Adrenergic Antagonists The adrenergic antagonists (also called blockers or sympatholytic agents) bind to adrenoceptors but do not trigger the usual receptor-mediated intracellular effects. These drugs act by either reversibly or irreversibly attaching to the receptor, thus preventing its activation by endogenous catecholamines. These drugs termed as adrenergic receptor antagonists , which inhibit the interaction of norepinephrine, epinephrine, and other sympathomimetic drugs with a and b receptors.

Adrenergic Antagonists
Dec 28, 2015
pharmacology
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Adrenergic Antagonists
The adrenergic antagonists (also called blockers or sympatholytic agents) bind to adrenoceptors but do not trigger the usual receptor-mediated intracellular effects.
These drugs act by either reversibly or irreversibly attaching to the receptor, thus preventing its activation by endogenous catecholamines.
These drugs termed as adrenergic receptor antagonists, which inhibit the interaction of norepinephrine, epinephrine, and other sympathomimetic drugs with a and b receptors.

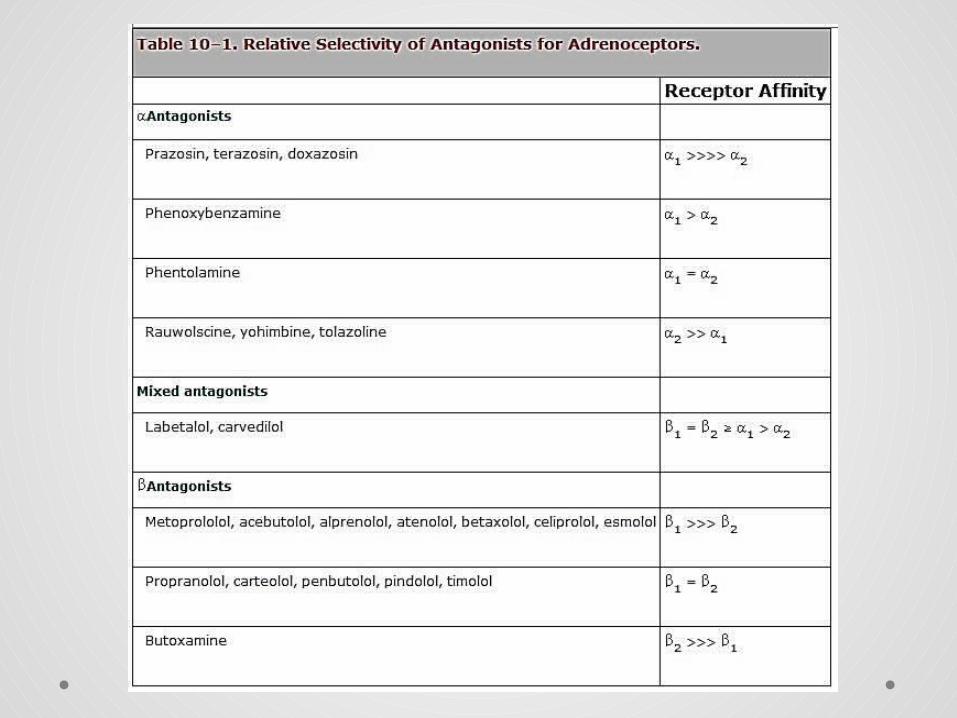
Classification of adrenoceptors Main pharmacological classification into α and β subtypes,
based originally on order of potency among agonists, later on selective antagonists.
Adrenoceptor subtypes:
• Two main α-receptor subtypes, α1 and α2, each divided into three further subtypes
• Three β-adrenoceptor subtypes (β1, β2, β3)
• All belong to the superfamily of G-protein-coupled receptors.
Second messengers:
• α1-receptors activate phospholipase C, producing inositol trisphosphate and diacylglycerol as second messengers
• α2-receptors inhibit adenylate cyclase, decreasing cAMP formation
• all types of β-receptor stimulate adenylyl cyclase.

