounds.
Inorganic stereochemistry deals with central atoms having coordination numbers from two to
twelve.
4.1 Coordination number 2
Coordination number 2 is confined to complexes of CuI, Ag
I, Au
I, and Hg
I. The complexes
are all linear. Examples include ammine complex of AgI,
[NH3 → Ag NH3]+, and cyano complex of Ag
I, [NC Ag CN]
-. These ions have d
10
configuration. Other examples are [AgCl2]-, [HgCl2], [AuCl2]
- and Au(CN)2
-.
4.2 Coordination number 3 This is rather rare among metal complexes. Many complexes which appear to be 3-
coordinate as judged by their stoichiometry are found upon examination to have higher
coordination numbers. For examples, Cs[CuCl3] has an infinite chains –Cl-CuCl2-Cl-, a

coordination number of 4; K[CuCl3] has an infinite double chains of Cl4-Cu2Cl2-Cl4, a
coordination number of 6, having a distorted octahedral structure; and K[Cu(CN)2] has a
chain [-CN-Cu(CN)-CN-Cu(CN)-CN-) and is an example of true 3-coordination.
Four other examples of truly 3-coordinate complexes are the triiodomercurate(II) anion, HgI3-
, bis(thiourea)copper(I) chloride, [Cu(tu)2]Cl, tris(trimethylphosphinesulphidecopper(I)
perchlorate, [Cu(SPMe3)3]+ClO4
- and tris(triphenylphosphine)platinum(0), [{(Ph)3P}3Pt]
0.
All the examples feature ligands with bulky groups.
4.3 Coordination number 4
This coordination number is very common. The structures formed with this coordination
number can be divided into two: tetrahedral and square planar. There are, however,
intermediate structures and distortions.
4.4 Tetrahedral complexes
Tetrahedral structures are not stabilized by large CFSE (see later). The structure is favoured
by large ligands like Cl-, Br-, I
- and small metal ions of three types:
Those with a noble gas configuration ns2np
6 such as Be
2+, Mn
VII (e.g. MnO4
-)
Those with a pseudo-noble gas configuration ns2np
6(n-1)d
10, such as Cu
I, Zn
II Ga
III
and Ni0.
Those transition metal ions, which do not strongly favour other structures by virtue of
the CFSE such as CoII, d
7.
Specific examples for transition metal ions are MnO4-, Ni(CO)4 and [Cu(py)4]
+. Tetrahedral
complexes do not exhibit geometric isomerism, although they are potentially optically active
just like tetrahedral carbon.
Example: bis(benzoylacetonato)beryllium.
4.5 Square planar complexes
They are formed by very few metal ions. The best known are d8 species such as Ni
2+, Pd
2+,
Pt2+
, and Au3+
. There are a few complexes of Co2+
(d7) with bidentate ligands that are square
planar, but otherwise such complexes are rather scarce. Chlorophyll and other bio-complexes
are important exceptions to this rule where the geometry is dictated by the rigid porphyrin
structure. Square planar structure is favoured by non-bulky, strong field ligands with
sufficiently good -bonders to compensate for the energy „lost‟ through 4- rather than 6-
coordination. For example [Ni(CN)4]2-
is square planar whereas Ni2+
forms octahedral
complexes with H2O and NH3, and tetrahedral complexes with Cl-, Br
- and I
-. For the heavier
d8 metals steric requirements are relaxed and the effective field strength of all ligands is
increased. Under such conditions [PdCl4]2-
, [PtCl4]2-+
and [AuCl4]- are square planar.
Account for why Pd2+
and Pt2+
have stronger tendency to form square planar complexes
than Ni2+
.
There is only a small difference in the energy of square planar and tetrahedral complexes.
Both structures can therefore inter-convert easily. A number of Ni2+
complexes do inter-
convert readily.
In the M2[CuX4] series of complexes of CuII, variation of M
I and X gives complex anions
with stereochemistries ranging from square planar (e.g. (NH4)2 [CuCl4]) to almost tetrahedral
(e.g. Cs2[CuBr4].

Square planar complexes, of the formula [MA2B2], exhibit cis-trans isomerism (see next
chapter). If such complexes are neutral molecules, they may be readily distinguished by the
presence of a dipole moment in the cis isomer but none in the trans isomer.
4.6 Coordination number 5 The structures of 5-coordinate complexes lie between two limiting geometries: trigonal
bipyramidal and square pyramidal. These limiting structures are not markedly different. The
conversion of one structure into other requires a relatively slight distortion.
Trigonal bipyramidal Square pyramidal
Examples: [CdCl5]3-
is almost an ideal trigonal bipyramidal; [NiCl5]3-
is almost an ideal
square pyramidal.
4.7 Coordination number 6 This is the commonest and most important coordination number for transition metal
complexes. The geometry usually corresponds to six coordinated atoms at the corners of an
octahedron or a distorted octahedron.
A regular geometry
The octahedral geometry is often subjected to tetragonal distortion leading to elongation or
contraction of the axial bonds. Complexes of the type [MA6] can have regular geometry
whereas complexes of the types [MA5B], [MA4B2], etc cannot have regular octahedral
geometry because not all the bonds will have the same length.
5 BONDING
5.1 Valence Bond Theory (VBT); Revision
Valence bond theory assumes that bonding in coordination compounds is solely covalent.
The formation of coordinate compounds is described by the hypothetical sequence:
Removal of electrons from the metal to give the appropriate cation.
Hybridization of those atomic orbitals which will provide a set of equivalent hybrid
orbitals directed towards the ligands.
Where necessary, rearrangement of the metal‟s electrons in order to ensure that the
hybrid orbitals are empty.
Formation of covalent bonds by overlap of the hybrid orbitals with the ligand
orbitals containing the lone pair of electrons.

The common types of hybrid orbitals are given below:
Coordination No. Atomic orbitals Hybrid orbitals Geometry
2
3
4
4
5
6
spx
spxpy
spxpypz
spxpydx2
-y2
dx2
- y2spxpypz
spxpypzdx2
-y2dz
2
sp
sp2
sp3
sp2d or dsp
2
dsp3 or sp
3d
sp3d
2 or d
2sp
3
Linear
Trigonal planar
Tetrahedral
Square planar
Square pyramidal
or trigonal
bipyramidal
Octahedral
[Hybridization is mixing of orbitals to get hybrid orbitals. Hybridization is not a
phenomenon, but a mathematical manipulation. Hybrid orbitals have no physical existence in
reality]
Examples
Consider CoII complexes in 4- and 6- coordinate complexes:
Co
Co2+
sp3
XX XX XX XX
sp3 hybridization
3d7
4s2 4p
04d
0
- tetrahedaral

dsp2
sp3d
2
d2sp
3
XX XX
XX
XX XXXX XX XX XX
XX XX XX XX XX
XX XX
dsp2 hybridization
sp3d
2 hybridization - octahedral
d2sp
3 hybridization - octahedral
?
*
*
XX Lone pairs of electrons from the ligand.
* Rearranged electrons to create empty hybrid orbitals.
? The remaining electron from the CoII ion promoted to an unknown orbital.
- square planar
Note: Two types of octahedral complexes are apparent: one which uses only 3d orbitals and
the other which makes use of 4d orbitals for bonding. These have been described as
“covalent” or “inner orbital” and “ionic” or “outer orbital” respectively. In the inner orbital
case note the promotion of a single electron to an unspecified outer orbital. This is in
agreement with the case with which Co2+
complexes are oxidized to Co3+
.
Secondly the two forms of 6-coordinate and 4-coordinate complexes differ in the number of
unpaired, non-bonding electrons present. Since the magnetic moments of complexes are
dependent on the number of unpaired electrons, magnetic moments can provide useful
indications of structure and bond type.
Shortcomings of the valence bond theory
Other aspects of magnetic behaviours, such as variation of magnetic moment with
temperature are not explained by VBT.
Does not explain origin of colour in transition metal compounds. This is a major
shortcoming.
Does not explain why some ligands give rise to high-spin and some to low-spin
complexes.
Cannot distinguish two complexes with same number of unpaired electrons but with
different structures.
Examples
1. Discuss bonding in [Co(NH3)6]Cl3 and [CoF6]3-
which are diamagnetic and
paramagneticrespectively.

Co
Co3+
XX XX XX XX
3d 4s 4p 4d
[Co(NH3)6] XX XX
d2sp
3 hybridization; inner orbital octahedral complex; XX = NH3;
lone pair on N
Note that all electrons are paired, hence the complex is diamagnetic.
[CoF6]3-
XX XX XX
sp3d
2 hybridization; outer orbital octahedral complex; XX = F
-
XX XX XX
3d 4s 4p 4d
Note 4 unpaired electrons, paramagnetic. Magnetic moments can therefore be used to
distinguish between the two complexes.
2. Consider [Fe(H2O)6]3+
with 5 unpaired electrons and Fe(CN)6]3-
with one unpaired
electron. Use the valence bond theory to explain these observations.
3. There are three groups of complexes of Ni2+
; one group is diamagnetic and two
are paramagnetic. By considering 4-coordinate and 6-coordinate complexes, account for the
three groups of complexes.
6 Crystal Field Theory
6.1 Octahedral complexes
Crystal field theory (CFT) assumes that bonding in coordination compounds is electrostatic
(i.e. ionic). The d-orbitals fall into two groups: those whose orbitals point along the axes (dx2
-
y2 and dz
2) and those whose orbitals point between axes (dxy, dyz, dxz). The shapes of the d-
orbitals are given (see page 25).
In gaseous metal ion, the 5 orbitals are degenerate (i.e. of the same energy). If a spherically
symmetric field of negative charges is placed around the metal ion, the energy of all the
orbitals will be raised equally. This is as a result of the repulsion between the negative field
and the electrons of the orbitals. When the field results from the influence of real ligands the
symmetry must be less than symmetrical because of the finite number (usually 4 or 6) of
ligands involved. When, for example, six ligands approach to form an octahedral complex,
there are two types of d-orbitals, the dx2
-y2 and dz
2, which have lobes point along the x-, y-,
and z- axes and dxy, dyz, and dxz whose lobes point between axes. If 6 ligands are imagined to
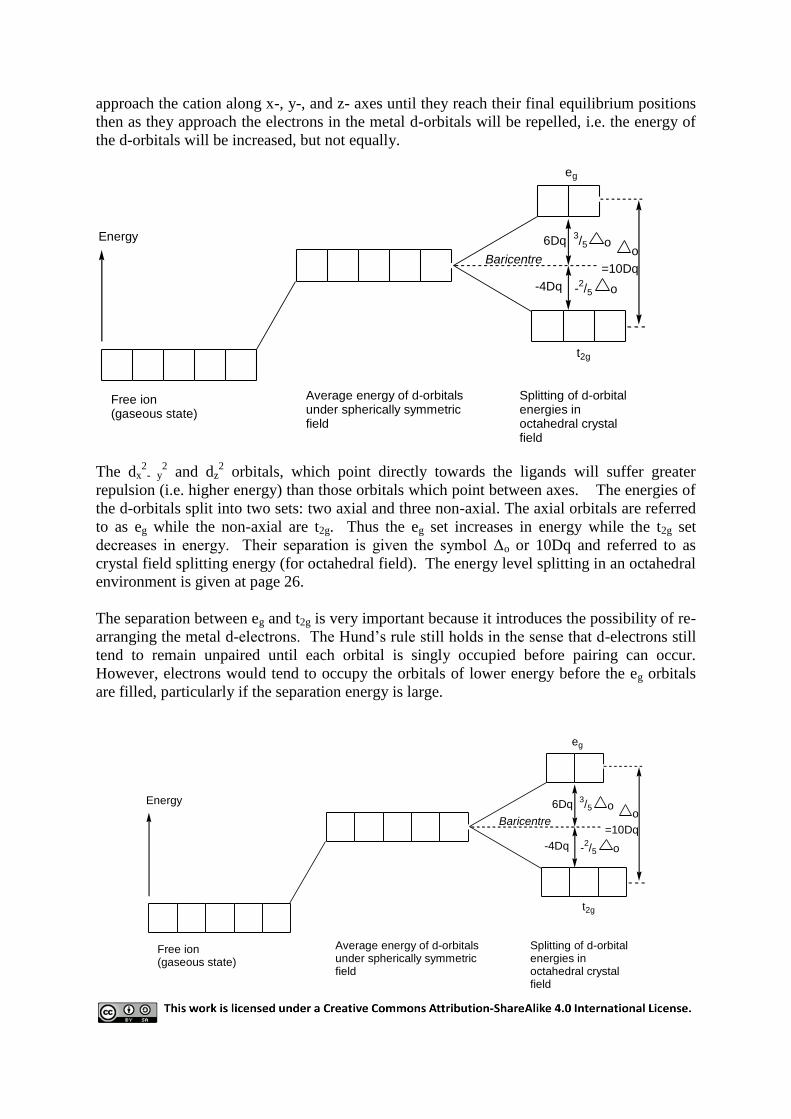
approach the cation along x-, y-, and z- axes until they reach their final equilibrium positions
then as they approach the electrons in the metal d-orbitals will be repelled, i.e. the energy of
the d-orbitals will be increased, but not equally.
6Dq
-4Dq
=10DqBaricentre
eg
t2g
Free ion(gaseous state)
Average energy of d-orbitalsunder spherically symmetric field
Splitting of d-orbital energies in octahedral crystal field
Energyo
o
o3/5
-2/5
The dx2
- y2 and dz
2 orbitals, which point directly towards the ligands will suffer greater
repulsion (i.e. higher energy) than those orbitals which point between axes. The energies of
the d-orbitals split into two sets: two axial and three non-axial. The axial orbitals are referred
to as eg while the non-axial are t2g. Thus the eg set increases in energy while the t2g set
decreases in energy. Their separation is given the symbol Δo or 10Dq and referred to as
crystal field splitting energy (for octahedral field). The energy level splitting in an octahedral
environment is given at page 26.
The separation between eg and t2g is very important because it introduces the possibility of re-
arranging the metal d-electrons. The Hund‟s rule still holds in the sense that d-electrons still
tend to remain unpaired until each orbital is singly occupied before pairing can occur.
However, electrons would tend to occupy the orbitals of lower energy before the eg orbitals
are filled, particularly if the separation energy is large.
6Dq
-4Dq
=10DqBaricentre
eg
t2g
Free ion(gaseous state)
Average energy of d-orbitalsunder spherically symmetric field
Splitting of d-orbital energies in octahedral crystal field
Energyo
o
o3/5
-2/5
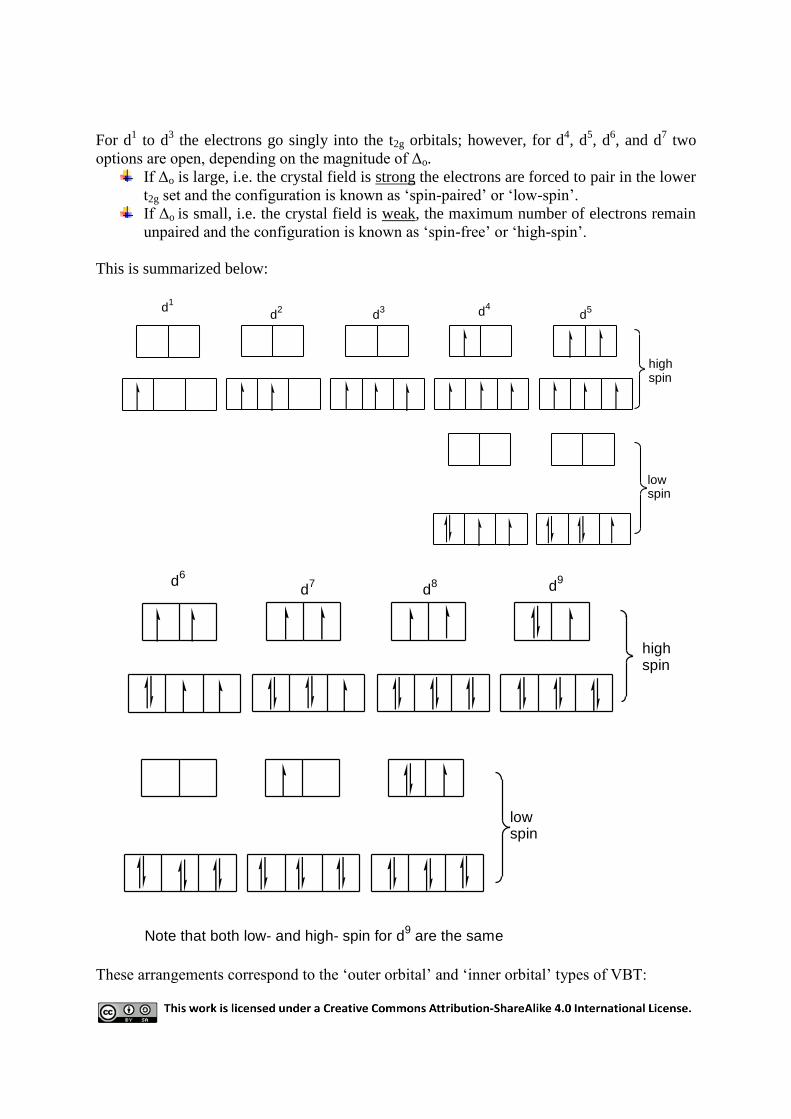
For d1 to d
3 the electrons go singly into the t2g orbitals; however, for d
4, d
5, d
6, and d
7 two
options are open, depending on the magnitude of Δo.
If Δo is large, i.e. the crystal field is strong the electrons are forced to pair in the lower
t2g set and the configuration is known as „spin-paired‟ or „low-spin‟.
If Δo is small, i.e. the crystal field is weak, the maximum number of electrons remain
unpaired and the configuration is known as „spin-free‟ or „high-spin‟.
This is summarized below:
d1
d2
d3 d
4d
5
lowspin
highspin
d6
d7
d8 d
9
lowspin
highspin
Note that both low- and high- spin for d9 are the same
These arrangements correspond to the „outer orbital‟ and „inner orbital‟ types of VBT:

Outer orbital High spin
Inner orbital Low spin
Thus the vacant orbital „created‟ to allow for vacant d-orbitals required for hybridization (i.e.
d2sp
3) in VBT is equivalent to the empty eg orbital in low-spin configuration in CFT.
For example, Co2+
octahedral complex in CFT would generate two configurations:
oo
eg
t2g
eg
t2g
Co
2+ d
7 high spin VBT outer orbital Co
2+ d
7 low spin VBT inner orbital
Also note that the electron promoted to unknown orbital in VBT is actually in the eg level in
CFT and that Δo for high-spin complexes is smaller than Δo for low-spin.
Other examples:
Use the crystal field theory to account for
(a) [Fe(H2O)6]3+
having 5 unpaired electrons whereas [Fe(CN)6]3-
has one unpaired
electron.
(b) [Co(NH3)6]3+
is diamagnetic whereas [CoF6]3-
is paramagnetic.
6.2 Formation of tetrahedral complexes
Bonding in tetrahedral complexes occurs essentially the same way as in octahedral complexes
except that electrons approaching between axis (now t2) will be more strongly repelled than
those along axis (now e). The d-orbital splitting is thus an inverted form of the octahedral.

Baricentre
Energy
t
t2
e
-3/5
t2/5
t
The separation energy between e and t2 levels is Δt. Both Δo and Δt are called crystal field
splitting energies for octahedral and tetrahedral geometries respectively. For the same metal,
at the same oxidation state, and the same ligands, Δt -4/9Δo.
The occurrence of high-spin and low-spin configurations for d3, d
4, d
5 and d
6 ions is also
possible in principle but rather rare to have low-spin tetrahedral complexes because of the
small crystal field splitting energy involved.
Reasons why Δt is smaller than Δo
Four ligands are involved in tetrahedral complexes as against six in octahedral.
Larger crystal field is therefore expected for six ligands than for four.
The ligands in tetrahedral complexes are less efficiently directed (between axes) than
they are in octahedral (along axes). The ligands are therefore not aimed at any of the
orbitals but exert a somewhat larger influence on the t2g orbitals than the eg orbitals.
6.3 Formation of square-planar complexes
The square-planar geometry is usually conceived as an octahedral complex where the axial
ligands have been elongated to infinity (i.e. totally removed). The effect of this is to reduce
the energy of the dz2 orbital (which points along the vertical axis). This would have the effect
of lowering the energy of the dxz
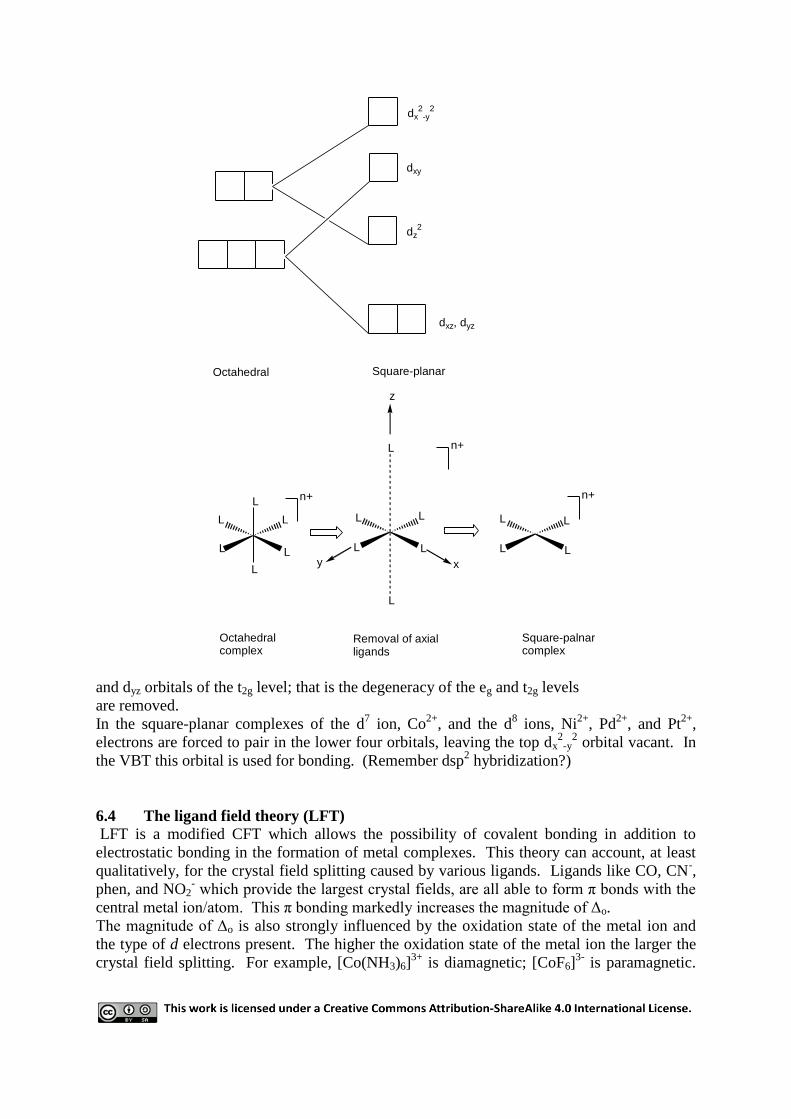
dx2
-y2
dxy
dz2
dxz, dyz
Octahedral Square-planar
n+
n+
n+
L
L
L
L
L
L
L
L
L
L
L
L
L L
L L
z
xy
Octahedralcomplex
Removal of axialligands
Square-palnarcomplex
and dyz orbitals of the t2g level; that is the degeneracy of the eg and t2g levels
are removed.
In the square-planar complexes of the d7 ion, Co
2+, and the d
8 ions, Ni
2+, Pd
2+, and Pt
2+,
electrons are forced to pair in the lower four orbitals, leaving the top dx2
-y2 orbital vacant. In
the VBT this orbital is used for bonding. (Remember dsp2 hybridization?)
6.4 The ligand field theory (LFT)
LFT is a modified CFT which allows the possibility of covalent bonding in addition to
electrostatic bonding in the formation of metal complexes. This theory can account, at least
qualitatively, for the crystal field splitting caused by various ligands. Ligands like CO, CN-,
phen, and NO2- which provide the largest crystal fields, are all able to form π bonds with the
central metal ion/atom. This π bonding markedly increases the magnitude of Δo.
The magnitude of Δo is also strongly influenced by the oxidation state of the metal ion and
the type of d electrons present. The higher the oxidation state of the metal ion the larger the
crystal field splitting. For example, [Co(NH3)6]3+
is diamagnetic; [CoF6]3-
is paramagnetic.

Ions with high charge and small sizes can be approached more strongly by ligands than larger
cations of smaller charge.
It is also known that the greatest crystal field splitting is observed in complexes of 5d metals
and decreases from 4d to 3d. Thus Δo([Ir(NH3)6]3+
) > Δo([Rh(NH3)6]3+
) > Δo([Co(NH3)6]3+
)
The arrangement of the ligands in the order of the magnitude of their Δo is known as the
spectrochemical series and follows the order:
Weak field ligands Intermediate field ligands Strong field ligands
I-<Br
-<Cl
-<OH
-<RCO2
-,F
-< Ox
-<H2O<NH3<en< NO2
-<phen<CN
-CO
To account for this order the extreme assumption of pure covalency in the VBT and pure
electrostatic model of CFT must be abandoned for the existence of both modes of bonding in
metal complexes.
Values of Δo for selected metal complexes
Complex Δ/cm-1
Complex Δ/cm-1
[TiF6]3-
[Ti(H2O)6]3+
[V(H2O)6]3+
[V(H2O)6]2+
[CrF6]3+
[Cr(H2O)6]3+
[Cr(H2O)6]2+
[Cr(NH3)6]3+
[Cr(CN)6]3-
[MnF6]2-
[Fe(H2O)6]3+
[Fe(H2O)6]2+
17 000
20 300
17 850
12 400
15 000
17 400
14 100
21 600
26 600
21 800
13 700
9 400
[Fe(ox)3]3-
[Fe(CN)6]3-
[Fe(CN)6]4-
[CoF6]3-
[Co(NH3)6]3+
[Co(NH3)6]2+
[Co(en)3]3+
[Co(H2O)6]3+
[Co(H2O)6]2+
[Ni(H2O)6]2+
[Ni(NH3)6]2+
[Ni(en)3]2+
14 100
35 000
33 800
13 100
22 900
10 200
24 000
18 200
9 300
8 500
10 800
11 500
Some applications of CFT
CFT provides explanations for
Colour in metal complexes;
Magnetic properties of metal complexes;
Determining the structure of complexes.
7 Colour
7.1 Origin of colour
Colour in metal complexes arises from
Transitions between metal-centred orbitals possessing d-character (d-d transitions).
Transitions between metal- and ligand-centred MOs which transfer charge from
metal-to-ligand or ligand-to-metal (charge-transfer transitions).
7.2 Selection rules
Electronic transitions obey the following selection rules:

Spin selection rule: ΔS = 0
Transitions may occur from singlet to singlet, or triplet to triplet states and so on, but a
change in spin multiplicity is forbidden.
Laporte selection rule: There must be a change in parity:
allowed transitions: g u
forbidden transitions: g g and u u
This leads to the selection rule: Δl = ± 1
and thus allowed transitions are s → p, p → d, d→ f;
forbidden transitions are s → s, p → p, d → d, f → f, s → d, p → f, etc.
7.3 d-d transitions
d-d transitions are generally weak with 100 and broad. This is because they are forbidden
transitions. The selection rule Δl = ±1 is broken because for d orbital l = 2; Δl = 0. Colour
is due to transitions between energy levels, e.g. between t2g and eg. Absorption of a particular
quantum of energy (Δ = hυ) causes promotion of an electron from t2g level to eg level. The
value of Δ is such that the energy lies in the visible part of the spectrum.
At 1kJ 83.7 cm-1
; Δo for [Ti(H2O)6]3+ 244 kJ. For this ion Δo = 10Dq = 20 300 cm
-1. Δ
has been measured for most ligands on a wide range of metals. Wavelengths () can be
converted to wavenumbers ( ) by the equation:
1
c=
Absorption bands are described in terms of max corresponding to the absorption Amax. The
unit of is nm while that of wavenumbers is cm-1
. If the electronic spectrum is done in a
solution, the extinction coefficient max of the absorption must also be reported. max indicates
the intensity of the absorption and is related to Amax.
max =Amax
c x l(max in dm
3 mol
-1cm
-1)
Generally the electronic spectra of
d1
, d4, d
6 and d
9 complexes consist of one absorption;
d2, d
3, d
7, and d
8 complexes consist of three absorptions;
d5 complexes consist of a series of very weak, relatively sharp absorptions.
7.4 What does Δ depend on?
It depends on the nature of metal within a row. In the first row transition metals Δo
for M2+
is 10 000 cm-1
and for M3+
17 000 cm-1
. Δo therefore depends on the oxidation
state of the metal ion. Δo for second row transition metals is 50% larger than those of
first row, i.e. Δ increases down a transition metal triad (3rd row TM‟s > 2nd row TM‟s >
1st row TM‟s.
For example: [M(NH3)6]3+
, Ir > Rh > Co
41 000 34 000 23 000 cm-1
Greatly on the nature of the ligand. Δ increases along the spectrochemical series: I-<
Br-< Cl
-< F
-< OH
-< Ox
-< H2O < NH3< en < phen < NO2
-< CN
-
All these facts enable the colours of complexes to be predicted.

[Ti(H2O)6]3+
, 490 nm; [TiCl6]3-
, >490 nm; [Ti(CN)6]3-
, <490 nm.
Hole formalism
d10-n
configuration behaves in CF similarly to dn configuration.
7.5 Charge-transfer transition
There are two types: metal-to-ligand (M→L) oxidation
or ligand-to-metal (M → L) reduction, which occurs usually as s → p or p → d or d → p.
These are allowed transitions and are therefore very intense.
[Ti(H2O)6]3+
, a d1 ion, is purple whereas [Ti(H2O)6]
4+, a d
0 ion, is yellow. The purple colour
in the former is due to d-d transition, whereas the yellow colour in the latter is due to charge-
transfer transition.
8 Thermodynamic effect on crystal field splitting
The separation of d-orbitals into t2g and eg levels translates into making the t2g orbitals more
stable and the eg orbitals less stable than the degenerate orbitals. Consequently a t2g electron
increases the stability of an octahedral complex by 2/5Δo and an eg electron decreases it by
2/5Δo. The net effect of all the d electrons represents the additional stability which may be
thought to accrue because the CF splits the d-orbitals. This is known as Crystal Field
Stabilization Energy, CFSE. This extra stability reflects in experimental values of such
thermodynamic quantities as hydration energies, lattice energies, and standard reduction
potentials. The most complete set of data is the hydration energies of bivalent ions: the
experimental values of ΔHo lie on an irregular double-humped curve.
If the CFSE is, in each case, subtracted from ΔHo, the „corrected‟ values fall very neatly on a
smooth curve, passing through the points for the spherically symmetrical d0, d
5, and d
10 ions.
CFSE, therefore, offers a ready explanation of the Irving-Williams order of stability, MnII<
FeII< Co
II< Ni
II< Cu
II> Zn
II.
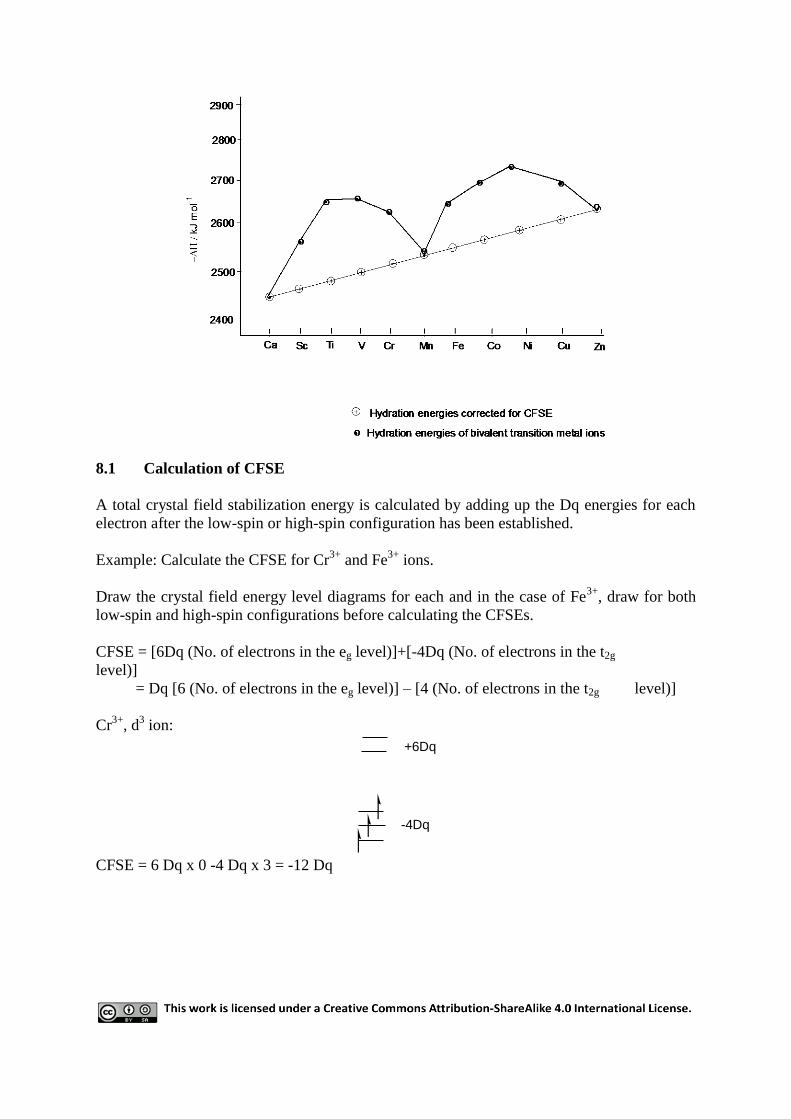
8.1 Calculation of CFSE
A total crystal field stabilization energy is calculated by adding up the Dq energies for each
electron after the low-spin or high-spin configuration has been established.
Example: Calculate the CFSE for Cr3+
and Fe3+
ions.
Draw the crystal field energy level diagrams for each and in the case of Fe3+
, draw for both
low-spin and high-spin configurations before calculating the CFSEs.
CFSE = [6Dq (No. of electrons in the eg level)]+[-4Dq (No. of electrons in the t2g
level)]
= Dq [6 (No. of electrons in the eg level)] – [4 (No. of electrons in the t2g level)]
Cr3+
, d3 ion:
-4Dq
+6Dq
CFSE = 6 Dq x 0 -4 Dq x 3 = -12 Dq

-4Dq
+6Dq
-4Dq
+6Dq
Weak field
Strong field For weak field
CFSE = 6 Dq x 2 – 4 Dq x 3 = 12 Dq – 12 Dq = 0 Dq
For strong field
CFSE = 6 Dq x 0 – 4 Dq x 5 = -20 Dq
The lower the CFSE, the more stable the complex. Thus low-spin Fe3+
is more stable than
high-spin Fe3+
. Also low-spin Fe3+
ion is more stable than Cr3+
ion.
9 Jahn - Teller distortion
A regular octahedral environment is the most stable one for a spherically symmetrical metal
ion surrounded by six donor atoms. For metal ions with certain d electron configurations
which are not spherically symmetric, the regular octahedral configuration is not the most
stable. This situation is expressed in Jahn – Teller theorem: Any non-linear molecule that is
in an electronically degenerate state will undergo distortion to lower the symmetry, remove
the degeneracy, and lower the energy. Jahn – Teller distortion occurs strongly in all cases
where the eg level is not uniformly occupied by electron. Much weaker distortion also occurs
if the t2g level is not uniformly occupied.
In the case of eg, any odd electron can occupy either the dz2 or the dx
2-y
2 orbital. If, however,
the complex undergoes distortion the eg level is split and the electron can occupy the lower of
the two orbitals (the dz2 orbital in the case of tetragonal elongation, or the dx
2-y
2 orbital in the
case of tetragonal compression).
In tetragonal elongation the ligands on the z axis move out and therefore interact less with
those orbitals which have a z component; i.e. the dz2, dxz, and dyz, and these orbitals attain
lower energy (i.e. stabilized). Those orbitals without a z component, i.e. dx2
-y2 and dxy, will
be raised a corresponding amount.
Elongation: dz2 attains lower energy
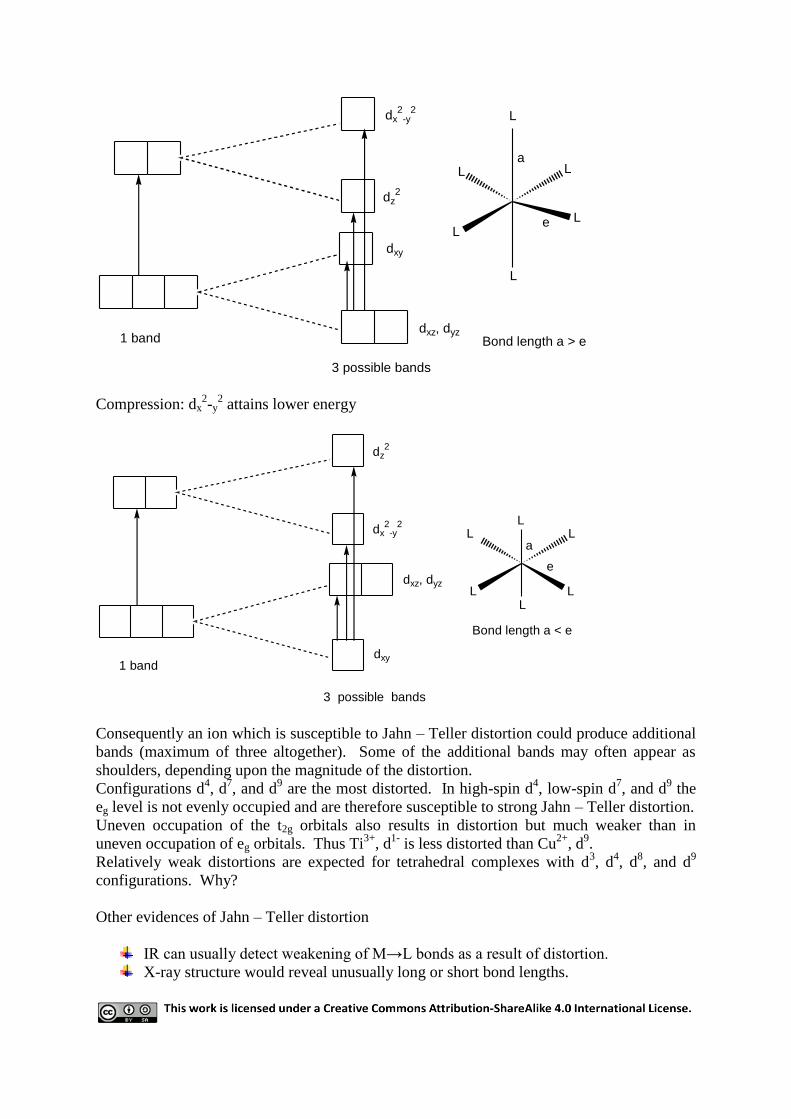
dx2
-y2
dz2
dxy
dxz, dyz1 band
3 possible bands
L
LL
L
L
L
a
e
Bond length a > e
Compression: dx2-y
2 attains lower energy
dx2
-y2
dz2
dxy
dxz, dyz
1 band
3 possible bands
L
L
L
L
L
L
a
e
Bond length a < e
Consequently an ion which is susceptible to Jahn – Teller distortion could produce additional
bands (maximum of three altogether). Some of the additional bands may often appear as
shoulders, depending upon the magnitude of the distortion.
Configurations d4, d
7, and d
9 are the most distorted. In high-spin d
4, low-spin d
7, and d
9 the
eg level is not evenly occupied and are therefore susceptible to strong Jahn – Teller distortion.
Uneven occupation of the t2g orbitals also results in distortion but much weaker than in
uneven occupation of eg orbitals. Thus Ti3+
, d1-
is less distorted than Cu2+
, d9.
Relatively weak distortions are expected for tetrahedral complexes with d3, d
4, d
8, and d
9
configurations. Why?
Other evidences of Jahn – Teller distortion
IR can usually detect weakening of M→L bonds as a result of distortion.
X-ray structure would reveal unusually long or short bond lengths.