The main effects of receptor activation are as follows.
• α1-receptors: vasoconstriction, relaxation of gastrointestinal smooth muscle, salivary secretion and hepatic glycogenolysis
• α2-receptors: inhibition of transmitter release (including noradrenaline and acetylcholine release from autonomic nerves), platelet aggregation, contraction of vascular smooth muscle, inhibition of insulin release
• β1-receptors: increased cardiac rate and force
• β2-receptors: bronchodilatation, vasodilatation, relaxation of visceral smooth muscle, hepatic glycogenolysis and muscle tremor
• β3-receptors: lipolysis
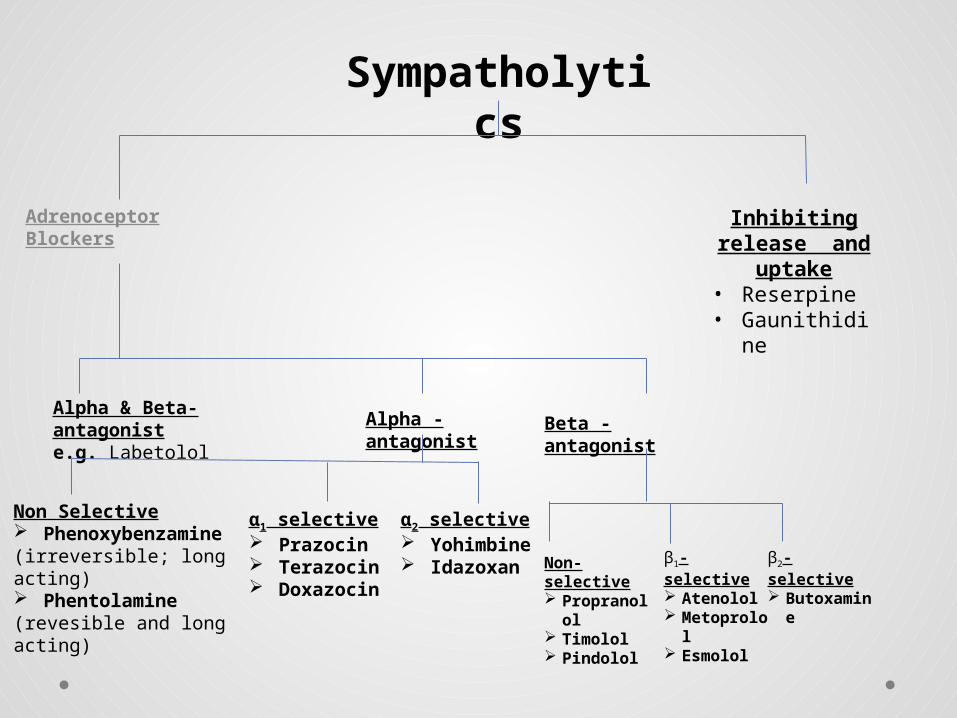
Sympatholytics
Alpha & Beta-antagoniste.g. Labetolol
Adrenoceptor Blockers
Alpha -antagonist
Non Selective Phenoxybenzamine(irreversible; long acting) Phentolamine(revesible and long acting)
α1 selective Prazocin Terazocin Doxazocin
α2 selective Yohimbin
e Idazoxan
Beta -antagonist
Non-selective Propranol
ol Timolol Pindolol
β1-selective Atenolol Metoprol
ol Esmolol
β2-selective Butoxami
ne
Inhibiting release and uptake
• Reserpine• Gaunithidine

CLINICAL PHARMACOLOGY OF THE ALPHA-RECEPTOR–BLOCKING DRUGS
1. Pheochromocytoma: This tumor is usually found in the adrenal medulla; it typically
releases a mixture of epinephrine and norepinephrine. Patients have many symptoms and signs of catecholamine
excess, including:• Intermittent or sustained hypertension• Headaches• Palpitations• Increased sweating Treatment:
Alpha-receptor antagonists are most useful in the preoperative management of patients with pheochromocytoma i.e., phenoxybenzamine and phentolamine

2. Hypertensive Emergencies: The alpha-adrenoceptor antagonist drugs have limited
application in the management of hypertensive emergencies, although labetalol has been used.
In theory, alpha-adrenoceptor antagonists are most useful when increased blood pressure reflects excess circulating concentrations of agonists,
• In pheochromocytoma• overdosage of sympathomimetic drugs• clonidine withdrawal. However, other drugs are generally preferable.

3. Chronic Hypertension
Members of the prazosin family of alpha1-selective antagonists are efficacious drugs in the treatment of mild to moderate systemic hypertension.
4. Local Vasoconstrictor Excess Phentolamine has been used to reverse the intense local
vasoconstriction caused by infiltration of agonists (e.g, norepinephrine) into subcutaneous tissue during intended intravenous administration. The antagonist is administered by local infiltration into the ischemic tissue.
5. Urinary ObstructionSeveral alpha1-receptor antagonists in patients with BPH(Benign prostatic hyperplasia). E.g. Prazosin, doxazosin, and terazosin are efficacious.

CLINICAL PHARMACOLOGY OF THE BETA-RECEPTOR–BLOCKING DRUGS
1. Hypertension: The beta-adrenoceptor-blocking drugs have proved to be
effective and well tolerated in hypertension. The drug is often used with either a diuretic or a vasodilator.
2. Ischemic Heart Disease: Beta-adrenoceptor blockers reduce the frequency of anginal
episodes and improve exercise tolerance in many patients with angina.
These actions relate to the blockade of beta-cardiac receptors, resulting in decreased cardiac work and reduction in oxygen demand.

3. Cardiac Arrhythmias: Beta antagonists are often effective in the treatment of both
supraventricular and ventricular arrhythmias.
4. Heart Failure: antagonists—metoprolol, bisoprolol, and carvedilol—are
effective in treating chronic heart failure in selected patients.
5. Glaucoma : Systemic administration of beta-blocking drugs was found to
reduce intraocular pressure in patients with glaucoma. It was found that topical administration also reduces intra-
ocular pressure. The mechanism appears to involve reduced production of
aqueous humor by the ciliary body, which is physiologically activated by cAMP.
E.g, Timolol, Betaxolol, carteolol, levobunolol, and metipranolol are approved for the treatment of glaucoma.

6. Hyperthyroidism: Excessive catecholamine action is an important aspect of the
pathophysiology of hyperthyroidism. They causes inhibition of peripheral conversion of thyroxine
to tri-iodothyronine. Propranolol has been used extensively in patients with
thyroid storm (severe hyperthyroidism). It is used cautiously in patients with this condition to control
supraventricular tachycardias that often precipitate heart failure.
7. Neurologic Diseases: Propranolol reduces the frequency and intensity of migraine
headache. Propranolol may contribute to the symptomatic treatment of
alcohol withdrawal in some patients.

PREPARATIONS AVAILABLE
1.Alpha Blockers:• Alfuzosin (Uroxatral)
Oral: 10 mg tablets (extended-release)
• Phenoxybenzamine (Dibenzyline)
Oral: 10 mg capsules• Phentolamine (generic, Regitine)
Parenteral: 5 mg/vial for injection• Tamsulosin (Flomax)
Oral: 0.4 mg capsule• Terazosin (generic, Hytrin)
Oral: 1, 2, 5, 10 mg tablets, capsules

2. Beta Blockers:• Atenolol (generic, Tenormin)
Oral: 25, 50, 100 mg tablets
Parenteral: 0.5 mg/mL for IV injection• Betaxolol
Oral (Kerlone): 10, 20 mg tablets
Ophthalmic (generic, Betoptic): 0.25%, 0.5% drops

Some Prototype Drugs:Alpha-Adrenergic Antagonists:1. Phentolamine:• An imidazoline derivative, is a potent competitive antagonist at both alpha1
and alpha2 receptors .
• Phentolamine causes a reduction in peripheral resistance through blockade of alpha1 receptors and possibly alpha2 receptors on vascular smooth muscle.
• The cardiac stimulation induced by phentolamine is due to sympathetic stimulation of the heart resulting from baroreflex mechanisms.
• Phentolamine has limited absorption after oral administration. Its pharmacokinetic properties are not well known;
• It may reach peak concentrations within an hour after oral administration and • It has a half-life of 5–7 hours. • The principal adverse effects are related to cardiac stimulation, which may
cause severe tachycardia, arrhythmias, and myocardial ischemia, nasal congestion, and headache
• Phentolamine has been used in the treatment of pheochromocytoma

2. Prazosin• It is a piperazinyl quinazoline effective in the management of
hypertension.
• It is highly selective for alpha1 receptors and typically 1000-fold less potent at alpha2 receptors.
• Prazosin is extensively metabolized in humans; because of metabolic degradation by the liver, only about 50% of the drug is available after oral administration.
• The half-life is normally about 3 hours.
3. Terazosin:• It is another reversible alpha1-selective antagonist that is effective in
hypertension.• it is also approved for use in men with urinary symptoms due to
benign prostatic hyperplasia (BPH). • Terazosin has high bioavailability but is extensively metabolized in
the liver, with only a small fraction of unchanged drug excreted in the urine.
• The half-life of terazosin is 9–12 hours.

Beta-Adrenergic Antagonists:
1. Propranolol : Propranolol is highly lipophilic and is almost completely absorbed
after oral administration. It is metabolized by the liver during its first passage through the
portal circulation; on average, only about 25% reaches the systemic circulation.
Propranolol has a large volume of distribution (4 liters/kg) and readily enters the CNS.
Approximately 90% of the drug in the circulation is bound to plasma proteins.
It is extensively metabolized, with most metabolites appearing in the urine.

Therapeutic Uses:• For the treatment of :
o Hypertension and anginao The prophylaxis of migraineo Myocardial infarctiono Pheochromocytoma
Drug interaction: It induce bronchospasm so contra-indicated in asthma.
It also may delay recovery from insulin-induced hypoglycemia so should be used with great caution in patients with diabetes.
Propranolol and Ca2+ channel blockers have additive effects on the cardiac conducting system.
Drugs such as phenytoin, rifampin, and phenobarbital, as well as smoking, induce hepatic biotransformation enzymes and may decrease plasma concentrations of propranolol.
Cimetidine and hydralazine may increase the bioavailability of agents such as propranolol and metoprolol.