10 Stability of coordination compounds
Most measurements of stability are done in aqueous solutions when the complex in question
is formed
by a ligand displacing water from the aquo complex of the metal ion. Metal complexes are
formed in solution by stepwise reaction, and equilibrium constants can be written for each
step. For example,
[Ag(H2O)x]+ [Ag(NH3)(H2O)x-1]
+ + H2O k1
[Ag(NH3)(H2O)x-1]+ + NH3
[Ag(NH3)2(H2O)x-2]+ + H2O k2
For simplicity we can ignore the water molecules that make up the hydration sphere of an
aqueous metal ion. Moreover, the solvent water molecules involved in the reaction are not
included in the equilibrium constants, k1 and k2 above are called stepwise-wise stability
constants.
k1 =[Ag(NH3)
+]
[Ag+][NH3]
; k2 =[Ag(NH3)2
+]
[Ag(NH3)+][NH3]
The larger the value of the constant, the greater the concentration of the complex species at
equilibrium. A second type of equilibrium constant, , called an overall stability constant can
be defined for the reactions above:
=[Ag(NH3)
+]
[Ag+][NH3]
; [Ag(NH3)2
+]
[Ag+][NH3]
2
Since the ks and s describe exactly the same chemical systems, they must be related to each
other:
[Ag(NH3)2
+]
[Ag+][NH3]
2=
[Ag(NH3)2+]
[Ag+][NH3][NH3]
.[Ag(NH3)
+]
[Ag(NH3)+]
[Ag(NH3)2+]
[Ag(NH3)+][NH3]
.[Ag(NH3)
+]
[Ag+][NH3]
=
= k2 . k1
Note that 1 = k1 and 2 = k2 . k1. By similar treatment it can be shown that
n = k1 k2 k3 …..kn.
The numerical value of stability constant describes the relative concentration of species at
equilibrium. Large stability constants indicate that the concentration of a complex is much
greater than the concentration of the components it is made.
A complex is said to be stable if the equilibrium constants describing its formation is large.
10.1 Factors that influence complex stability
The equilibrium constant of a reaction is a measure of heat released in the reaction and the
entropy change during the reaction.
-RT lnk = ΔG = ΔH – TΔS
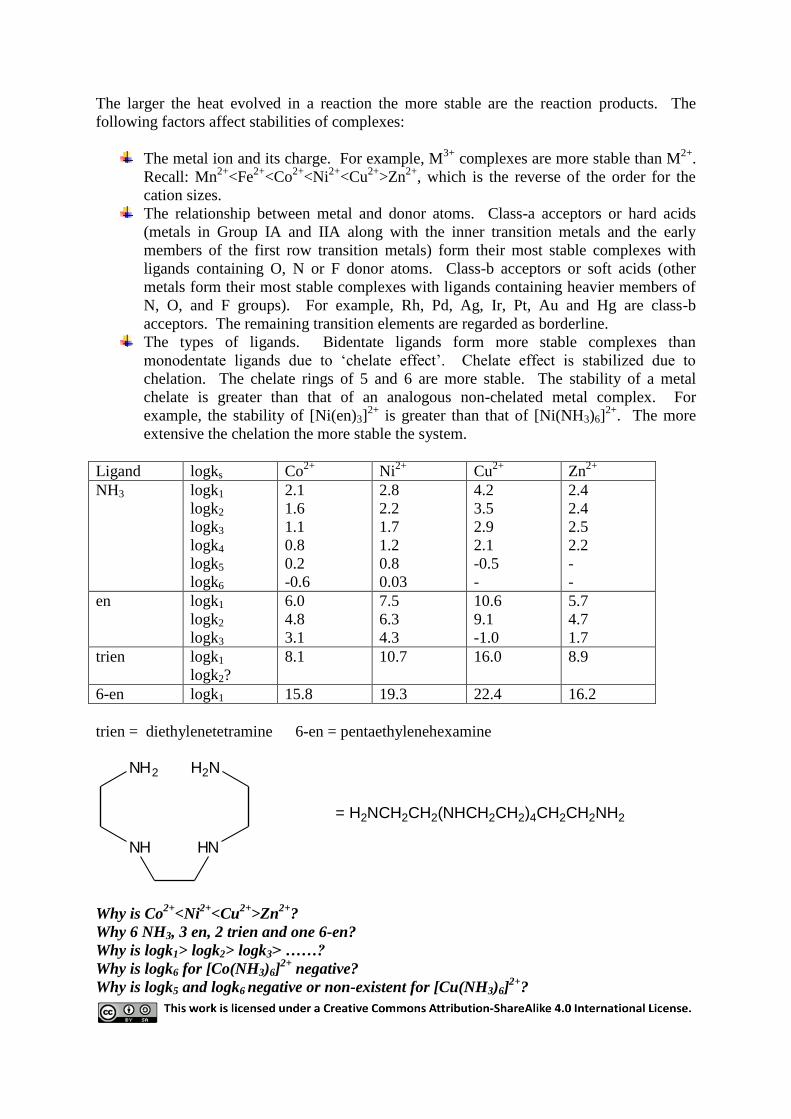
The larger the heat evolved in a reaction the more stable are the reaction products. The
following factors affect stabilities of complexes:
The metal ion and its charge. For example, M3+
complexes are more stable than M2+
.
Recall: Mn2+
<Fe2+
<Co2+
<Ni2+
<Cu2+
>Zn2+
, which is the reverse of the order for the
cation sizes.
The relationship between metal and donor atoms. Class-a acceptors or hard acids
(metals in Group IA and IIA along with the inner transition metals and the early
members of the first row transition metals) form their most stable complexes with
ligands containing O, N or F donor atoms. Class-b acceptors or soft acids (other
metals form their most stable complexes with ligands containing heavier members of
N, O, and F groups). For example, Rh, Pd, Ag, Ir, Pt, Au and Hg are class-b
acceptors. The remaining transition elements are regarded as borderline.
The types of ligands. Bidentate ligands form more stable complexes than
monodentate ligands due to „chelate effect‟. Chelate effect is stabilized due to
chelation. The chelate rings of 5 and 6 are more stable. The stability of a metal
chelate is greater than that of an analogous non-chelated metal complex. For
example, the stability of [Ni(en)3]2+
is greater than that of [Ni(NH3)6]2+
. The more
extensive the chelation the more stable the system.
Ligand logks Co2+
Ni2+
Cu2+
Zn2+
NH3 logk1
logk2
logk3
logk4
logk5
logk6
2.1
1.6
1.1
0.8
0.2
-0.6
2.8
2.2
1.7
1.2
0.8
0.03
4.2
3.5
2.9
2.1
-0.5
-
2.4
2.4
2.5
2.2
-
-
en logk1
logk2
logk3
6.0
4.8
3.1
7.5
6.3
4.3
10.6
9.1
-1.0
5.7
4.7
1.7
trien logk1
logk2?
8.1 10.7 16.0 8.9
6-en logk1 15.8 19.3 22.4 16.2
trien = diethylenetetramine 6-en = pentaethylenehexamine
NH HN
H2NNH2
= H2NCH2CH2(NHCH2CH2)4CH2CH2NH2
Why is Co2+
<Ni2+
<Cu2+
>Zn2+
?
Why 6 NH3, 3 en, 2 trien and one 6-en?
Why is logk1> logk2> logk3> ……?
Why is logk6 for [Co(NH3)6]2+
negative?
Why is logk5 and logk6 negative or non-existent for [Cu(NH3)6]2+
?

Why is log6 (for NH3 complexes) < log3 (for the en complexes) < log1 (for 6-
encomplexes) in each metal?
For steric reasons large bulky ligands form less stable metal complexes than do analogous
smaller ligands. For example H2NCH2CH2NH2 forms more stable complexes than
(CH3)2NCH2CH2N(CH3)2.
Examples:
Complex s
[Ni(NH3)6]2+
1 2 3 4 5 6
5 x 102
6 x 104
3 x 106
3 x 107 1.3 x 10
8 1.0 x 10
8
[Ni(en)3]2+
1 2 3
5 x 107 1.1 x 10
14 1.4 x 10
18
[Ni(dien)2]2+
1 2
6 x 1010
8 x 1018
dien = H2NCH2CH2NHCH2CH2NH2Ref.: Basolo & Johnson, Coordination Chemistry, p. 89.
For NH3 6s; for en 3s; for dien 2s. Why?
s [Ni(dien)2]2+
> [Ni(en)3]2+
> [Ni(NH3)6]2+
. Why?
The successive stability constants for [Cu(H2O)5NH3]2+
, [Cu(H2O)4(NH3)]2+
,
and[Cu(H2O)3(NH3)3]2+
are x, y, and z respectively. Write equations for each step
andcalculate the overall formation constant for [Cu(H2O)3(NH3)3]2+
.

11. Introduction to Magnetochemistry
11.1 Introduction
Magnetochemistry is the study of the magnetic properties of materials. By "magnetic
properties" refers to whether they will be attracted or repelled by a magnet. In this course we
are interested in the magnetic properties of metal complexes. A study of magnetic properties
of metal complexes could be used to derive the oxidation state of and the arrangement of
electrons in the central metal ion. It can also be used to derive the stereochemistry of the
complex. Magnetochemistry is therefore very informative in the study of coordination
chemistry,
Magnetism arises from moving charges, such as an electric current in a coil of wire. In a
material which does not have a current present, there are still magnetic interactions. Atoms
are made of charged particles (protons and electrons) which are moving constantly.
The processes which create magnetic fields in an atom are:
1. Nuclear spin. Some nuclei, such as a hydrogen atom, have a net spin, which creates a
magnetic field.
2. Electron spin. An electron has two intrinsic spin states (similar to a top spinning)
which we call up and down or alpha and beta.
3. Electron orbital motion. There is a magnetic field due to the electron moving around
the nucleus.
Each of these magnetic fields interacts with one another and with external magnetic fields.
However, some of these interactions are strong and others are negligible.
11.2 Magnetism
Lenz's Law (~1834), states that: when a substance is placed within a magnetic field, H, the
field within the substance, B, differs from H by the induced field, 4I, which is proportional
to the intensity of magnetization, I.
That is:
B = H + 4I ------------------------------------------------ (Eq. 1)
where B is the magnetic field within the substance, H is the applied magnetic field ,
and I is the intensity of magnetisation.
This can also be written as
B/H = 1 + 4I/H,
or B/H = 1 + 4
where B/H is called the magnetic permeability of the material and is the magnetic
susceptibility per unit volume, (I/H)
By definition, in a vacuum is zero, so that B=H.
It is usually more convenient to measure mass (gram) susceptibility, g, which is related to
the volume susceptibility through the density.
g =/ ----------------------------------------------------------- (Eq. 2)
where is the density.

Finally to get our measured quantity on a basis that can be related to atomic properties, we
convert to molar susceptibility
m =g * MW (MW = molecular weight of the sample)
Normal paramagnetic substances obey the Curie Law
= C/T ----------------------------------------------------------------- (Eq. 3)
where C is the Curie constant.
Thus a plot of 1/ versus T should give a straight line of slope 1/C passing through the origin
(0 K).
Many substances give a straight line that intercepts just a little above 0 K and these are said to
obey the Curie-Weiss Law:
= C/(T+) --------------------------------------------------------------- (Eq. 4)
where is known as the Weiss constant.
The constant C is given by the Langevin expression, which relates the susceptibility to the
magnetic moment:
m =N2/3kT ---------------------------------------------------------------- (Eq. 5)
where N is Avogadro number, k is the Boltzmann constant, and T the absolute temperature.
Re-writing Eq, 5 gives the magnetic moment as
= 2.828 √mT = 2.828(m.T)½
-------------------------------------------- (Eq. 6)
Many transition metal salts and complexes are paramagnetic due to partially filled d-
orbitals.
The experimentally measured magnetic moment () (Eq. 6) can provide some important
information about the compounds themselves:
No of unpaired electrons present
Distinction between high-spin and low-spin octahedral complexes
Spectral behaviour, and
Structure of the complexes.
11.3 Sources of Paramagnetism
Orbital motion of the electron generating ORBITAL MAGNETIC MOMENT (l)
Spin motion of the electron (on its own axis) generating SPIN MAGNETIC
MOMENT (s)
If a transition metal ion contains n unpaired electrons (multi-electron system), the total orbital
and total spin motions are, respectively, given by:

L = l1 + l2 + l3 + …………….
S = s1 + s2 + s3 + ……………
where l = orbital angular momentum, s = spin angular momentum
The magnetic moment due to the contribution of orbital and spin motions is given by
L+S = [4S(S+1)+ L(L+1)]½
B.M. ------------------------------------- (Eq. 7)
For TM-complexes, the magnetic properties arise mainly from the exposed d-orbitals. The d-
orbitals are perturbed by ligands.
The rotation of electrons about the nucleus is restricted which leads to L = 0; i.e. the
orbital contribution to magnetic moment is quenched; thus
s = [4S(S+1)]½
B.M. -------------------------------------------- (Eq. 8)
The spin-only formula can be derived as follows:
S = n (1/2) = n/2; n = no of unpaired electrons
Hence
s = [4S(S+1)]½
B.M.
= [4(n/2)(n/2+1)]½ B.M.
= [n(n+2)]½
B.M.
This is called Spin-Only Formula.
11.4 When does orbital angular momentum contribute?
There must be an unfilled/half-filled orbital similar in energy to that of the orbital occupied
by the unpaired electrons. If this is so, the electrons can make use of the available orbitals to
circulate or move around the centre of the complexes and hence generate L and L
Essential Conditions:
1. The orbitals should be degenerate (t2g or eg)
2. The orbitals should be similar in shape and size so that they are transferable into one
another by rotation about the same axis (e.g. dxy is related to dx2-y2 by a rotation of 45o
about the z-axis.
3. Orbitals must not contain electrons of identical spin.
For an octahedral complex:
Condition t2g set eg set
1 Obeyed Obeyed
2 Obeyed Not obeyed
3 Since 1 and 2 are satisfied
condition 3 dictates whether
t2g will generate l or not
Does not matter since
condition 2 is already not
obeyed.

These conditions are fulfilled whenever one or two of the three t2g orbitals contain an odd
no. of electrons.
Exercise: Work-out all possible dn LS and HS cases with orbital contribution.
HS:
LS:
11.5 Other Reasons for Orbital Contribution:
Although normally develops from GS, sometimes ES also may contribute, especially the
GS-ES energy difference is very small
Example:
Take Ni2+
octahedral; d8; GS: t2g
6eg
2 no l
ES: t2g5eg
3l contributes
Similarly,
Take Co2+
tetrahedral; d7 GS: e
4 t2
3nol
ES: e3t2
4l contributes
Therefore, obs>s for both Oh Ni2+
and Td Co2+
.
11.6 Magnetic Properties of lanthanides
4f electrons are too far inside 4fn5s
25p
6
(compared to the d electrons in transition metals)
Thus 4f normally unaffected by surrounding ligands
Hence, the magnetic moments of Ln3+
ions are generally well described from the
coupling of spin and orbital angular momenta ~ Russell-Saunders Coupling to give J
vector
spin orbit coupling constants are large (ca. 1000 cm-1)
ligand field effects are very small (ca. 100 cm-1)
o only ground J-state is populated
o spin-orbit coupling >> ligand field splittings
o magnetism is essentially independent of environment
Magnetic moment of a J-state is expressed by:
J = L+S, L+S-1,……L-S
For the calculation of g value, we use
minimum value of J for the configurations up to half-filled;
i.e. J = L-S for f0-f
7 configurations
maximum value of J for configurations more than half-filled;
i. e. J = L+S for f8-f
14 configurations
For f0, f
7, and f
14, L = 0, hence J becomes S
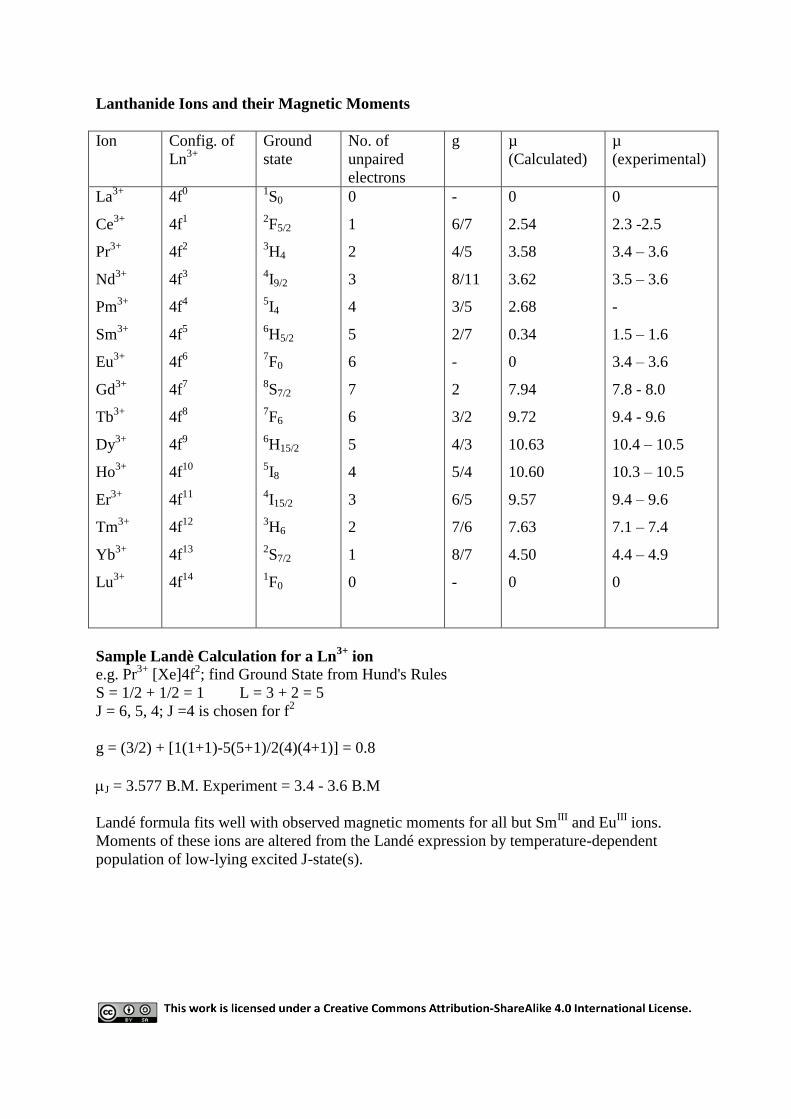
Lanthanide Ions and their Magnetic Moments
Ion Config. of
Ln3+
Ground
state
No. of
unpaired
electrons
g µ
(Calculated)
µ
(experimental)
La3+
Ce3+
Pr3+
Nd3+
Pm3+
Sm3+
Eu3+
Gd3+
Tb3+
Dy3+
Ho3+
Er3+
Tm3+
Yb3+
Lu3+
4f0
4f1
4f2
4f3
4f4
4f5
4f6
4f7
4f8
4f9
4f10
4f11
4f12
4f13
4f14
1S0
2F5/2
3H4
4I9/2
5I4
6H5/2
7F0
8S7/2
7F6
6H15/2
5I8
4I15/2
3H6
2S7/2
1F0
0
1
2
3
4
5
6
7
6
5
4
3
2
1
0
-
6/7
4/5
8/11
3/5
2/7
-
2
3/2
4/3
5/4
6/5
7/6
8/7
-
0
2.54
3.58
3.62
2.68
0.34
0
7.94
9.72
10.63
10.60
9.57
7.63
4.50
0
0
2.3 -2.5
3.4 – 3.6
3.5 – 3.6
-
1.5 – 1.6
3.4 – 3.6
7.8 - 8.0
9.4 - 9.6
10.4 – 10.5
10.3 – 10.5
9.4 – 9.6
7.1 – 7.4
4.4 – 4.9
0
Sample Landè Calculation for a Ln3+
ion
e.g. Pr3+
[Xe]4f2; find Ground State from Hund's Rules
S = 1/2 + 1/2 = 1 L = 3 + 2 = 5
J = 6, 5, 4; J =4 is chosen for f2
g = (3/2) + [1(1+1)-5(5+1)/2(4)(4+1)] = 0.8
J = 3.577 B.M. Experiment = 3.4 - 3.6 B.M
Landé formula fits well with observed magnetic moments for all but SmIII
and EuIII
ions.
Moments of these ions are altered from the Landé expression by temperature-dependent
population of low-lying excited J-state(s).
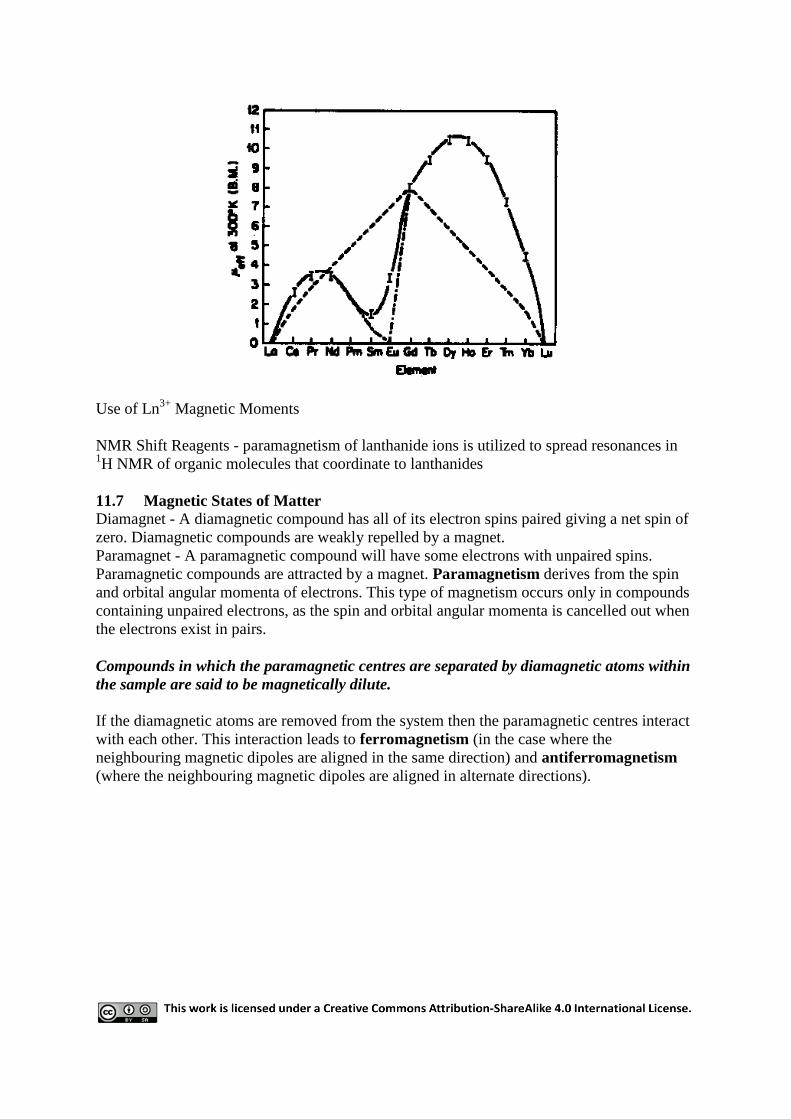
Use of Ln3+
Magnetic Moments
NMR Shift Reagents - paramagnetism of lanthanide ions is utilized to spread resonances in 1H NMR of organic molecules that coordinate to lanthanides
11.7 Magnetic States of Matter
Diamagnet - A diamagnetic compound has all of its electron spins paired giving a net spin of
zero. Diamagnetic compounds are weakly repelled by a magnet.
Paramagnet - A paramagnetic compound will have some electrons with unpaired spins.
Paramagnetic compounds are attracted by a magnet. Paramagnetism derives from the spin
and orbital angular momenta of electrons. This type of magnetism occurs only in compounds
containing unpaired electrons, as the spin and orbital angular momenta is cancelled out when
the electrons exist in pairs.
Compounds in which the paramagnetic centres are separated by diamagnetic atoms within
the sample are said to be magnetically dilute.
If the diamagnetic atoms are removed from the system then the paramagnetic centres interact
with each other. This interaction leads to ferromagnetism (in the case where the
neighbouring magnetic dipoles are aligned in the same direction) and antiferromagnetism
(where the neighbouring magnetic dipoles are aligned in alternate directions).

These two forms of paramagnetism show characteristic variations of the magnetic
susceptibility with temperature.
In the case of ferromagnetism, above the Curie point the material displays "normal"
paramagnetic behaviour. Below the Curie point the material displays strong magnetic
properties. Ferromagnetism is commonly found in compounds containing iron and in alloys.
For antiferromagnetism, above the Neel point the material displays "normal" paramagnetic
behaviour. Below the Neel point the material displays weak magnetic properties which at
lower and lower temperatures can become essentially diamagnetic. Antiferromagnetism is
more common and is found to occur in transition metal halides and oxides, such as TiCl3 and
VCl2.
A worked example
Account for the magnetic moments of (Et4N)2[NiCl4] recorded at 80 and 300 K.
80K 300K
3.25 3.89 B.M.
Ni2+
is a d8 metal ion.
The formula suggests a 4 coordinate complex and we can assume that the complex is
tetrahedral with a d electron configuration of e4 t2
4 therefore the spin-only magnetic moment
can be calculated as 2.83 B.M.
Why did we ignore the possibility of it being square-planar?
The free ion Russell-Saunders ground term is 3F (L=3 and S=1) which will give rise to a
lowest energy T term in a tetrahedral field and hence the resultant magnetic moment is
expected to be temperature dependent and have a direct orbital contribution.
The observed values may be quite different then to the calculated spin only magnetic
moment.
The value of S+L can be calculated as: S+L = [4S(S+1)+L(L+1)]
= (8+12) = 20 = 4.47 B.M.
If you use the spin-only formula s = n(n+2) = 8 = 2.8 BM
Now go back and check above the observed magnetic moments at the given temperatures.
What do you conclude?
From the observed values it can be seen that the magnetic moment of the d8 Ni
2+ complex is
intermediate between the s and S+L values (probably due to partial quenching of the orbital
angular momentum contribution) and is dependent on temperature.


12. LANTHANIDES AND ACTINIDES
Introduction
You would recall that elements in the Periodic Table could be classified into blocks: s-, p-, d-
and f-blocks. The d- and f-blocks are also called transition elements, while the term
transition metals are generally retained for the d-block metals, the f-block is generally
referred to as inner transition elements, which appears firstly after lanthanum and secondly
after actinium, in the Periodic Table. The elements from cerium to lutetium are known as
lanthanides, after lanthanum. Because of its chemical similarity to these elements, lanthanum
is usually included with the lanthanides. Scandium and yttrium also share similar chemical
properties with the lanthanides and are both included when discussing the chemistry of the
lanthanides.
The second series of f-block starts from thorium to lawrencium and are called actinide series.
For the same reason as in lanthanum, actinium is also considered with the actinides series
when discussing their chemistry.
The transition elements have similar characteristic properties including the following:
Solid (metals) except mercury, which is a liquid.
Conductors of electricity and heat
Form alloys with one another and with metallic main group elements.
High melting and boiling, except mercury.
Mostly reactive with mineral acids to form salts (some are inert to conc. oxidizing
acids
Display variable oxidation states, accessible by simple laboratory reactions.
However, there are exceptions in the lanthanides, where the chemistry is dominated
by the Ln3+
ions.
Like the d-block, the early members of the actinide series (protactinium to americium)
display variable oxidation states but the latter members behave like the lanthanides,
dominated by An3+
ions.
The presence of partially filled d and f orbitals in the f-block elements often results in
some transition element in having odd number of electrons that make them t be
paramagnetic.
Compounds are often coloured due to the presence of partially –filled d orbitals in the
d transition metals, and d or f in f transition elements.
12.1. The Lanthanides
General introduction
Position in the Periodic Table: Z = 58 – 71
Electron configuration
The electron configuration can be written as 4f2-14
5s25p
65d
0-16s
2 or [Xe]4f
2-145d
66s
2.
The third outer shell, (4f orbital) is being filled with electrons, while the number of electrons
in the outer shell (6s2) and in the penultimate (5s
25p
6) shell of most of the lanthanides is
identical. Even though the energies of the 4f and 5d are similar, the 4f states predominate in
the chemistry of the lanthanides.
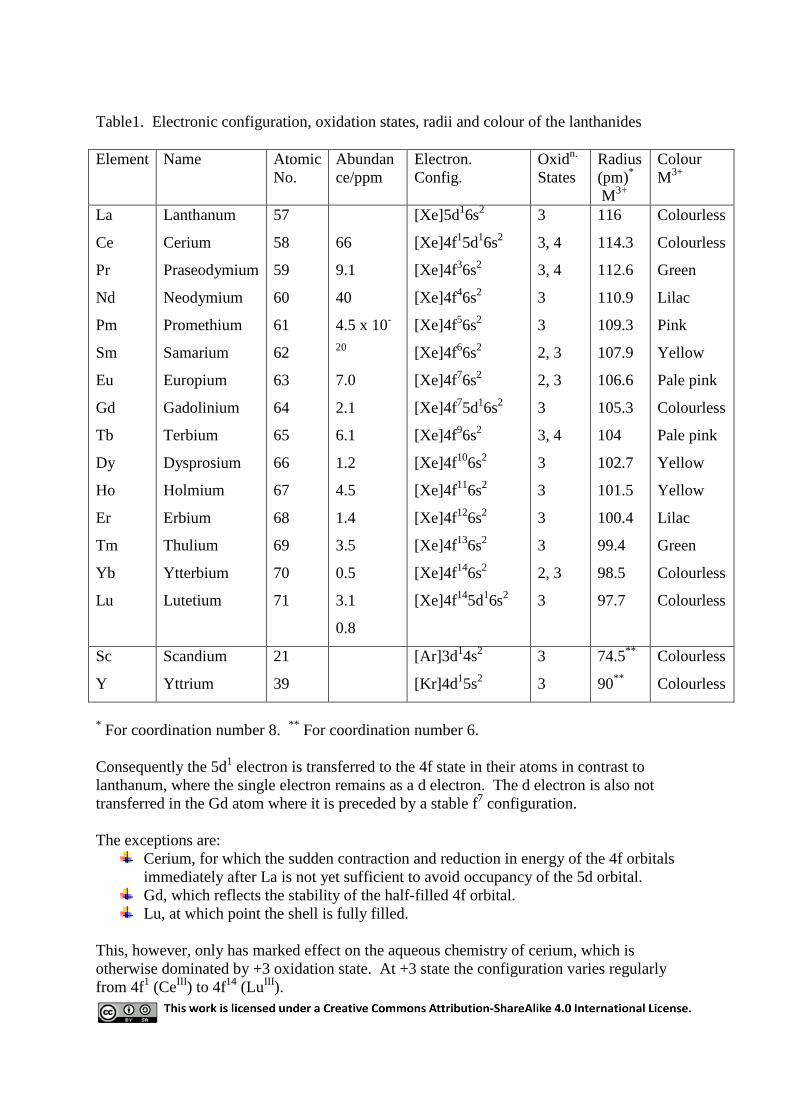
Table1. Electronic configuration, oxidation states, radii and colour of the lanthanides
Element Name Atomic
No.
Abundan
ce/ppm
Electron.
Config.
Oxidn.
States
Radius
(pm)*
M3+
Colour
M3+
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
Lanthanum
Cerium
Praseodymium
Neodymium
Promethium
Samarium
Europium
Gadolinium
Terbium
Dysprosium
Holmium
Erbium
Thulium
Ytterbium
Lutetium
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
66
9.1
40
4.5 x 10-
20
7.0
2.1
6.1
1.2
4.5
1.4
3.5
0.5
3.1
0.8
[Xe]5d16s
2
[Xe]4f15d
16s
2
[Xe]4f36s
2
[Xe]4f46s
2
[Xe]4f56s
2
[Xe]4f66s
2
[Xe]4f76s
2
[Xe]4f75d
16s
2
[Xe]4f96s
2
[Xe]4f10
6s2
[Xe]4f11
6s2
[Xe]4f12
6s2
[Xe]4f13
6s2
[Xe]4f14
6s2
[Xe]4f14
5d16s
2
3
3, 4
3, 4
3
3
2, 3
2, 3
3
3, 4
3
3
3
3
2, 3
3
116
114.3
112.6
110.9
109.3
107.9
106.6
105.3
104
102.7
101.5
100.4
99.4
98.5
97.7
Colourless
Colourless
Green
Lilac
Pink
Yellow
Pale pink
Colourless
Pale pink
Yellow
Yellow
Lilac
Green
Colourless
Colourless
Sc
Y
Scandium
Yttrium
21
39
[Ar]3d14s
2
[Kr]4d15s
2
3
3
74.5**
90**
Colourless
Colourless
* For coordination number 8.
** For coordination number 6.
Consequently the 5d1 electron is transferred to the 4f state in their atoms in contrast to
lanthanum, where the single electron remains as a d electron. The d electron is also not
transferred in the Gd atom where it is preceded by a stable f7 configuration.
The exceptions are:
Cerium, for which the sudden contraction and reduction in energy of the 4f orbitals
immediately after La is not yet sufficient to avoid occupancy of the 5d orbital.
Gd, which reflects the stability of the half-filled 4f orbital.
Lu, at which point the shell is fully filled.
This, however, only has marked effect on the aqueous chemistry of cerium, which is
otherwise dominated by +3 oxidation state. At +3 state the configuration varies regularly
from 4f1 (Ce
III) to 4f
14 (Lu
III).

The elements can be subdivided into two sub-families according to the filling of the 4f
orbitals:
The first seven (Ce-Gd) in which each of 4f orbital is filled by one electron are called
the cerium sub-family
The other seven (Tb-Lu) in which the 4f orbitals are filled by a second electron are
called terbium sub-family.
In Gd and Lu, as in La, the extra electron over the stable f7 and f
14 configurations respectively
is located on the 5d state.
The f electrons have no significant effect on the chemical properties of most lanthanides,
except in few instances where, when slightly excited, one (seldomly 2) electron is transferred
to the 5d state. Their properties are therefore mostly determined by the 5d16s
2 electrons.
Consequently they have great similarity to Group III d-elements (Sc, Y, and La). The
greatest similarity to the lanthanides is in Y and La whose atomic and ionic radii are close to
those of the lanthanides.
As evident above, the difference in electron structure on the atoms is in the third outermost
shell, which does not greatly affect their chemical properties; all the lanthanides are
extremely alike. Because of this special similarity of properties the lanthanides are classed
with lanthanum, yttrium and scandium.
Despite their similarity, the elements have their differences.
Their uniform properties are due to lanthanide contraction, which accounts for the gradual
decrease in atomic and ionic sizes in the Ce-Lu series. For any particular property in which
4fn configuration is maintained across the series a regular variation is observed. However,
variations in properties where the 4fn configuration is not maintained are highly irregular.
12.2. Lanthanide contraction
Lanthanide contraction consists of a significant and steady decrease in size of atoms and ions
with increasing atomic number. This is due to the poor shielding of the 4f and 5f electrons,
as a result of the shapes of the orbitals, which results in a steady increase in effective nuclear
charge and consequent reduction in size of atoms and ions as one proceeds from La to Lu.
The successive shrinkages is called the lanthanide contraction. This has remarkable effect on
the radii of subsequent elements, which are usually smaller than expected. For example ZrIV
and HfIV
have almost the same radii despite the atomic number of 40 and 72 respectively.
The importance/consequences of lanthanide contraction
The normal +3 oxidation state resembles one another much more closely than
members of any series of elements and the separation of some of the lanthanides were
used to be very difficult.
The reduction in size from one LnIII
to the next makes their separation possible, but
the smallness and regularity of the reduction makes the separation difficult.
By the time Ho is reached the LnIII
radius has been sufficiently reduced to be almost
identical with that of YIII
which is why this much larger element is always found
associated with the heavier lanthanides.

As mentioned when we discussed the second and third row transition metals, the
comparable size of metals in the second row and the corresponding ones in the third
row of transition metals is due to lanthanide contraction.
Lanthanide contraction and actinide contraction
The actinide contraction initially parallels that of lanthanides; however, the
contraction is smaller than expected from curium on. This is probably due to poorer
shielding by 5f electrons in the elements.
The lanthanide curve consists of two very shallow arcs with a discontinuity at the
spherically symmetrical Gd3+
(4f7) ion. A similar discontinuity is not observed at
Cm3+
.
The 4f electrons appear to be more deeply buried within the atom that they are not affected
by the environment to any great degree. On the other hand the 5f electrons, at least in the
earlier elements of the series, Th to Bk, are available for bondng, allowing oxidation state up
to +7. These elements therefore resemble d electrons of the transition metals.
12.3. Names:
Rare earths, used originally to describe almost any naturally occurring but unfamiliar oxide,
reflecting the difficulty encountered in separating the elements.
Lanthanons or lanthanoids, which arises from their relationship to lanthanum, element
57,(Group IIIA).
The elements are far from being rare, however, and hence the preference for lanthanides,
lanthanons or lanthanoids. We will adopt lanthanides in this course, with general symbol Ln
to refer to the 14 elements Ce – Lu, inclusive.
Both lanthanides and actinides are also collectively known as inner transition metals. La
and Ac, strictly Group III elements, are classified with lanthanides and actinides respectively
because they share similar properties with lanthanides and actinides respectively.
The lanthanides comprise the largest naturally-occurring group in the Periodic Table.
“Yttria” was isolated in 1794 by J Gadolin. The discovery of the lanthanides is summarized
in Table 2.
12.4 Terrestrial abundance and distribution
The only rare of the lanthanides is the unstable 147
Pm (promethium), t½, 2.62 y), which occurs
as traces in uranium ores. Cerium is the 26th
most abundant of all elements. It is half as
abundant as chlorine and 5 times as abundant as lead. Thulium, the rarest after Pm, is more
abundant than iodine and as common as bismuth. Although there are over 100 minerals
known to contain lanthanides, the only two of commercial importence are monazite (a
mixture La, Th, Ln orthophosphate), and bastnaesite (a La, Ln fluorocarbonate, MIII
CO3F).
Monazite is widely but sparsely distributed in many rocks but, because of its high density and
inertness, it is concentrated by weathering into sands on beaches and river beds. Ilmenite
(FeTiO3) and cassiterite (SnO2) are often found with monazite. La, Ce, Pr and Nd make up
about 90% of monazite with Y and the heavier elements taking about 10%.