2. Atenolol : Atenolol (TENORMIN, others) is a beta1-selective antagonist.
Atenolol is very hydrophilic and appears to penetrate the CNS only to a limited extent.
Its half-life is somewhat longer than that of metoprolol. Atenolol is incompletely absorbed (about 50%), but most of
the absorbed dose reaches the systemic circulation. The drug is excreted largely unchanged in the urine, and the
elimination half-life is about 5 to 8 hours. The drug accumulates in patients with renal failure, and
dosage should be adjusted for patients whose creatinine clearance is less than 35 ml/minute.

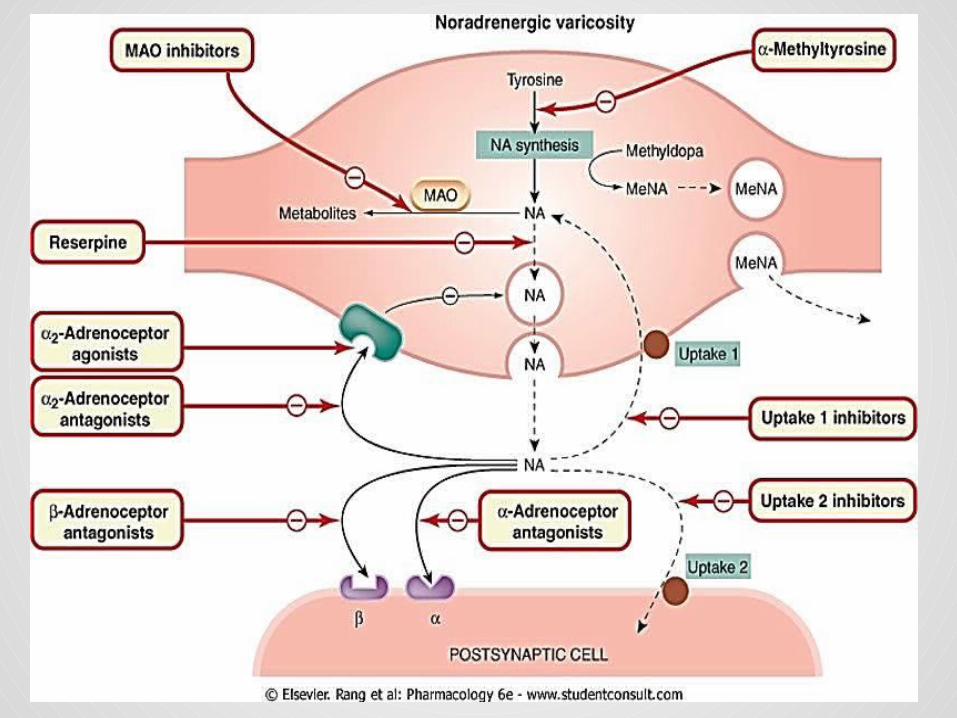
Indirect acting sympatholytics:
1. Reserpine: Reserpine is an alkaloid from the shrub Rauwolfia, which has
been used in India for centuries for the treatment of mental dis orders.
At very low concentration, blocks the transport of noradrenaline and other amines into synaptic vesicles, by blocking the vesicular monoamine transporter.
It is now used only experimentally, but was at one time used as an antihypertensive drug.
Its central effects, especially depression, which probably result from impairment of noradrenergic and 5-HT-mediated transmission in the brain are a serious disadvantage.
It has a slow onset, a long duration of action, and effects that persist for many days after discontinuation.

2. Guanethidine : It was first discovered in the mid-1950s It inhibits the release of noradrenaline from sympathetic nerve
terminals. It has little effect on the adrenal medulla. Guanethidine is also concentrated in synaptic vesicles by means
of the vesicular transporter, possibly interfering with their ability to undergo exocytosis, and also displacing noradrenaline.
It causes a gradual and long-lasting depletion of noradrenaline in sympathetic nerve endings, similar to the effect of reserpine.
Given in large doses, it causes structural damage to noradrenergic neurons.
they produce severe side effects associated with the loss of sympathetic reflexes.
The most troublesome are postural hypotension, diarrhea, nasal congestion and failure of ejaculation.

REFRENCES Pharmacology by Rang and Dale Katzung Pharmacology Tenth Ed GOODMAN & GILMAN'S THE
PHARMACOLOGICAL BASIS OF THERAPEUTICS - 11th Ed. (2006)
Lippincott's Illustrated Reviews Pharmacology, 4th Edition

THANK YOU
Related Documents