Monazite and other minerals carrying lanthanides in the +3 state are usually poor in Eu,
which has a relatively strong tendency to assume the +2 state and therefore found with the
calcium group of minerals.
12.5. Preparation of the elements
The ores are dressed to yield minerals of better than 90% purity. They are then broken down
(“opened”) by either acidic or alkaline attack. Variations can be introduced depending on the
extent to which the metals are to be separated from each other.
Separations are enhanced by
Different solubility of Ln2(SO4)3.Na2SO4.xH2O for the light and heavy lanthanides;
Low solubility of the hydrous oxide of thorium.
Because only small amounts of thorium and heavy lanthanides are present in bastnaesite, its
chemical treatment is less complex.
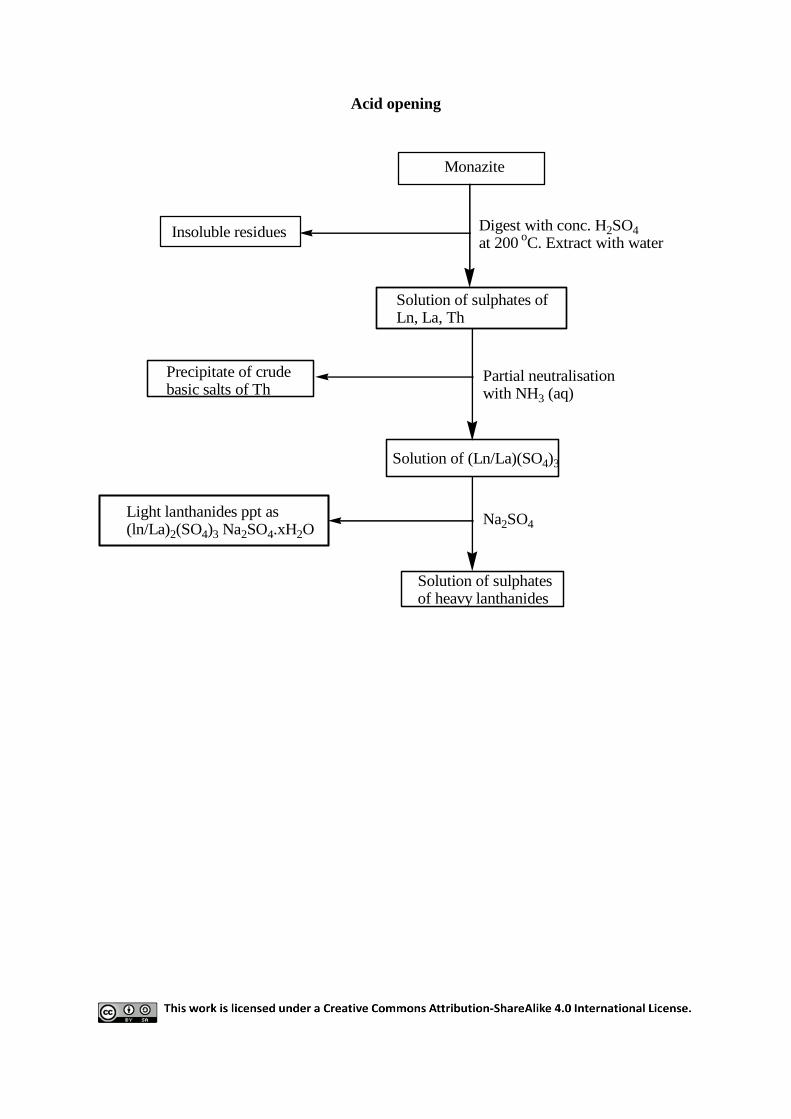
Acid opening
Monazite
Digest with conc. H2SO4
at 200 o
C. Extract with waterInsoluble residues
Solution of sulphates of Ln, La, Th
Precipitate of crude basic salts of Th
Partial neutralisation with NH3 (aq)
Solution of (Ln/La)(SO4)3
Light lanthanides ppt as (ln/La)2(SO4)3 Na2SO4.xH2O
Solution of sulphates of heavy lanthanides
Na2SO4
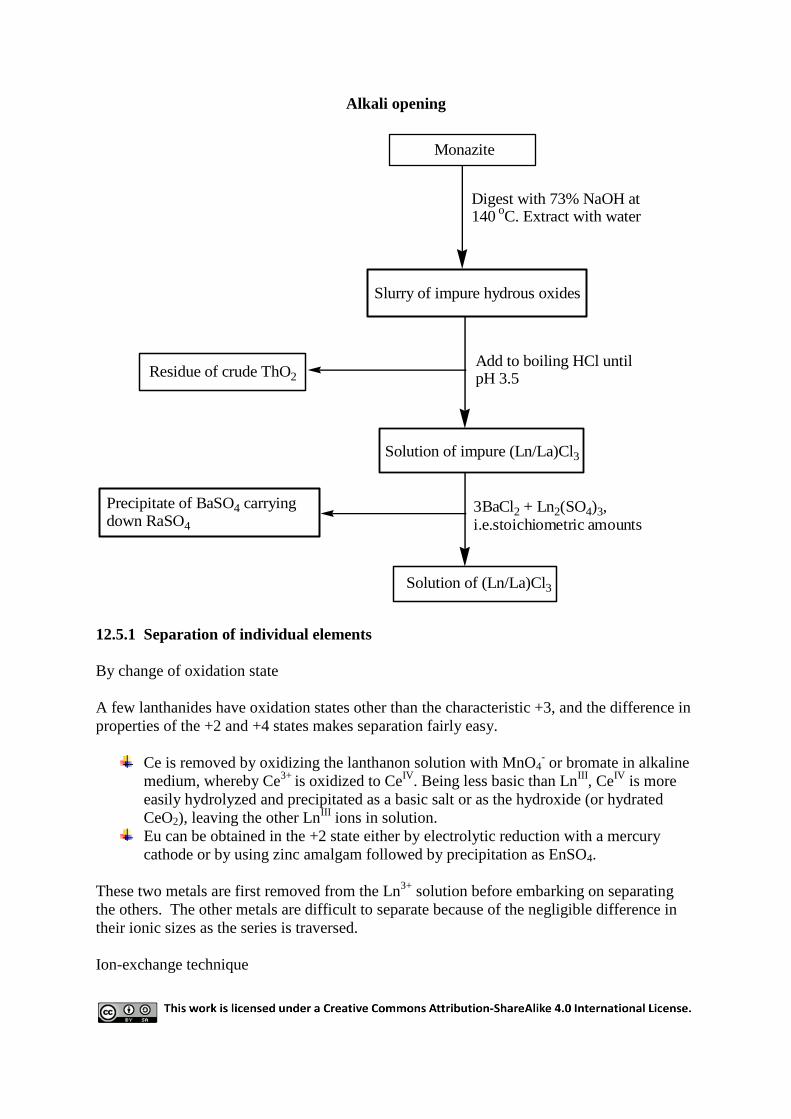
Alkali opening
Monazite
Digest with 73% NaOH at140
oC. Extract with water
Residue of crude ThO2
Add to boiling HCl until pH 3.5
Solution of impure (Ln/La)Cl3
Solution of (Ln/La)Cl3
3BaCl2 + Ln2(SO4)3, i.e.stoichiometric amounts
Slurry of impure hydrous oxides
Precipitate of BaSO4 carrying down RaSO4
12.5.1 Separation of individual elements
By change of oxidation state
A few lanthanides have oxidation states other than the characteristic +3, and the difference in
properties of the +2 and +4 states makes separation fairly easy.
Ce is removed by oxidizing the lanthanon solution with MnO4- or bromate in alkaline
medium, whereby Ce3+
is oxidized to CeIV
. Being less basic than LnIII
, CeIV
is more
easily hydrolyzed and precipitated as a basic salt or as the hydroxide (or hydrated
CeO2), leaving the other LnIII
ions in solution.
Eu can be obtained in the +2 state either by electrolytic reduction with a mercury
cathode or by using zinc amalgam followed by precipitation as EnSO4.
These two metals are first removed from the Ln3+
solution before embarking on separating
the others. The other metals are difficult to separate because of the negligible difference in
their ionic sizes as the series is traversed.
Ion-exchange technique

Separation by this technique is carried out commercially on a large scale.
The ion-exchange behaviour depends primarily on the hydrated ionic radius. The smaller the
hydrated size of the ion the more tightly it bounds to ligands. La has the largest ionic size
and Lu the smallest. La therefore has the smallest hydrated ion and Lu the largest hence the
order of elution is Lu → La. The separation is effected by use of appropriate complexing
agents at appropriate pHs. The ion of smallest radius also forms the strongest complexes and
hence the preference for the aqueous phase is enhanced. One of the best complexing agents
is α-hydroxyisobutyric acid, (CH3)2CH(OH)COOH, at 25 o and pH 3.55, but EDTA and other
hydroxo or amino carboxylic acids can also be used.
A typical ion-exchange resin is sulphonated polystyrene, which may be denoted by H-Resin,
since it corresponds to an insoluble strong acid. A solution containing the lanthanides is
applied, and the acid formed is washed down,
3 H-Resin + Lnaq3+
Ln(Resin)3 + 3 H3O+
The column is eluted with a weakly acidic solution of the complexing agent, e.g. citric acid
(H-Cit), buffered with ammonia to a constant pH = 5, when the new equilibrium,
Ln(Resin)3 + 3 H-Cit 3 H-Resin + LnCit3
is set up. As the buffered citric acid flows down the column, the concentration of the
lanthanide ions changes and the equilibrium reverses several times. The heavier ions (smaller
hydrated ions) are more strongly complexed by the citrate ion and so will tend to spend more
times in solution and less on the resin. Because the complexes possess a lower positive
charge than the initial Ln3+
, they are less tightly held by the resin than Ln3+
and are washed
down the column first and will eventually be eluted. The elements are usually dilute. They
are concentrated by precipitation of the oxalate and the exchange repeated to give pure
samples.
Other methods of separation
Precipitation
The substance with the lowest solubility product is precipitated most rapidly and most
completely. Addition of OH- ions to the aqueous solution of the lanthanide nitrates results in
the precipitation of the hydroxides, the weakest base, Lu(OH)3 being precipitated first and the
strongest base La(OH)3 last. Only partial separation is achieved using this method.
Thermal reaction
On heating the nitrates there is a temperature when the least basic would be converted to the
oxide. The mixture is leached with water when the insoluble oxide remains. The oxide is
then converted to the nitrates again when the process is repeated.
Fractional crystallization of their simple salts, e.g. nitrates, sulfates, bromates, perchlorates
and oxalates, including some double salts of the type 2L(NO3)3.3Mg(NO3)2.24H2O.
Solubility decreases from La to Lu. The re-crystallization process is repeated several times.
Complex formation
The oxalates of the lanthanons are insoluble, but they can be held in solution by a chelating
agent, e.g. EDTA. The stability of the M-EDTA complexes varies. The metal-complexes are
destroyed in order of increasing stability by addition of acid which then precipitate as
insoluble oxalates. Separation is not complete hence the oxalates are re-dissolved and the
process is repeated several times.
Solvent extraction
The ratios of the partition coefficients of La(NO3)3 and Gd(NO3)3 between a solution of the
metal ions in strong HNO3 and tributylphosphate is 1:1.06. Though this difference is small a
very large number of partition can be performed using a continuous counter – current
apparatus. Kg quantities of 95% pure Gd have been obtain by this method.
Change in oxidation state
As mentioned earlier a few lanthanons have oxidation states other than the characteristic +3
and the difference in properties of the +2 and +4 states makes separation fairly easy. For
example Ce can be removed form lanthanide mixtures by oxidizing the solution with MnO4-
or bromate in alkaline medium. CeIV
is smaller and has higher charge and less basic than
Ce3+
. It is therefore precipitated as Ce(OH)4, CeO2, or a basic salt, leaving the M3+
ions in
solution.
Alternatively Ce3+
can be readily extracted from the other M3+
lanthanides in HNO3 solution
using tributylphosphate. 99% pure Ce can be obtained in one stage from a mixture containing
40% Ce.
Europium can be obtained in the +2 state either by electrolytic reduction with a mercury
cathode or zinc amalgam, followed by the precipitation of EuSO4.
Table 2. The discovery of the oxides of Group IIIA and the Lanthanide elements (Earnshaw
& Greenwood, p 1229)
Element Discoverer Date Origin of name
Ce Cerium
La Lanthanum
Pr Praeseodymium
Nd Neodymium
Sm Samarium
Eu Europium
Y Yttrium
Tb Terbium
Er Erbium
Yb Ytterbium
C G Mosander
C G Mosander
C A von Welsbach
C A von Welsbach
L de Boisbaudran
E A Demarcay
C G Mosander
C G Mosander
C G Mosander
J C G de Marignac
1839
1839
1885
1885
1879
1901
1843
1843
1843
1878
The steroid, Ceres
Greek lanthanein, to escape
notice.
Greek praseos + didymos, leek
green + twin or green twin (from
its green salts)
Greek neos + didymos, new twin
The mineral, samarskite
Europe
Ytterby, Sweden
Ytterby, Sweden
Ytterby, Sweden
Ytterby,Sweden

Sc Scandium
Ho Holmium
Tm Thulium
Gd Gadolinium
Dy Dysprosium
Lu Lutetium
Pm Promethium
L F Nilson
P T Cleve
P T Cleve
J C G de Marignac
L de Boisbaudran
G Urban
C A von Welsbach
C James
1879
1879
1879
1880
1886
1907
1947
Scandinavia
Latin Holmia: Stockholm
Latin Thule, “most northerly
land
Finnish chemist, J Gadolin
Greek dysprositos, hard to get.
Latin Lutetia: Paris
Greek, dysprositos, hard to find
Latin Lutetia: Paris
After Prometheus, the only Ln
that has never been found in
nature.
12.6. Chemical reactivity
The lanthanides are remarkably similar in chemical properties because of their near identical
configurations of 6s2+
+ an additional lightly held electron in either 5d or 4f orbital (the
energies of the 5d and the 4f orbitals are very nearly equal for te lanthanides of low atomic
numbers).
The elements are highly electropositive with M3+
/M potential varying from -2.25 V (Lu) to -
2.52 V (La). The chemistry is therefore predominantly ionic and of M3+
ions.
They are more reactive than the d-transition metals and are therefore closer to alkali or
alkaline earth metals than to most of the transition metals.
They all react with water with evolution of hydrogen
Ionic compounds are common and the coordination chemistry is quite different from, and less
extensive than, that of the d-transition metals.
Coordination numbers are generally high and stereochemistries are frequently ill-defined and
the complexes are labile.
Only strongly chelating ligands yield products that can be isolated from aqueous solution in
association with coordinated water.
Coordination numbers below 6, considered to be unusual, are found only with very bulky
ligands. The complexes therefore are prepared in non-aqueous systems.
Coordination numbers of 10 and over require chelating ligands with small “bites”, such as
NO3- or SO4
2-. Such complexes are found in large, lighter lanthanides.
The chemical properties of the lanthanides, however, differ

Because the successive addition of electrons to the 4f (or 5d) orbitals of the atoms on the
aufbau principle while maintaining the electrical neutrality of the atom, causes variations in
the distribution of the negative charge and
Because the effective nuclear charge (Zeff)ion of the M3+
increases steadily with increasing
atomic number and hence the electronegativities of the ions increase, and the radii of the ions
decrease.
The effect of ionic size on properties of compounds is noticeable as one traverses the series.
Salts become somewhat less ionic as the Ln3+
radius decreases across the series. The
hydroxides become less ionic and therefore become less basic. At the end of the series
Yb(OH)3 and Lu(OH)3, though mainly basic, can be made to dissolve with difficulty in hot
conc. NaOH.
The hydrated ions, [Ln(H2O)x]3+
, become increasingly susceptible to hydrolysis as one goes
across the series and need to be stabilized by addition of acids to their solutions.
There are no consistent trends noticeable in their aqueous or non-aqueous solutions. Some
distinctions can, however, be made between the cerium sub-family and the terbium sub-
family. The oxalates, double sulphates, and double nitrates of the cerium sub-family are
rather less soluble and the basic nitrates are more soluble than those of the terbium sub-
family.
Other oxidation states of +2 and +4 occur in Eu2+
and CeIV
. The two oxidation states are
stable in water and, even though they are strongly reducing and strongly oxidizing agents
respectively, have well established aqueous chemistry. LnIV
(Ln = Pr, Tb) and LnII (Ln = Nd,
Sm, Dy, Tm, and Yb) are also known in the solid state but are not stable in water.
Europium and ytterbium are particularly similar to the alkaline earth elements. They have the
lowest enthalpies of vaporization and the largest atomic radii of the lanthanides, more similar
to barium than to typical lanthanides.
One difference between the lanthanides and the transition metals lies in the sum of the first
three ionization energies which is 3 500 - 4 200 kJ mol-1
for the lanthanides compared to 5
230 kJ mol-1
for Cr3+
and 5 630 kJ mol-1
for Co3+
. The heat of atomization necessary to break
up the metal lattice is higher in the d transition series (because the d electrons are available
for bonding) than in the alkali, alkaline earth, and lanthanide metals.
12.6.1. Oxidation states
Since the 5d16s
2 electrons are mainly valence electrons in the lanthanides their most stable
oxidation state is +3. The elements adjacent to lanthanum (4f0), gadolinium (4f
7) and
lutenium (4f14
), however, have variable oxidation states. Thus in addition to +3 state, Ce has
+4 state arising from the transition of the 4f2 electrons to the 5d level; praseodymium with
4f36s
2 may also display +4 (by promoting 2 electrons to the d orbital) oxidation state although
this is less characteristic than in Ce. Eu with configuration 4f76s
2 can also have oxidation
state of +2.
Table 3. Oxidation states
La

+3
Ce
+3, +4
Pr
+3, +4
Nd
+3
Pm
+3
Sm
+3, +2
Eu
+3, +2
Ga
+3
Tb
+3, +4
Dy
+3, +4
Ho
+3
Er
+3
Tm
+3, (+2)
Yb
+3, +2
Lu
+3
Summary of the some basic reactions of the lanthanides
Ln
H2300 - 400
0C
X2
HeatLnX3
LnH2; LnH3
N2 (C, Si, P, S, and other non metals)
LnN
Heat
H2O
Ln(OH)3 + H2
O2
Heat
Ln2O3
(but CeO2
Ln2(CO3)3 + H2
H2O + CO2
Ln = Lanthanides
X = Halides
Reactions of Lanthanides
Complexes
The lanthanide ions have high charge but they are rather large (85 -100 pm) compared with
the normal transition elements (Cr3+
= 60, Fe3+
= 64 pm) and consequently they do not form
complexes very readily. The most common and stable complexes are those with chelating
oxygen ligands such as citric acid, oxalic acid, EDTA, and acetyacetone. These complexes
have water/solvent molecules attached to the central metal ion, and coordination numbers 7,
8, and 9 are very common. A variety of stereochemistries and coordination numbers are
found, e.g.,
Coordination number 7: [Er(NCS)6]3-
octahedral
Coordination number 10 [Ce(NO3)5]2-
trigonal bipyramidal; each NO3- is bidentate.

Complexes with monodentate ligands are much less stable than the chelates and undergo
dissociation easily in aqueous solution. Only en and NCS- complexes of N-donor ligands are
known and they are readily decomposed by water.
Ce4+
is smaller and has greater charge density. [Ce(NO3)6]2-
is formed in non-aqueous solvent
N2O4. It is 12 coordinate.
They have no complexes with π-bonding ligands, and this is attributable to the non-
availability of the f-orbital for bonding.
However, the existence of high coordination numbers would suggest that it is either that the f-
orbitals are involved in some bonding or that bond orders are less than one since involvement
of s, p, d orbitals would give a maximum of coordination number of 9.
12.7. Spectral and magnetic properties
Colour
The lanthanide ions that have unpaired electrons are coloured and are paramagnetic. Note
from Table 1 that the colour of 4fn ≈ 4f
14-n.
There is a fundamental difference between the spectra of the f-elements and those of the d-
elements. The difference arises from the fact that the 4f electrons are effectively shielded
from the influence of the external forces by the overlap of 5s2 and 5p
6 orbitals whereas the d-
electrons of the d transition metals are exposed directly to the influence of neighbouring
groups.
Consequently the 4fn configurations are only slightly affected by the chemical environments
of the ions and remain practically invariant for a given ion in all of its compounds.
On the other hand the free ion ground term of the d metals ions are subjected to the influence
of the chemical environment. It is therefore subjected to the effect of crystal field first before
any spin coupling comes into play.
As a result, electronic transitions between f orbitals give rise to extremely narrow absorption
bands, quite unlike the broad bands resulting from d-d transitions, and the magnetic
properties of the ions are little affected by their chemical surroundings.
Absorptions are observed in the visible or near UV regions of the spectrum except La3+
with
no f electrons and Lu3+
with no empty f orbitals.
Neither of the two ions shows any absorption bands in the UV/visible region.
The colours observed are due to transitions between f levels, f-f transitions.
Since the f-levels lie deep enough in the atom to be shielded from much perturbation by the
environment these transitions appear in the visible and near UV as sharp bands.
The sharp bands are useful for characterizing the lanthanides and for quantitative estimations.
The bands are weak because they are forbidden and the transitions only result in electron re-
distributions within the f orbitals.

The positions of the bands shift as the configurations changes giving rise to the visible
colours of the different ions given in Table 1.
Although Ce4+
is intensely coloured, the colour is not due to f-f transitions but to the charge
transfer transition between the ion and coordinated ligands.
Paramagnetism
In all cases where the f orbital is partially occupied the compounds are paramagnetic. These
elements differ from the d-block elements in that their magnetic moments do not obey the
simple spin-only formula:
µ = √n(n+2) B.M. 1
n = number of unpaired electrons or
µ = g√s(s+1) 2
s is the absolute value of the spin quantum number and g is a constant called the
gyromagnetic ratio ≈ 2.00.
The magnetic effect arising from the motion of the electron in its orbital (orbital contribution)
as well as that arising from the electrons spinning on its axis contribute to the paramagnetism
of the lanthanides (in the normal transition metal this orbital contribution, to a first
approximation, is usually quenched out by interaction with the electric fields of the
environment).
The magnetic moments of the lanthanides calculated by incorporating the orbital contribution
is given by:
µ = g√J(J+1) 4
g = 1 +S(S + 1) - L(L+1) + J(J+1)
2J(J+1)where
5
3
2 2J(J+1)
+ S(S+1) - L(L+1)=
The orbital moment is given by
µ = √L(L+1) 6
and the spin moment is given in equation 2.
The moment, taking care of orbital contribution is given by
µ = √4S(S+1) + L(L+1) 7
If the ground states of transition metal ions or lanthanides ions are S states (L = 0), there is no
orbital contribution, hence
µ = √4S(S+1) 8

The calculated magnetic moments of the lanthanide ions using equation 5 are given in Table
4.
Example: Calculate the magnetic moment of Ce3+
given that the ground term is 2F5/2
Ce3+
has configuration 4f1, S = ½, L = 3, J = L - S = 5/2
From equation 5 calculate g = 6/7
µ = 6/7√5/2(7/2) = 6/7√35/4 = 18/7
Table 4. Magnetic moments of lanthanide ions (Ln3+
)
Ion Ground state g µ (Calculated) µ
(experimental)
La3+
Ce3+
Pr3+
Nd3+
Pm3+
Sm3+
Eu3+
Gd3+
Tb3+
Dy3+
Ho3+
Er3+
Tm3+
Yb3+
Lu3+
1S0
2F5/2
3H4
4I9/2
5I4
6H5/2
7F0
8S7/2
7F6
6H15/2
5I8
4I15/2
3H6
2S7/2
1F0
-
6/7
4/5
8/11
3/5
2/7
-
2
3/2
4/3
5/4
6/5
7/6
8/7
-
0
2.54
3.58
3.62
2.68
0.34
0
7.94
9.72
10.63
10.60
9.57
7.63
4.50
0
0
2.3 -2.5
3.4 – 3.6
3.5 – 3.6
-
1.5 – 1.6
3.4 – 3.6
7.8 - 8.0
9.4 - 9.6
10.4 – 10.5
10.3 – 10.5
9.4 – 9.6
7.1 – 7.4
4.4 – 4.9
0
Note that except Sm3+
and Eu2+
the calculated values agree with experimental values.
Russell Saunders coupling
Spin-orbit coupling, which gives rise to a resultant angular momentum associated with an
overall quantum number J is much larger in the lanthanides than the crystal field and the
effect must be considered first.
J can take values J = L+S, L+S-1, ---L-S (or S-L if S<L), each corresponding to a different
energy, so that a term (defined by a pair of L and S values) is said to be split into a number of
component states (each defined by the same S and L values plus a value of J). The ground
state of the ion is that with J = L – S (or S – L) if the f shell is less than half-filled, and that
with J = L + S if the f shell is more than half filled. It is indicated simply by adding this
value of J as a subscript to the symbol for the ground term.
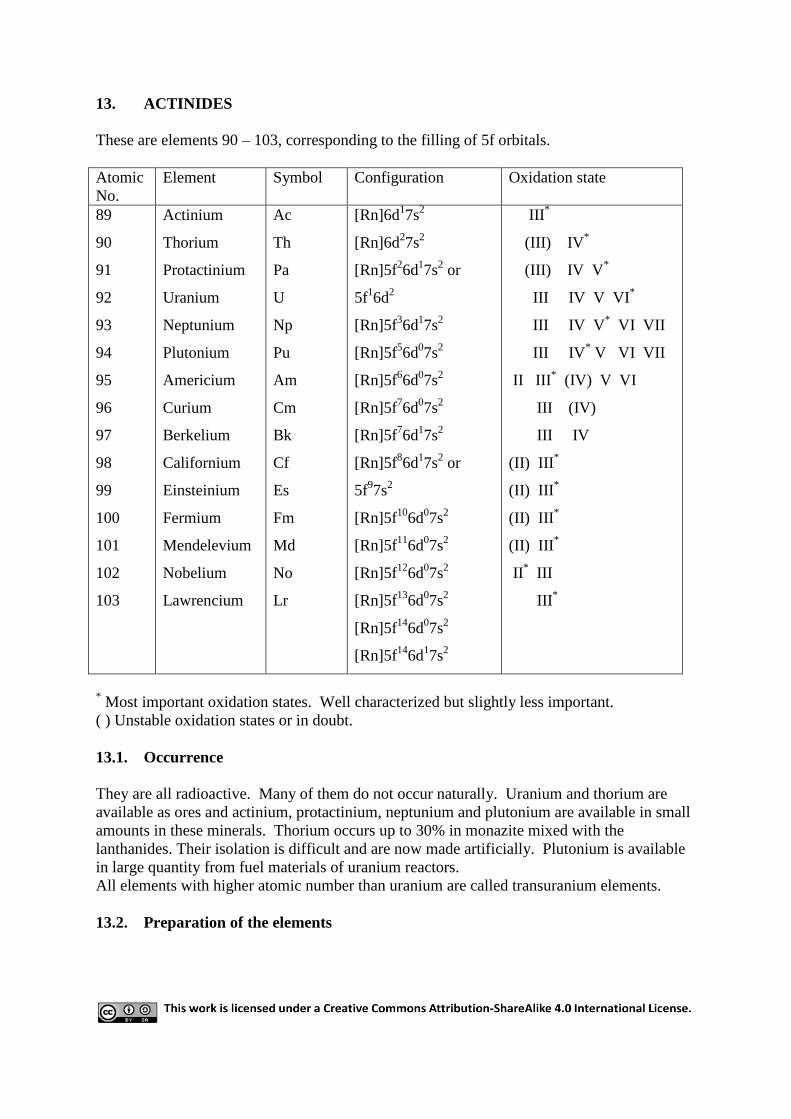
13. ACTINIDES
These are elements 90 – 103, corresponding to the filling of 5f orbitals.
Atomic
No.
Element Symbol Configuration Oxidation state
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
Actinium
Thorium
Protactinium
Uranium
Neptunium
Plutonium
Americium
Curium
Berkelium
Californium
Einsteinium
Fermium
Mendelevium
Nobelium
Lawrencium
Ac
Th
Pa
U
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
[Rn]6d17s
2
[Rn]6d27s
2
[Rn]5f26d
17s
2 or
5f16d
2
[Rn]5f36d
17s
2
[Rn]5f56d
07s
2
[Rn]5f66d
07s
2
[Rn]5f76d
07s
2
[Rn]5f76d
17s
2
[Rn]5f86d
17s
2 or
5f97s
2
[Rn]5f10
6d07s
2
[Rn]5f11
6d07s
2
[Rn]5f12
6d07s
2
[Rn]5f13
6d07s
2
[Rn]5f14
6d07s
2
[Rn]5f14
6d17s
2
III*
(III) IV*
(III) IV V*
III IV V VI*
III IV V* VI VII
III IV* V VI VII
II III* (IV) V VI
III (IV)
III IV
(II) III*
(II) III*
(II) III*
(II) III*
II* III
III*
* Most important oxidation states. Well characterized but slightly less important.
( ) Unstable oxidation states or in doubt.
13.1. Occurrence
They are all radioactive. Many of them do not occur naturally. Uranium and thorium are
available as ores and actinium, protactinium, neptunium and plutonium are available in small
amounts in these minerals. Thorium occurs up to 30% in monazite mixed with the
lanthanides. Their isolation is difficult and are now made artificially. Plutonium is available
in large quantity from fuel materials of uranium reactors.
All elements with higher atomic number than uranium are called transuranium elements.
13.2. Preparation of the elements

Neptunium and plutonium, named after planets, were made in 1940 by McMillan and
Abelson, and by Seaborg, McMillan, Kennedy, and Wahl respectively. Both are now
obtained from spent uranium fuel from nuclear reactors.
Two general methods that are to some extent complementary have been used for preparing
the transuranium elements:
Capture of neutrons, followed by β-decay which increases the atomic numbers by one
and
Capture of the nuclei of light elements, ranging from He to Ne, which increases
atomic number by several units in one step.
Successive neutron capture-β-decay sequences
The discovery of neptunium was based on the following sequence:
234
U (n,
U
23 min.
239Np
2.3d
239Pu
With the discovery of high power nuclear reactors an extension of the above sequence has
been used to produce other elements:
238U
(n, 239U
239Np
(n,
239Pu
(n,
240Pu
241Pu
24 360 y
241Am
The most stable isotope of Np,
237Np, is obtained by:
235
U
238U
(2n,
(n,2n)
237U
6.75d
93Np
237(2.2 x 10
6 y)
Only Pu is normally recovered since 239
Pu has fission properties similar to 235
U and can be
used as a fuel or in nuclear weapons. Some 237
Np is used to prepare 238
Pu (86.4 y), which is
used as a power source for satellites.
This method suffers from diminishing returns as each element has to be made from the one
before. For example in the irradiation of plutonium-239 less than 1% of the original sample
appears as californium-252 after the capture of 13 neutrons,

239Pu →
241Pu→
245Cm →
247Cm →
251Cf →
252Cf
% of original sample 100 30 10 1.5 0.7 0.3
The yield of heavier elements is controlled by:
The half lives of the various isotopes;
The ability to absorb neutrons.
The yield of heavier nucleus falls off sharply because successive neutron capture depends on
the build-up of intermediate elements and also because of decrease in nuclear stability with
increasing atomic weight. The heaviest elements are therefore best obtained by means of
cutting down on the step-by-step addition of neutrons. One means was provided by H-bomb.
A staring material can be bombarded with species that contains several nuclear particles. For
example the α-particle bombardment is the easiest way, and many actinides, such as 248
Cm, 249
Bk, 249
Cf, and 256
Md were first made in this way: e.g. 253
Es99 + 4He2 →
256Md101 +
1n0
α-particle bombardment requires the target to be the element with an atomic number of two
less than the desired element.
Both methods discussed above suffer from limitations, in so far as in both cases the yields
decrease rather rapidly as the atomic number increases. In the α-particle bombardment the
decreasing yields of successive elements arise first because the amounts of available target
materials are smaller, and also the probability of fission in the compound nuclei increases
quite rapidly with its charge.
Two modifications of the above methods have been used to produce isotopes of elements up
to 105:
Nuclear explosions
There is a vast flux of fast moving neutrons in an atomic explosion that can lead to
simultaneous addition of a number of neutrons before the intermediate nuclei can decay.
Thus einsteinium and fermium were first discovered in the fall out products of the first
atomic bomb.
Similar process to nuclear explosion occurs in certain stars called supernovae. Supernovae
arise as a result of a gigantic nuclear explosion in the star, creating neutron fluxes many
orders of magnitude greater than even the most powerful man-made devices.
Bombardment involving heavy ions (pioneered by the Russians)
Ions involved include B5+
, C6+
, N7+
, or O8+
. For example,
238U +
12C (
250Cf)
*
246Cf + 4 n
fission products In this way it is possible to „leap up‟ several elements in one step:
238
U92 + 12
C6 → 246
Cf98 + 4 1n0
238U92 +
16O8 →
250Fm100 + 4
1n0

246Cm96 +
12C6 →
254No102 + 4
1n0
252Cf98 +
11B5 →
257Lw103 + 6
1n0
13.3 General Chemical Properties of the Actinides
As discussed above there appears to be a competition between 5fn7s
2 and 5f
n-16d
17s
2
configurations. For the elements in the first half of the f shell it appears that less energy is
required for the promotion of 5f → 6d than for 4f → 5d promotion in the lanthanides; there is
thus a greater tendency to supply more bonding electrons with the corollary of higher
valences in the actinides. The second half resembles the lanthanides more closely.
Furthermore the 5f orbital is more accessible to bonding than the 4f. In the actinide series a
situation arises in which the energies of 5f, 6d, 7s, and 7p orbitals are about comparable over
a range of atomic numbers (especially U to Am). Bonding can therefore involve any or all of
them. In the chemistry this situation is indicated by the fact that the actinides are much more
prone to complex formation than are the lanthanides, where the bonding is almost extremely
ionic. Indeed the actinides can even form complexes with certain π-bonding ligands as well
as forming complexes with halides, sulphate, and other ions.
Since the energies of 5f, 6d, 7s, and 7p levels are comparable the energies involved in an
electron shifting from one to another may lie within the range of chemical binding energies.
It is therefore difficult to place the electronic structure of the elements in compounds and in
solutions as the ligands vary. It is also impossible to say which orbitals are being utilized in
bonding or to decide meaningfully whether the bonding is covalent or ionic.
Roughly the series falls into two families:
The first seven members have properties that are similar to the d-transition series while the
latter seven are similar to the lanthanides.
13.3.1 Oxidation states
The most stable oxidation state of the elements up to uranium is the one involving all the
valence electrons. Thus Np forms Np(VII), using all its 5f and 7s electrons. This oxidation
state is oxidizing and the most oxidation state is Np(V). Pu also forms Pu(VII) and Am up to
Am(VI) but the most stable oxidation state drops to Pu(IV) and Am(III) respectively. This
variable oxidation states make these early members of the series similar to the 3d-transition
series. (Recall that for the 4d and 5d series the highest oxidation states are more stable and
for the 3d series the lower oxidation states are more stable and dominate the aqueous
chemistry).
The latter elements tend to be most stable in the III state like the lanthanides.
This pattern of higher oxidation state stabilities has more in common with the d-series than
with the lanthanides. Compare Mn(VII) or Ru and Os(VIII) which states become more
oxidizing across the series.
However the III state becomes predominant from Cm → Lr, similar to the lanthanides.
Note that although Bk(IV) is strongly oxidizing it is more stable that Cm(IV) and Am(IV),
thus showing a parallel to Tb where the IV state corresponding to the f7 configuration has
some stability. Am(II) is not formed in aqueous solution but known in chloride melts. This

shows some slight resemblance to Eu which attains f7 configuration in its fairly stable II state.
No(II) is stable due to the attainment of the f14
configuration, analogous to Yb2+
in the
lanthanides.
The trend in ionic radii is similar to that shown by the lanthanides hence one can refer to the
actinide contraction arising from similar increase in effective nuclear charge due to poor
shielding by the f electrons.
13.3.2. Difference between 4f and 5f orbitals
The chief difference between the two depends upon the relative energies and spatial
distribution of the orbitals. The 4f orbitals populated in the lanthanides are sufficiently low in
energy that the electrons are seldom ionized or shared (hence the rarity of the LnIV
species).
Furthermore the 4f electrons are buried so deeply within the atom that they are unaffected by
the environment to any great degree. In contrast the 5f electrons, at least in the earlier
elements of the actinides, Th to Bk, are available for bonding allowing oxidation states up to
+7. In this respect these electrons resemble d electrons of the normal transition elements.
However, the heavier members resemble the lanthanides in displaying mainly oxidation state
of +3.
13.3.4. Electronic spectra of the lanthanides and actinides
As discussed earlier the electronic spectra of the lanthanides are typically sharp. The
absorption of actinides may be conveniently divided into two groups:
Am3+
and heavier actinides which have spectra that resemble the lanthanides;
Pu3+
and lighter actinides that have spectra similar in some ways but which have a
tendency towards broadening of absorption peaks, somewhat like the broadening seen
in the d-block metal ions.
Apparently the greater exposure of the 5f orbitals in the lighter actinide elements results in
greater ligand-metal orbital interaction and some broadening from vibrational effects. As the
nuclear charge increases the 5f orbitals behave more like the 4f orbitals in the lanthanides and
the spectra of the heavier actinides become more lanthanide-like.
13.3.5. Magnetic properties
The magnetic properties of the actinide ions are considerably harder to interpret than those of
the lanthanide ions. The experimental magnetic moments are usually lower than the values
calculated by using Russell-Saunders coupling and this appears to be due both to ligand field
effects similar to those operating in the d transition series and to inadequacy of this coupling
scheme. Since 5f can participate to some extent in covalent bonding ligand effects are to be
expected.
13.3.6. Electronic spectra
The electronic spectra of the actinide compounds originate from three types of electronic
transitions:
(i) 5f → 5f transitions:
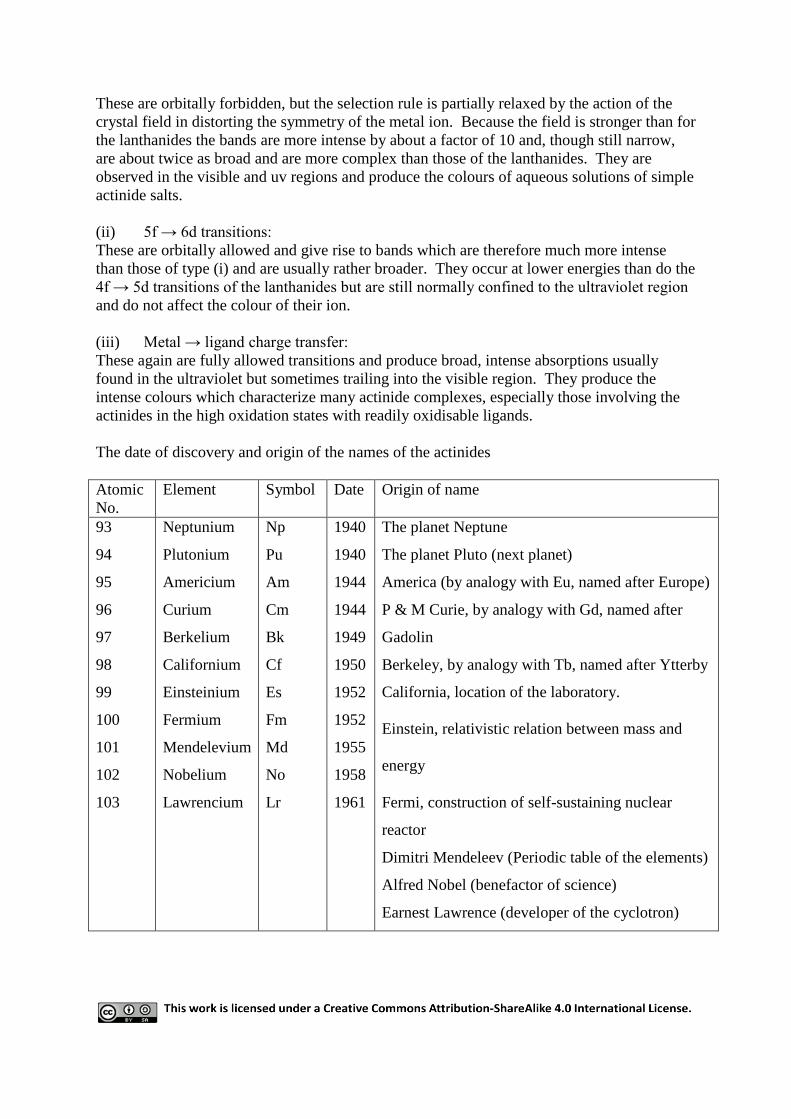
These are orbitally forbidden, but the selection rule is partially relaxed by the action of the
crystal field in distorting the symmetry of the metal ion. Because the field is stronger than for
the lanthanides the bands are more intense by about a factor of 10 and, though still narrow,
are about twice as broad and are more complex than those of the lanthanides. They are
observed in the visible and uv regions and produce the colours of aqueous solutions of simple
actinide salts.
(ii) 5f → 6d transitions:
These are orbitally allowed and give rise to bands which are therefore much more intense
than those of type (i) and are usually rather broader. They occur at lower energies than do the
4f → 5d transitions of the lanthanides but are still normally confined to the ultraviolet region
and do not affect the colour of their ion.
(iii) Metal → ligand charge transfer:
These again are fully allowed transitions and produce broad, intense absorptions usually
found in the ultraviolet but sometimes trailing into the visible region. They produce the
intense colours which characterize many actinide complexes, especially those involving the
actinides in the high oxidation states with readily oxidisable ligands.
The date of discovery and origin of the names of the actinides
Atomic
No.
Element Symbol Date Origin of name
93
94
95
96
97
98
99
100
101
102
103
Neptunium
Plutonium
Americium
Curium
Berkelium
Californium
Einsteinium
Fermium
Mendelevium
Nobelium
Lawrencium
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
1940
1940
1944
1944
1949
1950
1952
1952
1955
1958
1961
The planet Neptune
The planet Pluto (next planet)
America (by analogy with Eu, named after Europe)
P & M Curie, by analogy with Gd, named after
Gadolin
Berkeley, by analogy with Tb, named after Ytterby
California, location of the laboratory.
Einstein, relativistic relation between mass and
energy
Fermi, construction of self-sustaining nuclear
reactor
Dimitri Mendeleev (Periodic table of the elements)
Alfred Nobel (benefactor of science)
Earnest Lawrence (developer of the cyclotron)

14 INORGANIC REACTION MECHANISM
14.0 Preliminary comments
14.1 Inert and labile compounds:
Compounds that undergo substitution reactions at room temperature spontaneously are
said to be kinetically labile.
Examples: Addition of NH3 to aqueous solution of copper(II) in excess results in
instantaneous formation of a deep blue solution.
[Cu(H2O)6]2+
+ 4 NH3 → [Cu(NH3)4(H2O)2]2+
+ 4 H2O
blue deep blue
Addition of a thiocyanate solution to an iron(III) solution results in a change in colour from
pale blue to red:
[Fe(H2O)6]3+
+ SCN- → [Fe(H2O)5(SCN)]
2+ + H2O
very pale blue red
Compounds like these are said to be labile.
Taube suggested a reaction half-life of one minute or less as the criterion for lability.
Those where substitution takes hours/days are said to be kinetically inert.
Inert here does not suggest that reaction will not take place; it simply implies that the reaction
is very slow.
An example is the acid hydrolysis of [Co(NH3)6]3+
:
[Co(NH3)6]3+
+ 6 H3O+ → [Co(H2O)6]
3+ + 6 NH3
This reaction is very slow and hence [Co(NH3)6]3+
is an inert complex.
14.2. Inertness and stability:
[Cu(H2O)6]2+
, Fe(H2O)6]3+
and [Co(NH3)6]3+
are all thermodynamically stable because their
equilibrium constants for formation are very large. However, whereas the Cu2+
and Fe3+
are

labile (because they undergo substitution reactions readily, [Co(NH3)6]3+
is inert because it
undergoes substitution very slowly.
14.3. Differences between stability and inertness:
thermodynamically stable complexes have large, positive free energies of
reaction, G.
Inert complexes have large positive free energies of activation, G*
14.4. General rules guiding lability/inertness:
Labile complexes:
Complexes with central metal atom having d-electrons in the eg orbitals, e.g.
[Ga(C2O4)3]3-
, d10
; [Co(NH3)6]2+
, d7; [Cu(H2O)6]
2+, d
9; [Ni(H2O)6]
2+, d
8 (weak field) and
[Fe(H2O)6]3+
, d5 (high spin).
Complexes containing less than 3 electrons in the d orbitals, e.g. [Ti(H2O)]3+
, d1;
[V(phen)3]3+
, d2; and [Ca(EDTA)]
2+, d
0.
Inert complexes:
Octahedral low-spin d4, d
5, and d
6 complexes, e.g. [Fe(CN)6]
3-, d
5; [Co(NO2)6]
3-, d
6;
[PtCl6]2-
, d6.
Octahedral d3 complexes. e.g. [Cr(H2O)6]
3+, d
3.
Complexes with d8 configurations generally react somewhat faster but slower than the d
7,
d9, or d
10 complexes. Many square planar d
8 complexes are inert.
If you recall the Ligand Field Theory discussed last year and this year, you will realise that
the LFSE helps to see the picture clearly.
Summary:
Slow reactions (inert) Intermediate Fast reaction (labile)
d3, low spin d
4, d
5, and d
6,
strong field d8 (square
weak field d8
d1, d
2, high spin d
4, d
5, and
d6
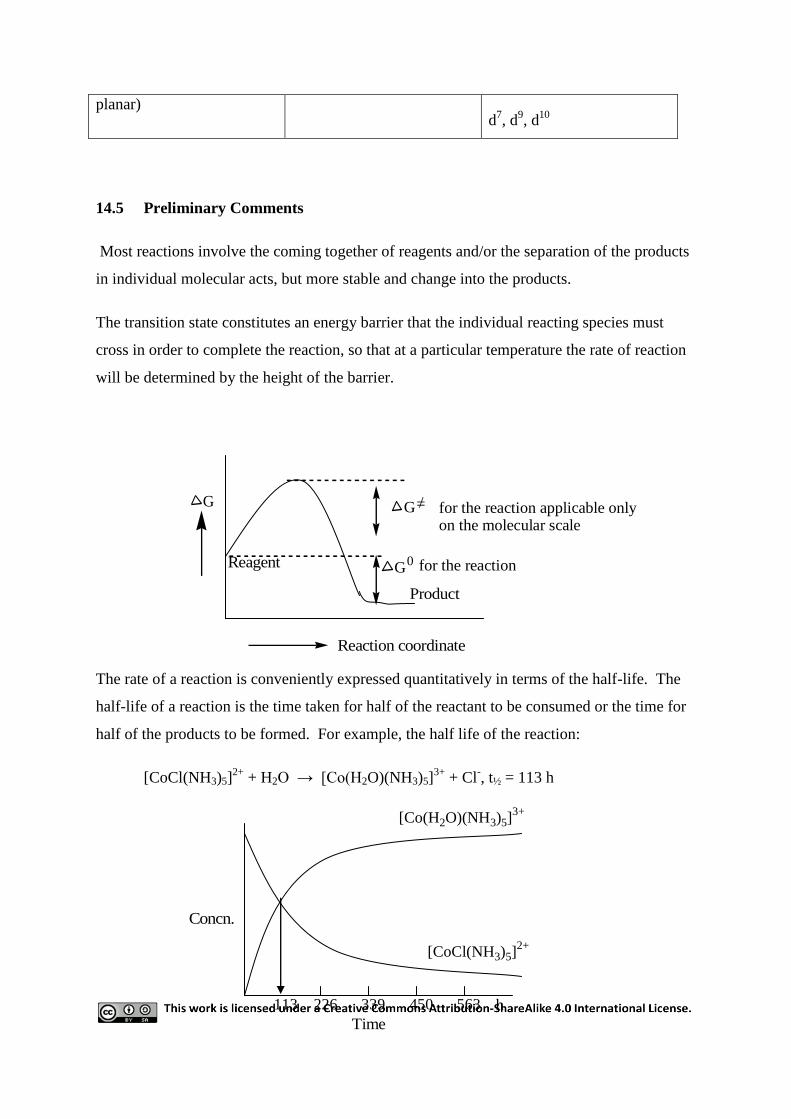
planar) d
7, d
9, d
10
14.5 Preliminary Comments
Most reactions involve the coming together of reagents and/or the separation of the products
in individual molecular acts, but more stable and change into the products.
The transition state constitutes an energy barrier that the individual reacting species must
cross in order to complete the reaction, so that at a particular temperature the rate of reaction
will be determined by the height of the barrier.
The rate of a reaction is conveniently expressed quantitatively in terms of the half-life. The
half-life of a reaction is the time taken for half of the reactant to be consumed or the time for
half of the products to be formed. For example, the half life of the reaction:
[CoCl(NH3)5]2+
+ H2O → [Co(H2O)(NH3)5]3+
+ Cl-, t½ = 113 h
G0 for the reaction
G=/ for the reaction applicable only on the molecular scale
G
Reagent
[Co(H2O)(NH3)5]3+
[CoCl(NH3)5]2+
113 226 339 450 563 h
Concn.
Time
Reaction coordinate
Product

14.6 Definitions
Reaction rate is the rate of change of concentration of a substance involved in the reaction
(being either substrate or product or both).
Rate constants are expressed as a function of temperature (or time) factor.
Rate expression is the functional relation between rate and concentration; it provides the
important clue about the mechanism.
14.7 Elucidation of mechanism
The most important evidence in the elucidation of a reaction mechanism is the experimentally
determined rate equation.
For reaction
A + B → C + D
the rate law is given as
= ka[A] + kab[A][B] + kab'[A][B][H
+]
-1
Other additional information are, however, very essential too. In particular, attention should
be paid to
the exact nature, including the stereochemistry, of the reactants and products.
the presence of equilibria.
the stoichiometry of the reaction.
14.8 Confirmation of a mechanism from other than kinetic evidence
Confirmation of a mechanism suggested on the basis of a rate equation may be obtained
from:
Detailed knowledge of the nature of the reactants.
Detection (direct or indirect) of suspected intermediates.
Detailed knowledge of the nature of the products.

One example only:
The copper(I) reduction of iron(III),
Cu(I) + Fe(III) → Cu(II) + Fe(II)
The rate law experimentally determined is
][
)]()][([)([
H
ICuIIFed
dt
IIIFed
The inverse dependence on [H+] suggests that a hydrolysis product is the reagent. It is known
that iron(III) solution at a given pH contains the hydroxopentaaquairon(III) ion and that Cu(I)
does not undergo any measurable hydrolysis under the given conditions indicates that it is the
hydrolysed iron(III) species which is the reactant. The mechanism is then represented as:
[Fe(H2O)6]3+
+ H2O → [Fe(H2O)5OH]2+
+ H3O+
[Fe(H2O)5OH]2+
+ Cu(I) Products
15. Substitution in Octahedral Complexes
Simple substitution is defined as the replacement of a ligand in the coordination sphere by
another coming from the environment by a path that involves nothing more complicated than
a temporary change in the coordination number of the reaction centre. Thus making and
breaking of bonds are involved.
Substitution reactions are generally classified as having either dissociative or associative
mechanisms.
Consider: [A5MX] + Y- [A5MY] + X
-
Two reaction pathways are possible:
Extreme dissociative mechanism, the D or SN1 mechanism, can be represented as:
or
Co
X
A A
A
AA
-XCoA A
A
AA
Co
Y
A A
A
AA
+Y
Slow Fast
Intermediate

[A5MX]n+
[A5M](n+1)+ + X
-
Y-
[A5MY]n+
-X
slow
fast
This involves a rate-determining (slow) loss of X- to give an intermediate with a coordination
number reduced by one. Subsequent addition of Y to form the product is fast.
SN1 = means substitution nucleophilic unimolecular.
The extreme associative mechanism, the A or SN2 mechanism, can be represented as:
This involves the rate-determining slow addition of Y- to form an intermediate with a
coordination number increased by one. Subsequent loss of X- is fast.
SN2 = Substitution nucleophilic bimolecular.
Both D and A mechanisms consist of two steps: bond breaking and bond making.
Most reactions are, however, intermediate in character: both bond breaking and bond
making occur simultaneously. Such concerted processes are said to occur by I (for
Interchange) mechanism. Here, a cooperative interchange occurs in which X- leaves as
Y- arrives. Bond breaking takes place as bond formation is being executed.
The I mechanism is further divided into two: ID and IA
Dissociative Interchange (ID) mechanism: breaking of metal-ligand bonds has a greater
effect on the rate and activation energy than formation of new bonds.
Co
X
A A
A
AA
CoA
AAA
Co
Y
A A
A
AA
Slow Fast
Intermediate
X
A
Y
+Y -X
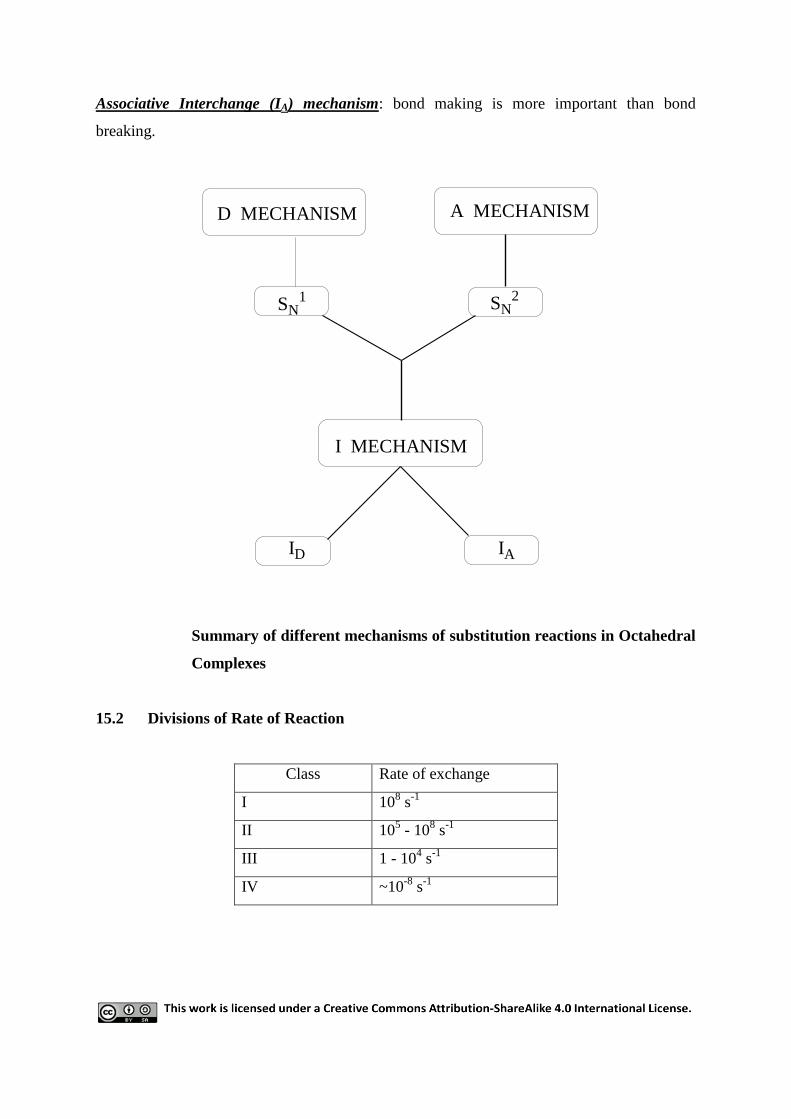
Associative Interchange (IA) mechanism: bond making is more important than bond
breaking.
Summary of different mechanisms of substitution reactions in Octahedral
Complexes
15.2 Divisions of Rate of Reaction
Class Rate of exchange
I 108 s
-1
II 105 - 10
8 s
-1
III 1 - 104 s
-1
IV ~10-8
s-1
D MECHANISM A MECHANISM
SN1 SN
2
I MECHANISM
ID IA

Class I: Rate constant ~10-8
s-1
. Here, purely electrostatic forces bind complexes. Ions
of alkali and alkali earth metals fall in this class. Ratio r
Z 2
for these ions
range up to
~10 x 1028
C2 m
-1. This class is difficult to study.
Class II Exchange of H2O is fast and first order rate constant. This class can be studied
using relaxation techniques. Here the equilibrium is perturbed by a fast
variation of pressure and temperature, and the response of the system is used
to estimate the rates of reaction. Dipositive transition metal ions, Mg2+
, and
Ln3+
are known. Here bonding is stronger than in Class I; but LFSE is
relatively small. r
Z 2
for these ions ranges
10 - 30 x 10-28
C2 m
-1
Class III H2O exchange is relatively slow than in I and II. First order rate constant is 1 -
104 s
-1. Reactions can be followed by more or less conventional kinetic
technique. Tripositive transition metal ions are examples, and stabilised, to
some extent, by LFSE. r
Z 2
ratio is greater than 30 x 10-28
C2 m
-1.
Class IV H2O exchange is slow, an example of inert complexes' behaviour. First order
rate constants range from 10-1
- 10-9
s-1
. Metal ions are comparable in size to
Class III ions and exhibit considerable LFSE, e.g. Cr3+
(d3) with CFSE of
12Dq; Co3+
(d6 low spin) with LFSE 24Dq and low spin d
8 ions like Pt
2+.
NOTE:
It is not the LFSE that makes reaction to be very slow but the loss upon formation of the
intermediate (activated complex). For example, if LFSE of the initial complex is >> than
that of the activated complex then the complex will be inert; but if the difference is small
then the complex will be labile.
The rate is also affected by the charge of the Mn+
ion, Mn3+
< M2+
.
Lability increases: Co3+
< Cr3+
< Mn3+
< Fe3+
etc.
Lability decreases: [AlF6]3-
> [SiF6]2-
> [PF6]-> SF6 (inert).
15.3 Complications
Solvent interaction

Ion pair formation
Nature of the ligands (Leaving and entering ligands)
15.4 The Rate Laws
D Mechanism:
][][
55
MXAkdt
MXAd
A Mechanism
]][[][
55
YMXAkdt
MXAd
Unfortunately, however, a particular rate law does not prove that the reaction is SN1 or SN
2.
For example, where there is solvent interaction, which is very common in reactions
conducted in aqueous solution, the keq value will not be a true value, and will have to be
corrected. Consequently for a reaction like
the rate law is
]][[][
255
OHMXAkdt
MXAd
= k/[A5MX]
since [H2O] is constant because water is the solvent. This reaction can then be SN1 or SN
2.
Generally for Co3+
, which is the ion of interest, substitution follows the D mechanism.
Ion pair formation:
Common when reacting cations and anions; and also in non-aqueous solvents.
So, the equilibrium constant, K1p, should be taken into consideration (in certain cases).
[A5MX] + H2OSlow
[A5M(H2O)] + X-
[A5M(H2O)] + Y-
Fast
[A5MY] + H2O
[A5MX]n+
K1p
k-1
{[A5MX]Y}n-m ....
kK1p[A5MX][Y]
1+ k-1 [Y]-d[A5MX] =

Rate is dependent on whether k-1[Y] << 1 or not.
15.5 Hydrolysis
Hydrolysis is generally classified according to the conditions. In acid solution the
process is termed acid hydrolysis.or aquation and is illustrated by the equation:
[A5MX] + H2O [A5M(H2O)] + X-
In a basic solution the process is termed base hydrolysis and illustrated by the equation:
[A5MX]n+
+ OH-
[A5M(OH)]n+
+ X-
Depending on pH of the reaction mixture it follows that the product of a given hydrolysis
can be a mixture of both the aqua and the hydroxo complex. For a typical complex for
which the base hydrolysis is observable the rate law is:
The first term (kA) refers to the acid hydrolysis and the second term (kB) refers to the base
hydrolysis.
15.5 Acid hydrolysis
At pH 0-3 base hydrolysis is negligible and the second term vanishes. The behaviour is then
first order.with respect to the complex and independent of acid concentration.
The rate law provides no information as to the role of water and does not enable a distinction
between a dissociative, associative or concerted process to be obtained. Mechanistic
information must, therefore, be obtained from other than kinetic sources.
For example, the rate of hydrolysis decreases with increase in the thermodynamic bond
strength of the Co-X bond indicating that this bond is broken initially before Y comes in.
Acid hydrolysis is easy to follow and pH dependent.
Substitution in octahedral complexes is predominantly dissociative, except probably when
the ion is large (like in the 2nd
and 3rd
row transition metal ions, where a dominant
associative character can be acquired).
In such larger ions there is more room for attack or lower nucleophilic attack and hence
permit association.
-d[A5MX]
dt= kA[A5MX] + kB[A5MX][OH
-]
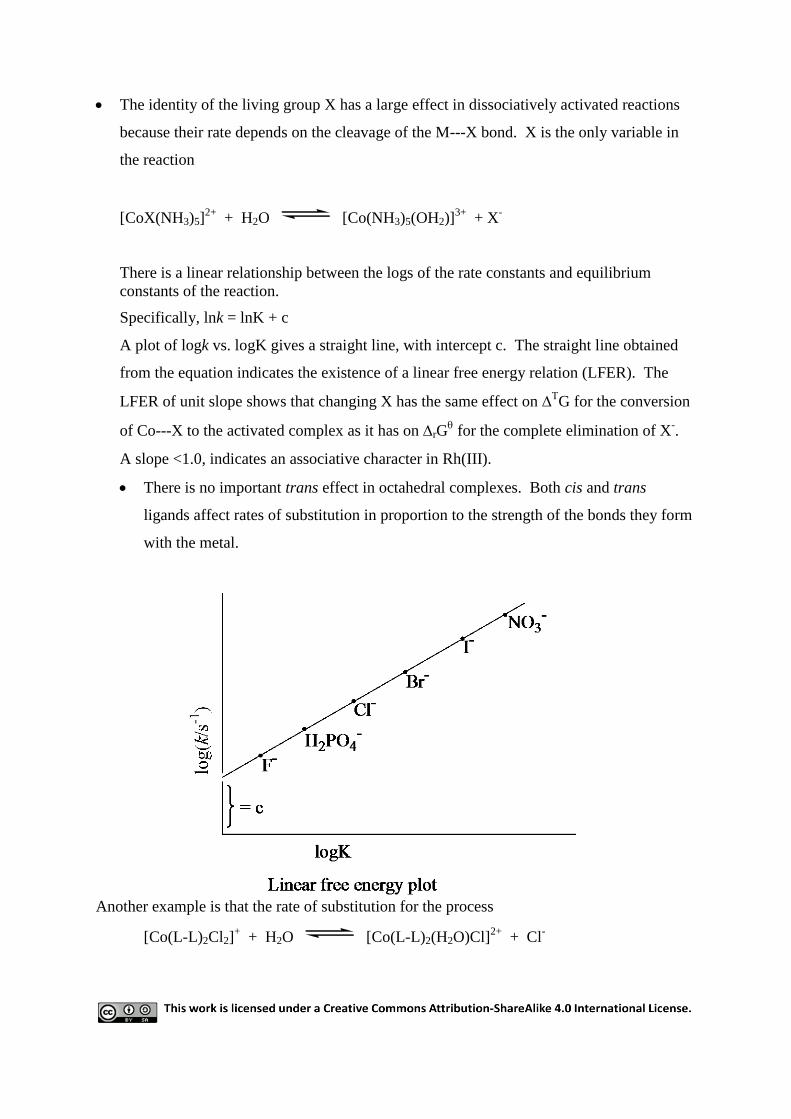
The identity of the living group X has a large effect in dissociatively activated reactions
because their rate depends on the cleavage of the M---X bond. X is the only variable in
the reaction
[CoX(NH3)5]2+
+ H2O [Co(NH3)5(OH2)]3+
+ X-
There is a linear relationship between the logs of the rate constants and equilibrium
constants of the reaction.
Specifically, lnk = lnK + c
A plot of logk vs. logK gives a straight line, with intercept c. The straight line obtained
from the equation indicates the existence of a linear free energy relation (LFER). The
LFER of unit slope shows that changing X has the same effect on G for the conversion
of Co---X to the activated complex as it has on rG for the complete elimination of X
-.
A slope <1.0, indicates an associative character in Rh(III).
There is no important trans effect in octahedral complexes. Both cis and trans
ligands affect rates of substitution in proportion to the strength of the bonds they form
with the metal.
Another example is that the rate of substitution for the process
[Co(L-L)2Cl2]+ + H2O [Co(L-L)2(H2O)Cl]
2+ + Cl
-

increases with an increase in size of the ligand, L-L. This is explained in terms of
dissociative process when an increase in size of the non-involved ligand discourages
association to give a seven-coordinate intermediate.
Note:
By increasing the bulkiness of the ligand, one decreases the solvation effect, and
then the transition state slows down the reaction rate.
It must go through a D-mechanism.
The inductive effect rises due to increase of alkyl group, by increasing charge
effect.
H2NCH2CH2NH2
L L k x 105 s
-1
3.2
H2NCH2CHNH2
CH3
6.2
H2N CH CHNH2
CH3
CH3
15.0
H2N CH CH
CH3 CH3
NH242.0
H2N C C
CH3 CH3
NH2
CH3 CH3
3300

Since the base strengths of the substituents do not change by more than a 1.5
factor, the increase in rates is not due to inductive effect and solvation effect, but
possibly due to steric strains.
15.7 The conjugate base mechanism
The behaviour of base hydrolysis appears to be associative mechanism, but
subsequently has been found to have a conjugate base mechanism (called SN1CB for
substitution, nucleophilic, unimolecular, conjugate base). The reactions depend on
amine, ammine, or aqua ligands that can lose protons to form amido or hydroxo
species that are then more likely to lose one of the orher ligands.
K
1. [Co(NH3)5Cl]2+
+ OH-
[Co(NH3)4(NH2)Cl]+ + H2O
(pre-equilibrium)
k2
2. [Co(NH3)4(NH2)Cl]+
[Co(NH3)4(NH2)]2+
+ Cl- (slow)
3. [Co(NH3)4(NH2)]2+
+ H2O [Co(NH3)5(OH)]2+
(fast)
Overall:
[Co(NH3)5Cl]2+
+ OH- [Co(NH3)5(OH)]
2+ + Cl
-
In the first step, an NH3 ligand acts as a BrØnsted acid, resulting in the formation of its
conjugate base, the NH2- ion, as a ligand. Because NH2
- is a strong -donor it greatly
accelerates the loss of Cl- ion.
Octahedral substitution is greatly accelerated by OH- ions when ligands with acidic protons
are present.
Basolo revealed that the role of OH- is catalytic, by removing a proton from the base ligand.
The rate law is
Rate = k[CoCl(NH3)52+
][OH-]
The presence of [OH-] in the rate equation shows that it plays a rate-determining role.
However, it is not because [OH-] attacks the metal centre but rather because it deprotonates a
coordinated NH3 ligand to form a conjugate base; hence the name of the mechanism. A pre-

equilibrium is first established, followed by loss of Cl- to give a reactive amido species, and
finally formation of the product in a fast step.
If the equilbrium constant for equation (1) is K then the rate law consistent with mechanism
is given by
Rate = ][1
]][)([ 2
532
OHK
OHClNHCoKk
Two supporting evidences for this mechanism are
If NH3 is replaced by pyridine or another tertiary amine, base hydrolysis is very much
slower.
The exchange of H (in NH3) for D in alkaline D2O is much faster than the rate of base
hydrolysis.
The isotope ratio (18
O/16
O) in the product in 18
O-enriched water is the same as that in
water regardless of the leaving group (X = Cl-, Br
-, NO3
-). If an incoming water molecule
had a large influence (associative mechanism), the concentration of 18
O should be larger
in the product;
RNH2 compounds react faster than NH3 compounds, showing that steric crowding
favours the 5-coordinate intermediate (eq. 2 above).
The rate constants and dissociation constants for the compounds form a linear free energy
relationship (LFER), where a plot of lnkOH versus lnKOH is linear.
15.8. Anation reactions
Involve the replacement of coordinated water:
[Co(NH3)5(H2O)]3+
+ X- [Co(NH3)5X]
2+ + H2O
They may undergo ion-pairing formation, leading to a slow rate of water
displacement, or
Purely undergoing dissociative pathway.
e.g.:

the intermediate has long enough half-life to distinguish it from ion-paring process.
Rate = -dt
OHCNCod ])([ 2
25
= ][
]][)([
21
2
2521
Xkk
XOHCNCokk
once X-is in large concentration, the rate law approximates a first order, i.e. pseudo-
first order reaction condition and the kobsd resolves as following:
][
][][
][1
21
21
Xkk
Xkksubstratek
dt
substratedobsd
By taking series of runs with varying [X-], k1 will tend to be
= ~1.6 x 10-3
s-1
and therefore the ratio of k2/ k-1 is a measure of the nucleophilic
power of the different substituents, and k-1 is independent of the substituents.
k2 k1/k-1 can be found from graph.
The order of k2 is as follows:
OH-> N3
-> NCS
-> I
-> Br
->NH3> H2O at 40
oC and = 1.0.
16. Substitution Reactions in Square Planar Complexes
A large number of relatively inert square planar complexes have been known for a
long time, e.g. PtII, Pd
II, Ni
II, Au
III, Rh
I and Ir
I. They all have d
8 configuration, which
gives stability to square planar complexes. We will concentrate our efforts on PtII,
which shows the general features which are common to all the others.
Let us consider [PtA2LX], where X = leaving group, L = cis or trans to X (i.e. X
defines geometry).
The general process:
[Co(CN)5H2O]2- k1
k-1
[Co(CN)5]2-
+ H2O
+ X-
[Co(CN)5X]3-
k2
[PtA2LX] + Y-
[PtA2LY]SN
2
+ X-

PtII mechanism is invariably SN
2
This is predictable:
Add Y-; it is easy! The 5
th position is open, there is no steric hindrance to the
approach of Y-.
There is strong evidence for the existence of a 5-coordinate intermediate, e.g.
There is usually a retention of configuration:
cis-[PtA2LX] + Y-cis-[PtA2LY] + X
- (100% retention)
trans-[PtA2LX] + Y- trans-[PtA2LY] + X
- (100% retention)
16.1 Mechanism
The SN2mechanism is nota one step process. The rate is independent of X, i.e. it is the
rate of bond formation, which is important, not the rate of bond breaking.
16.2. Kinetics of the reaction:
In general a 2-term rate law is observed:
Rate = k1[complex] + k2[complex][Y-]
1st order term 2
nd order term
16.3 Explanation of the two terms
XPt
Y-
Ni(CN)4]2-
+ excess CN-
H2O[Ni(CN)5]
3-
Evidence from Uv-Vis
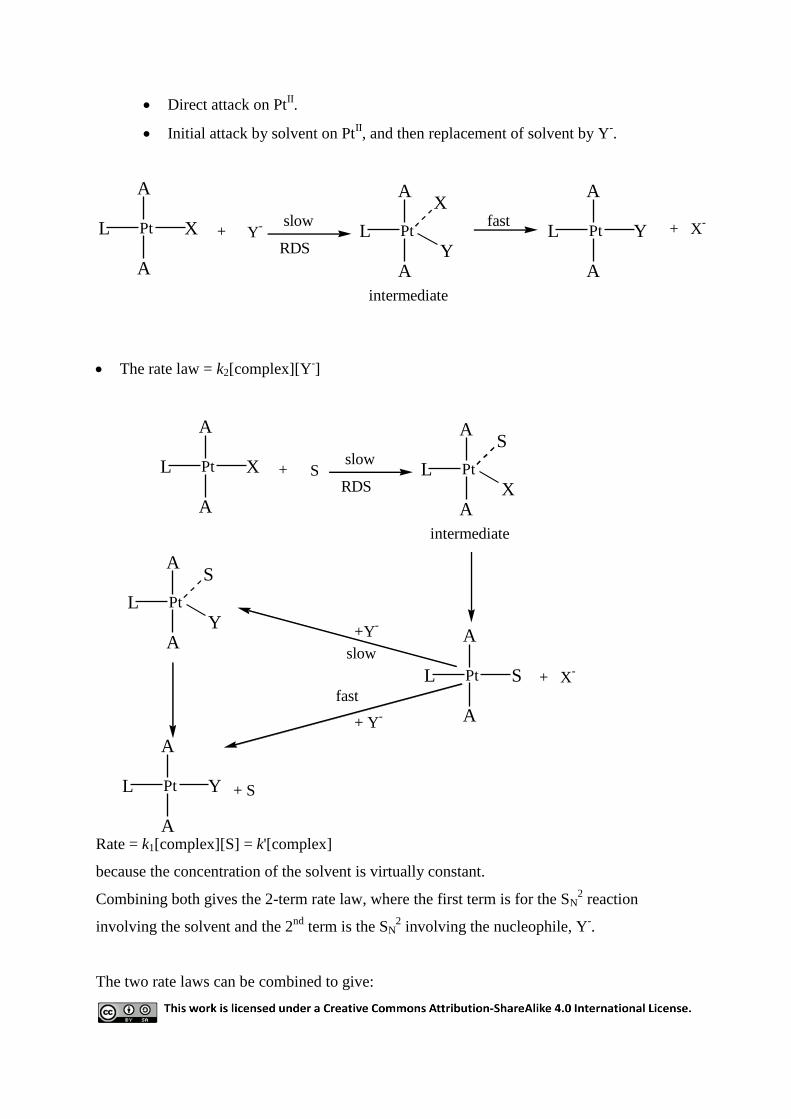
Direct attack on PtII.
Initial attack by solvent on PtII, and then replacement of solvent by Y
-.
The rate law = k2[complex][Y-]
Rate = k1[complex][S] = k'[complex]
because the concentration of the solvent is virtually constant.
Combining both gives the 2-term rate law, where the first term is for the SN2 reaction
involving the solvent and the 2nd
term is the SN2 involving the nucleophile, Y
-.
The two rate laws can be combined to give:
Pt
A
A
L X + Y- slow
RDSPtL
A
AY
X
Pt
A
A
L Yfast
+ X-
intermediate
Pt
A
A
L X + Sslow
RDSPtL
A
AX
S
Pt
A
A
L S + X-
intermediate
+Y-
slow
+ Y-
fast
Pt
A
A
L Y + S
PtL
A
AY
S

Rate = dt
complexd ][ = {k1 + k2[Y
-]}[complex]
Summary
Both reactions proceed via an associative process (A), involving a trigonal bipyramid
transition state. Chemical justification for this transition state includes:
Many five coordinate transition metal complexes are known, e.g., Fe(CO)5,
[CoL2(CO3)] +, [Ni(CN)5]
3-
ML3X complexes are sterically and electronically unsaturated and have space for Y
to coordinate.
Evidence
Rate law is consistent with associative mechanism
Charge on the metal centre - has no effect on the rate of reaction
Steric effect-significant increase in rate was observed for less hindered ligands; trans-
is faster than cis
C6H5->2-Me-C6H4
->2,4,6-Me3-C6H2
-
Note trans>cis for the substitution reaction
[Pt(PEt3)RCl] + Y- → [Pt(PEt3)RY] + Cl
-
16.4 The trans effect
It is observed that during the substitution reactions of square planar metal complexes, some
ligands preferentially direct the substitution trans to themselves. i.e., the choice of leaving
group is determined by the nature of ligand trans to it.
“The trans effect is defined as the effect of a coordinated ligand upon the rate of substitution
of ligands opposite to it” or
“The Trans effect can be defined as the effect of a ligand over rate of substitution of another
ligand positioned trans to it in the square planar complexes”.
Where 'T' is the trans directing group and „Nu‟ is the nucleophilic ligand which preferentially
substitutes the ligand 'X' which is trans to ligand 'T'.
For Pt(II) compounds the order of trans effect H2O ~ OH- ~ NH3 ~ NR3< Cl
- ~ Br
-< SCN
- ~ I
-
~ NO2- ~ C6H5
-< CH3
-< PR3 ~ AsR3 ~H
-< olefins ~ CO ~ CN
-.
trans-effect in synthesis
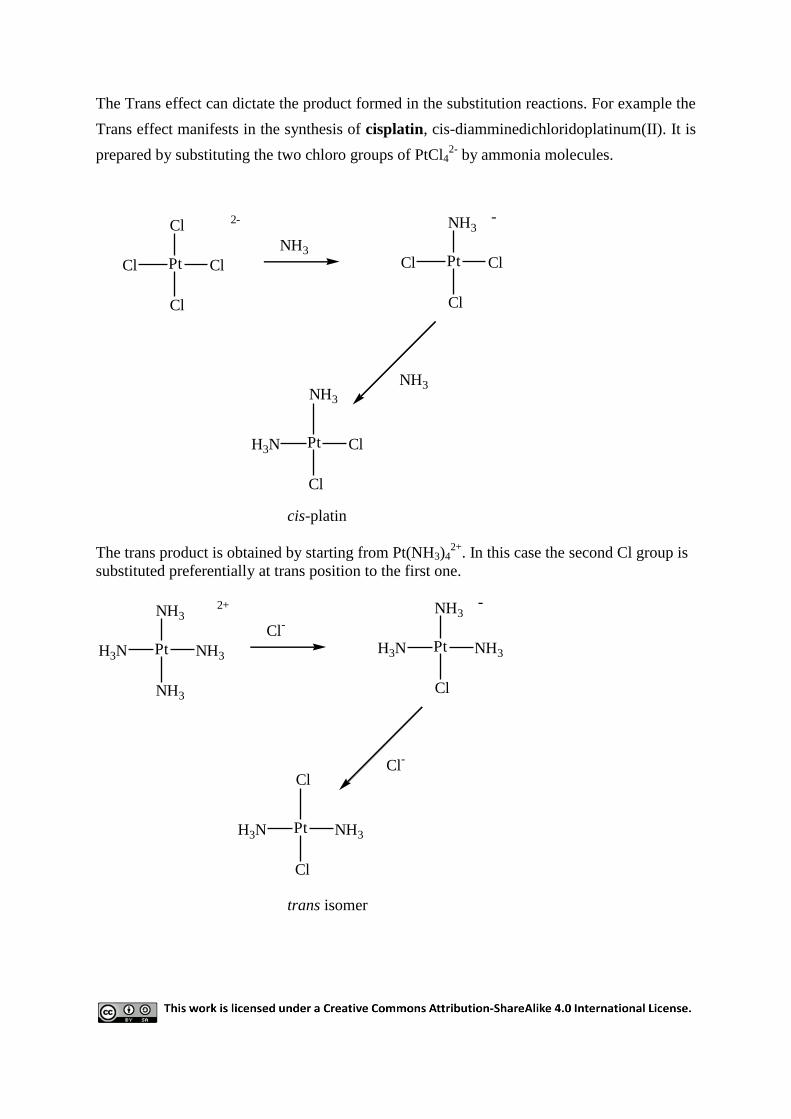
The Trans effect can dictate the product formed in the substitution reactions. For example the
Trans effect manifests in the synthesis of cisplatin, cis-diamminedichloridoplatinum(II). It is
prepared by substituting the two chloro groups of PtCl42-
by ammonia molecules.
Pt
Cl
Cl
Cl
Cl
NH3Pt
NH3
Cl
Cl
Cl
2- -
NH3
PtH3N
Cl
Cl
NH3
cis-platin
The trans product is obtained by starting from Pt(NH3)42+
. In this case the second Cl group is
substituted preferentially at trans position to the first one.
Pt
NH3
H3N
NH3
NH3
Cl-
Pt
NH3
H3N
Cl
NH3
2+ -
Cl-
PtH3N
Cl
NH3
Cl
trans isomer
Related Documents











